TRPV1 Activation Exacerbates Hypoxia/Reoxygenation-Induced Apoptosis in H9C2 Cells via Calcium Overload and Mitochondrial Dysfunction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. Results
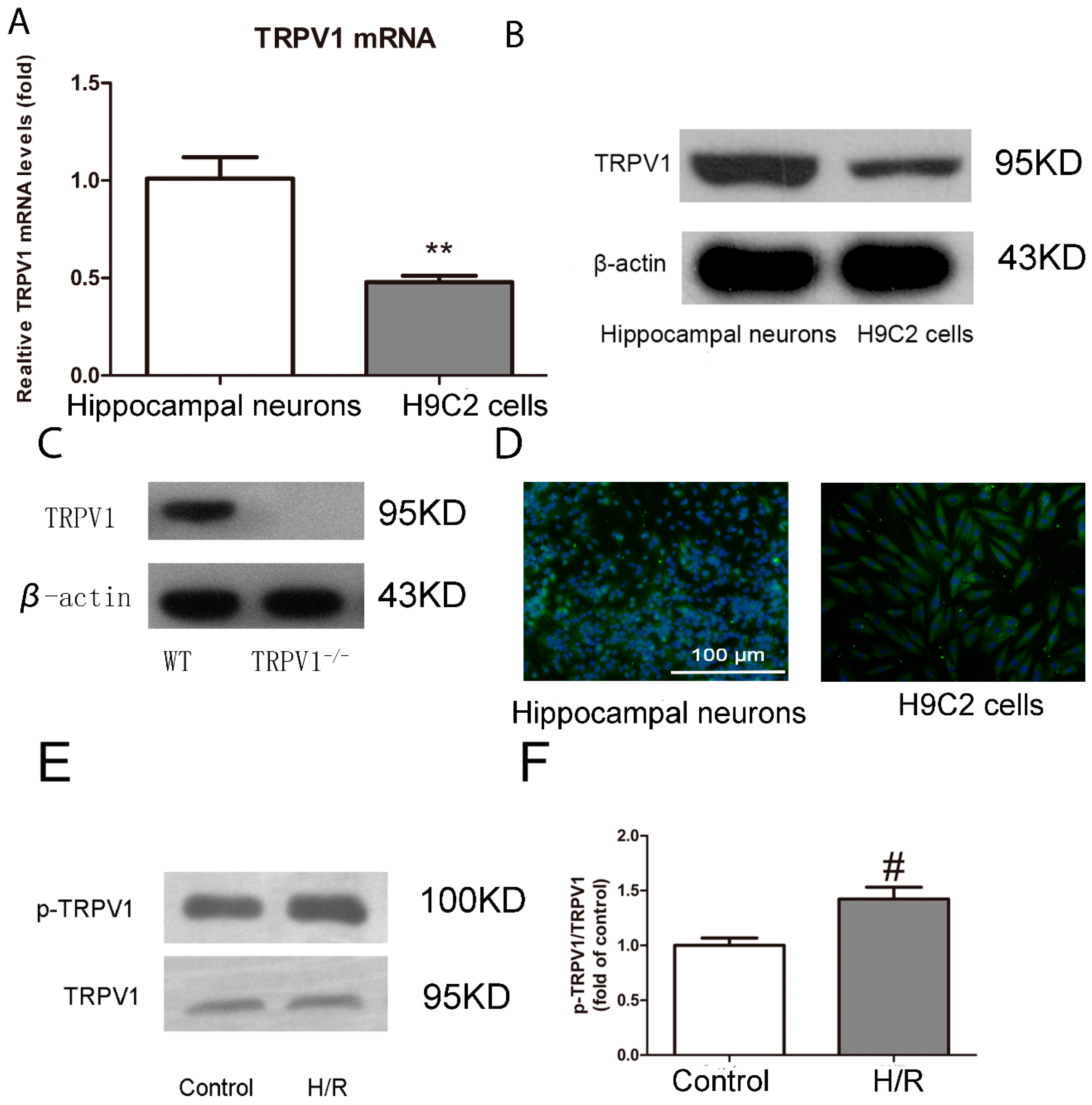
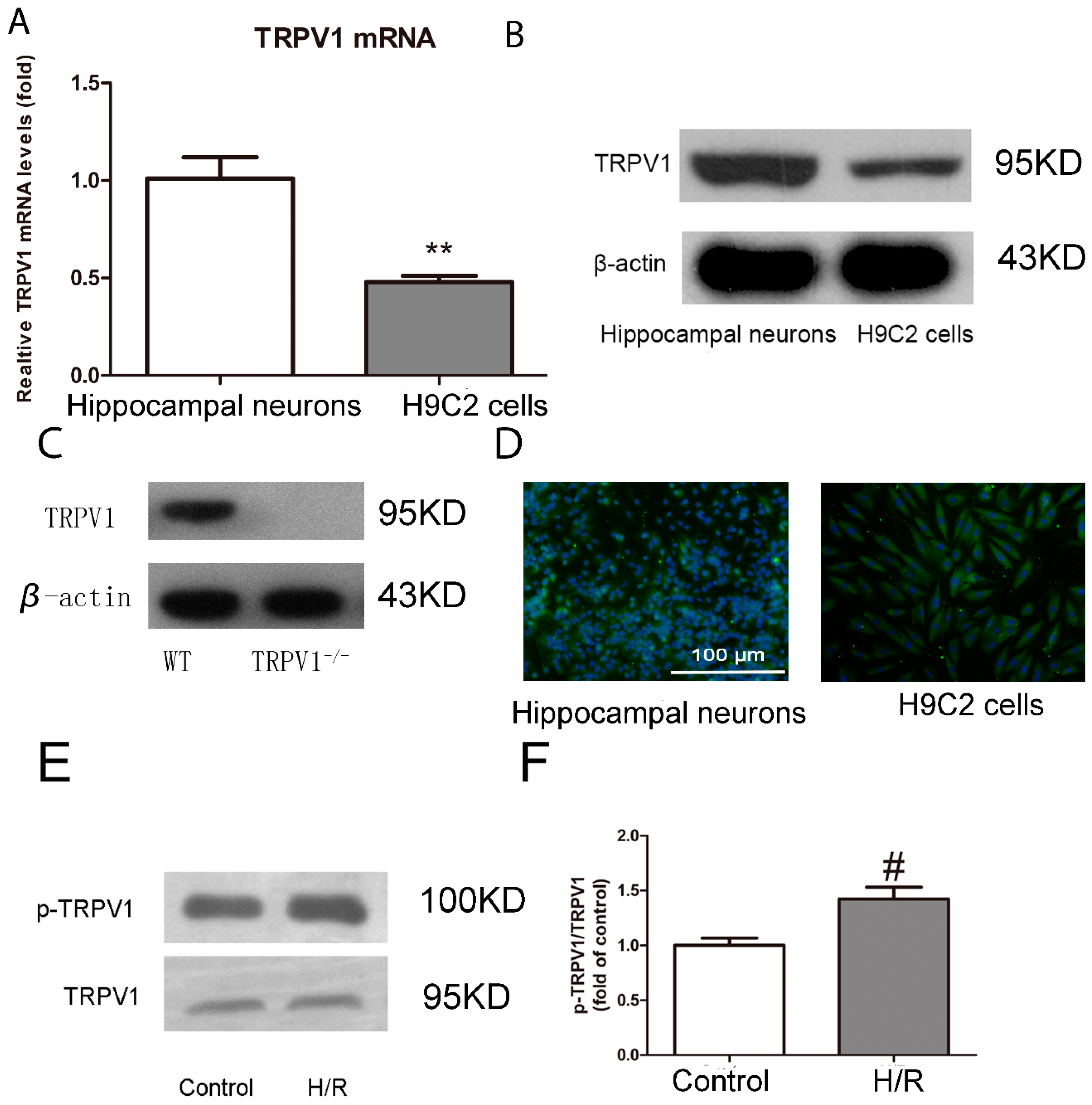
2.1.1. TRPV1 Is Expressed on H9C2 Cells and Activated during H/R
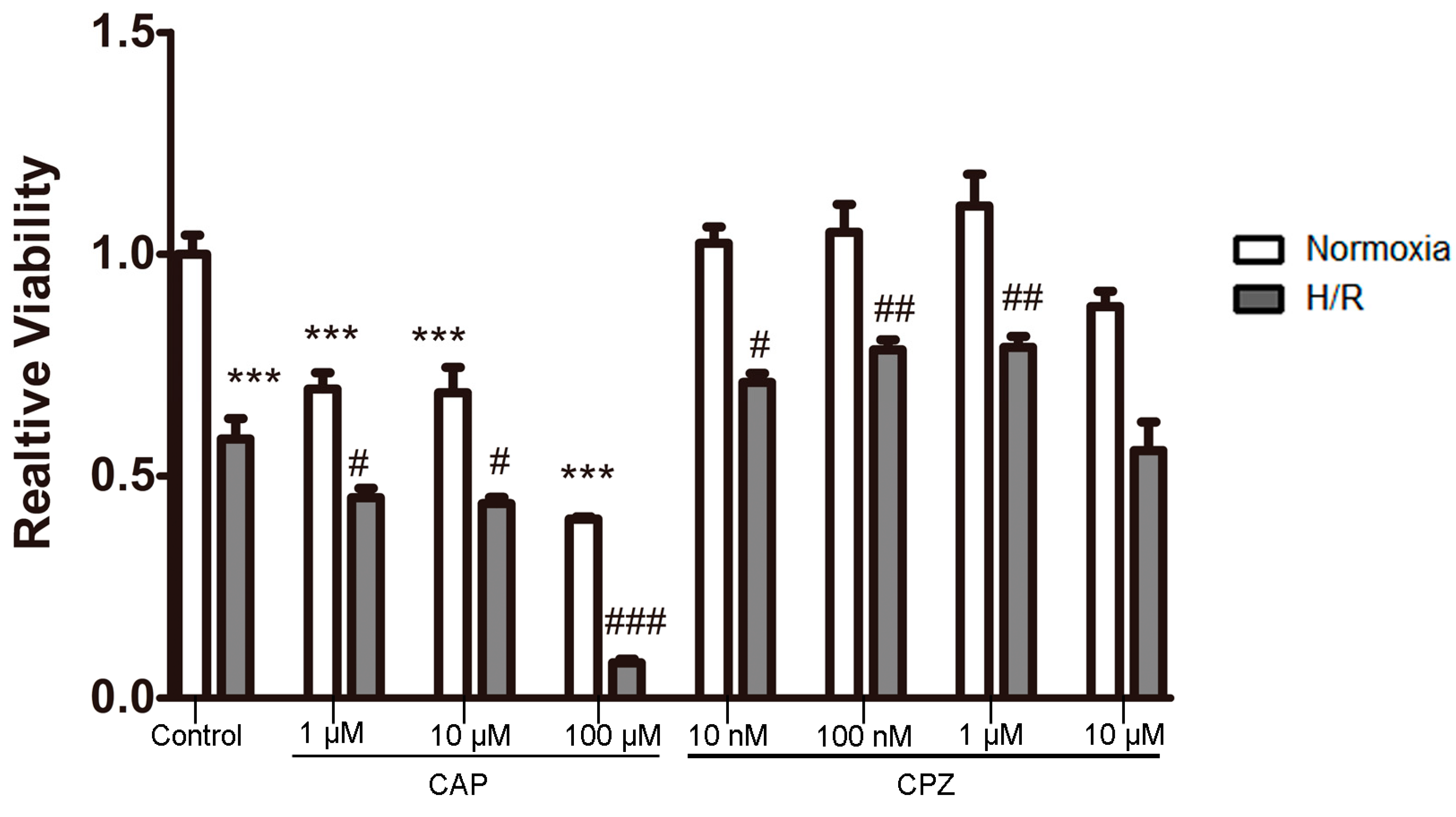
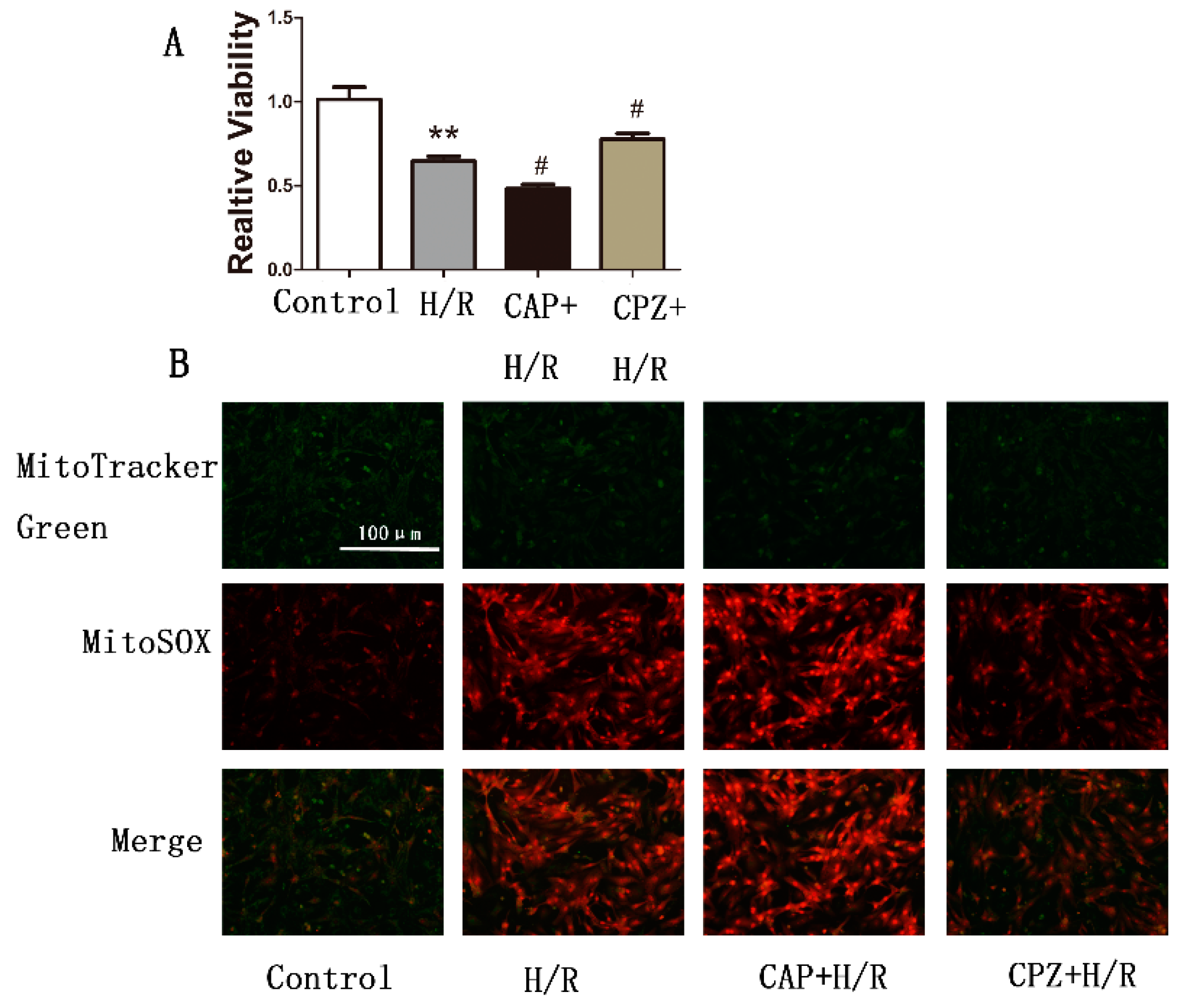
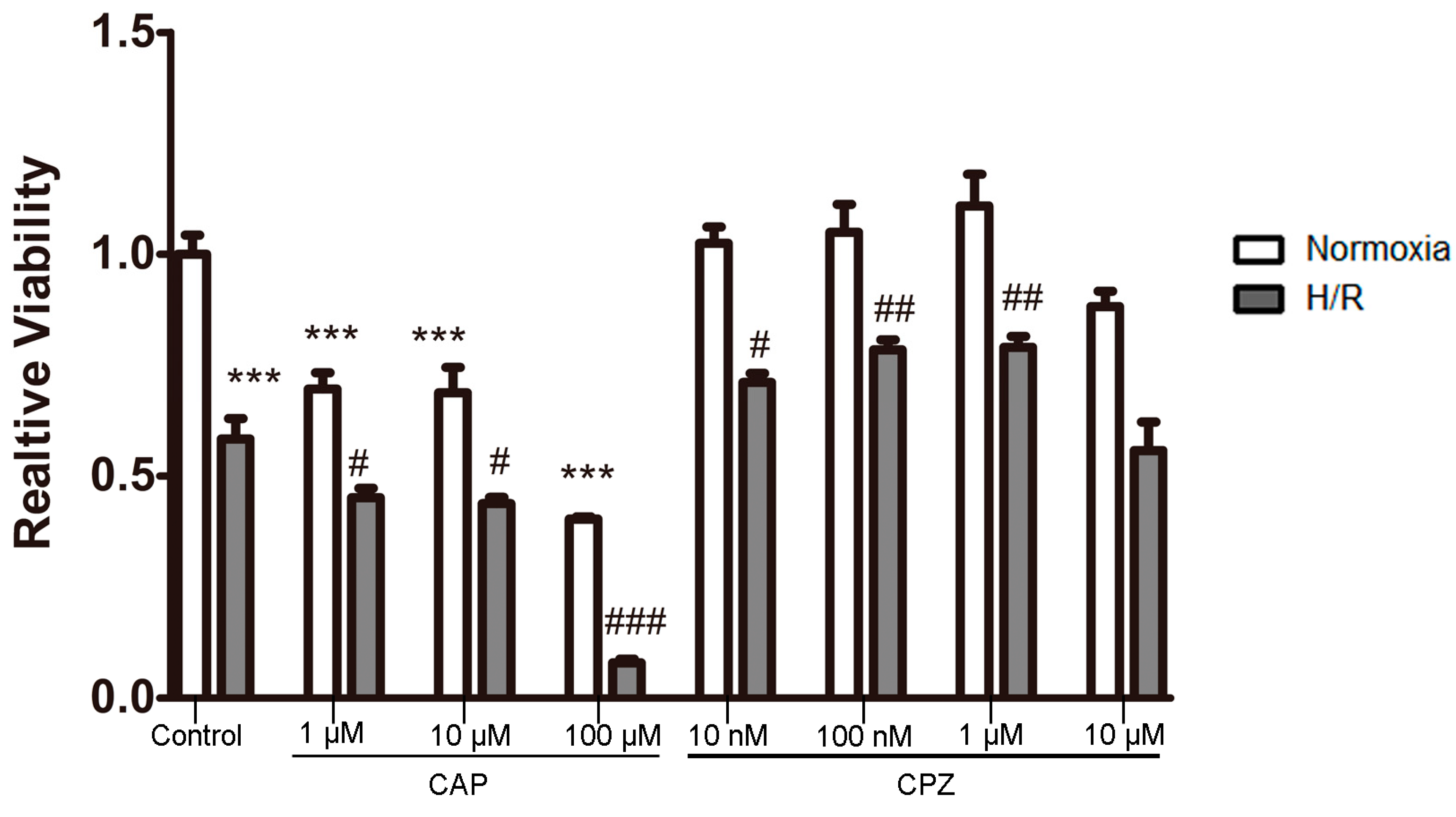
2.1.2. Activation of TRPV1 Leads to the Loss of Cell Viability
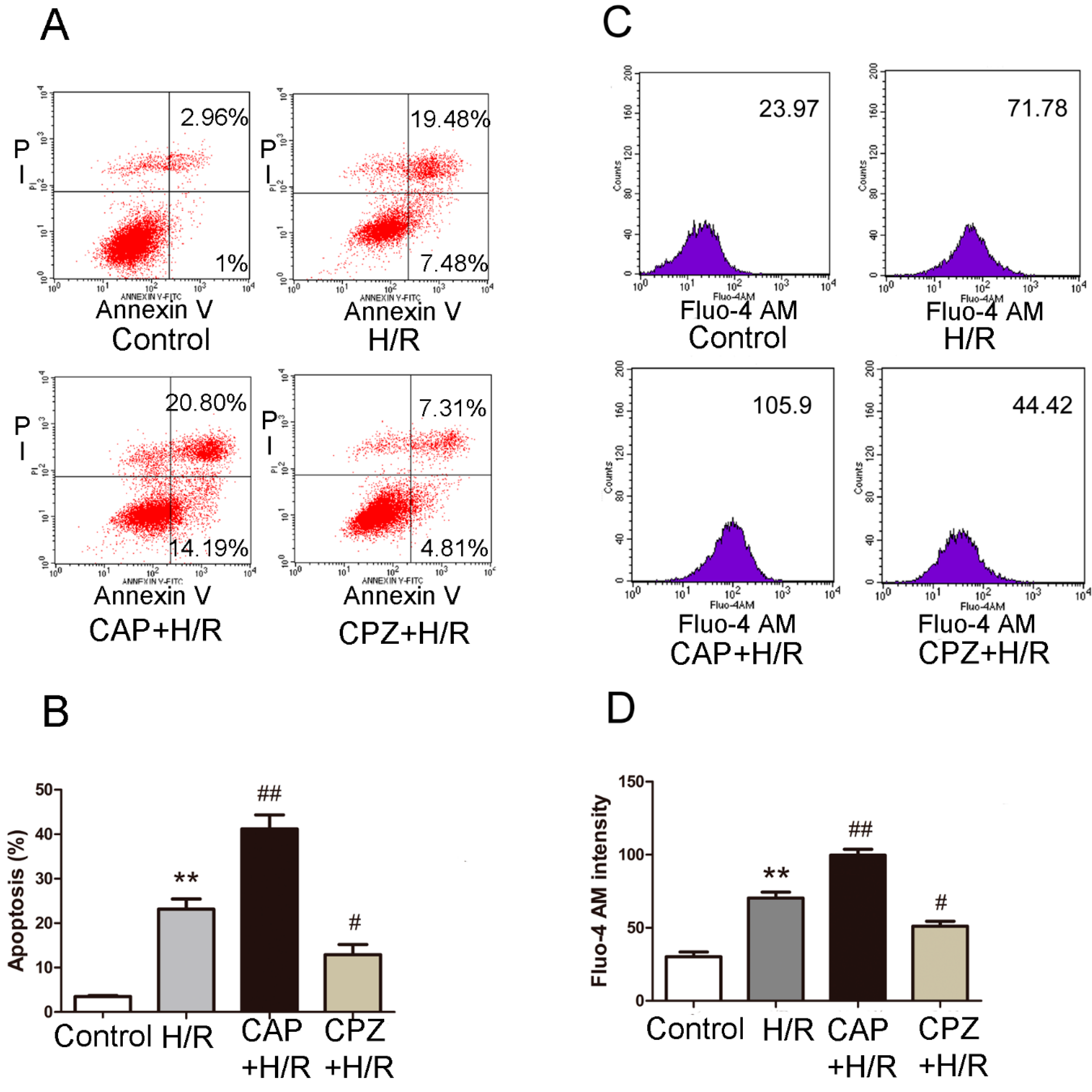
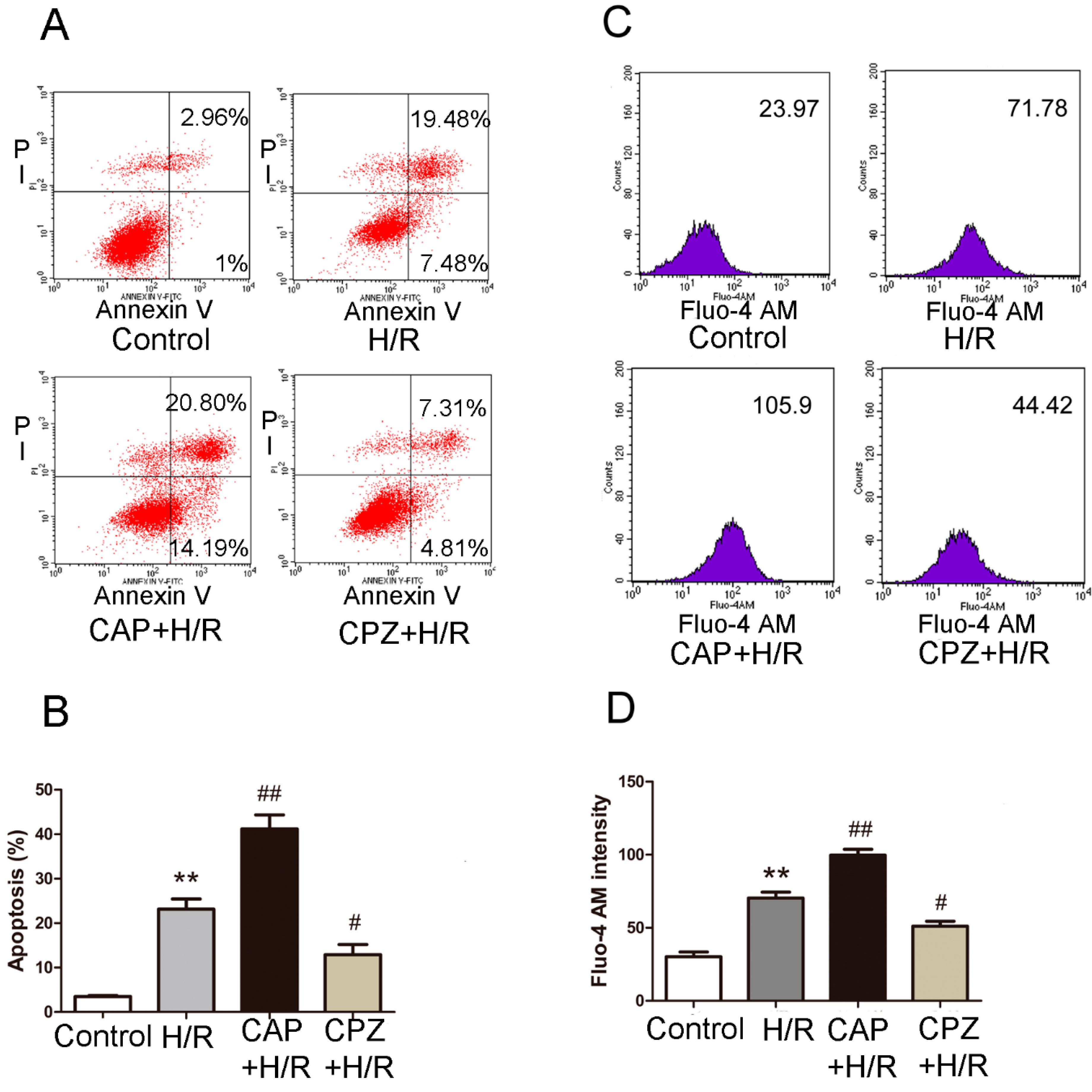
2.1.3. Activation of TRPV1 Induces Apoptosis and Elevates Intracellular Calcium Levels Following H/R
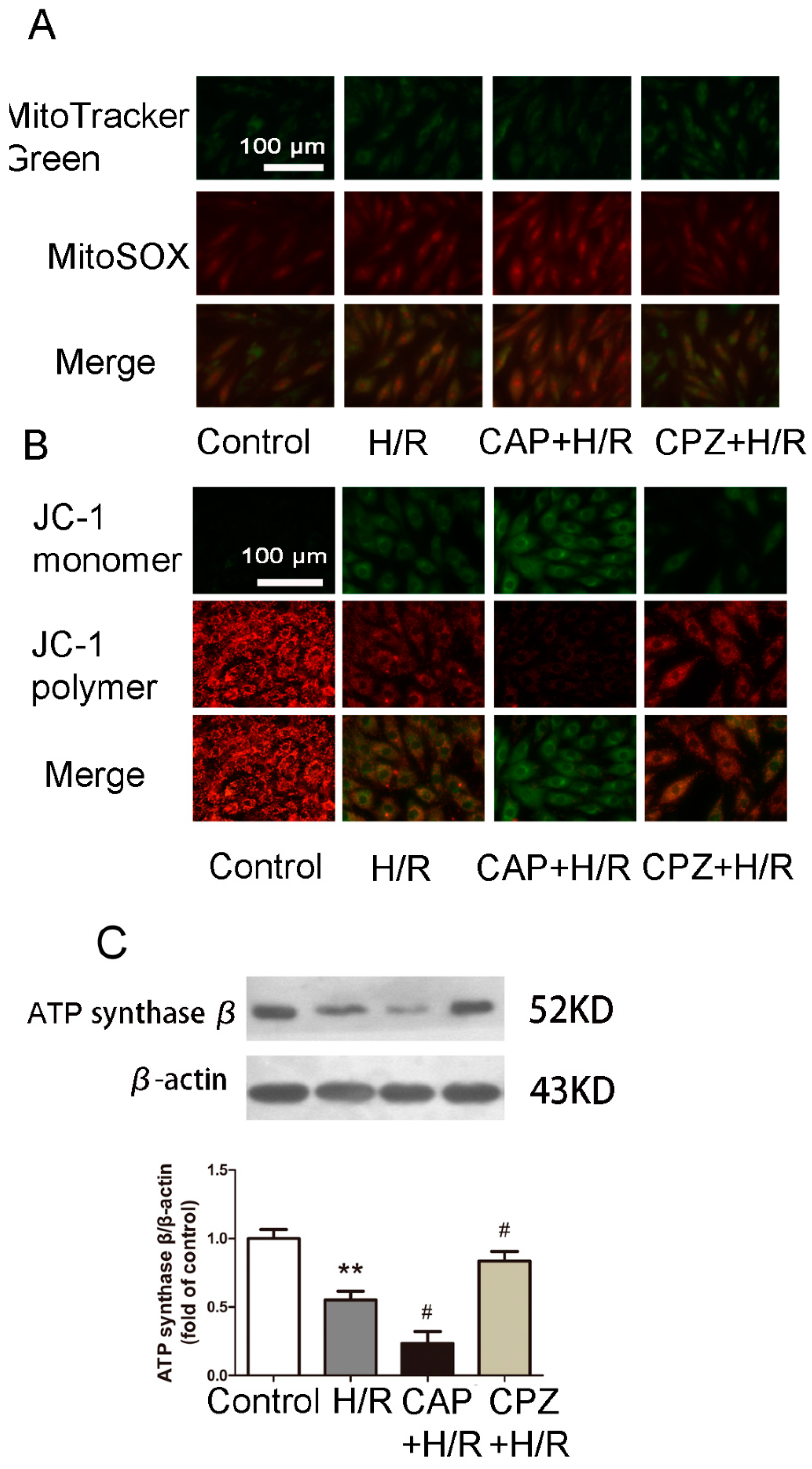
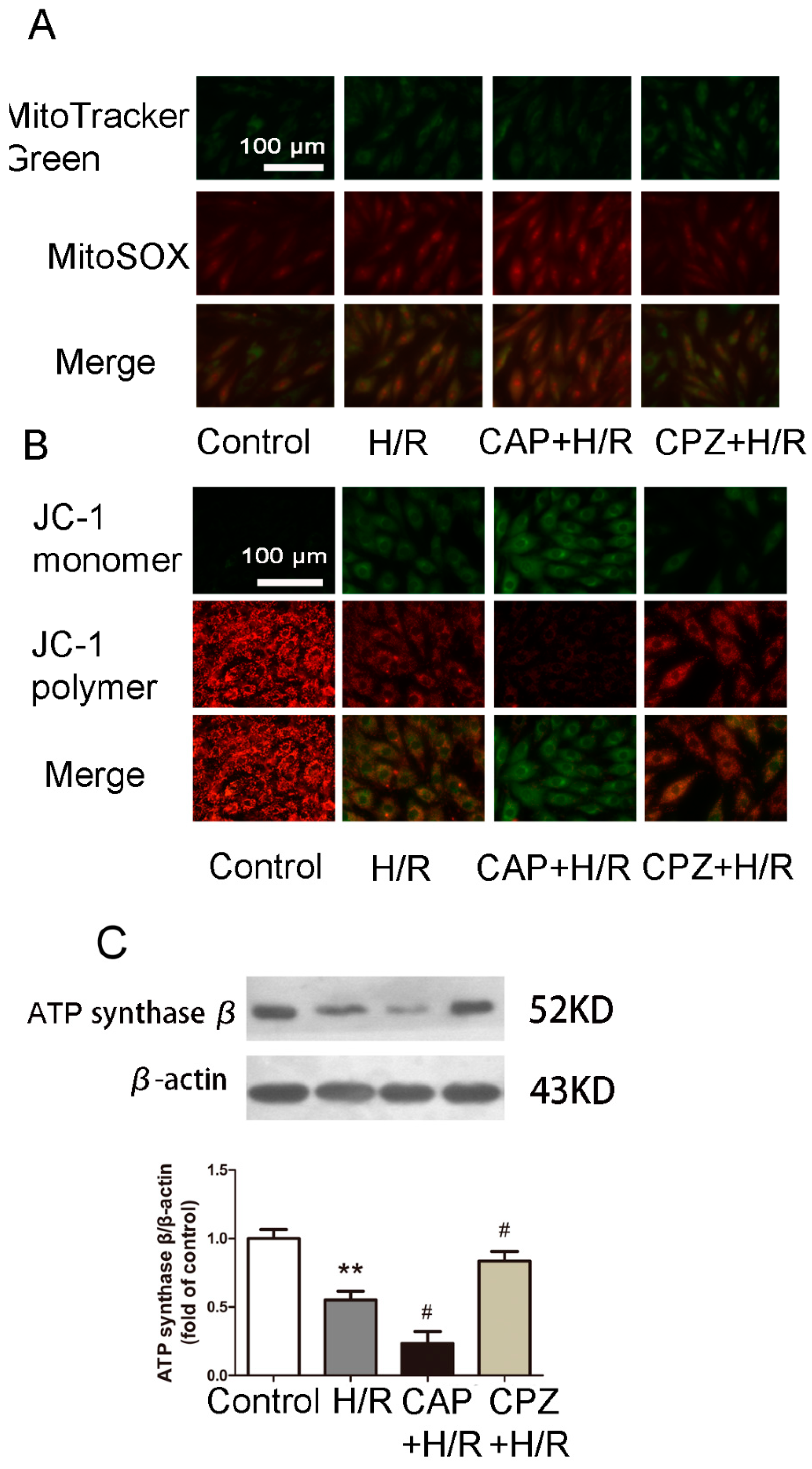
2.1.4. Activation of TRPV1 Induces Mitochondrial Superoxide Production and Mitochondrial Membrane Depolarization
2.1.5. Activation of TRPV1 Inhibited Mitochondrial Biogenesis
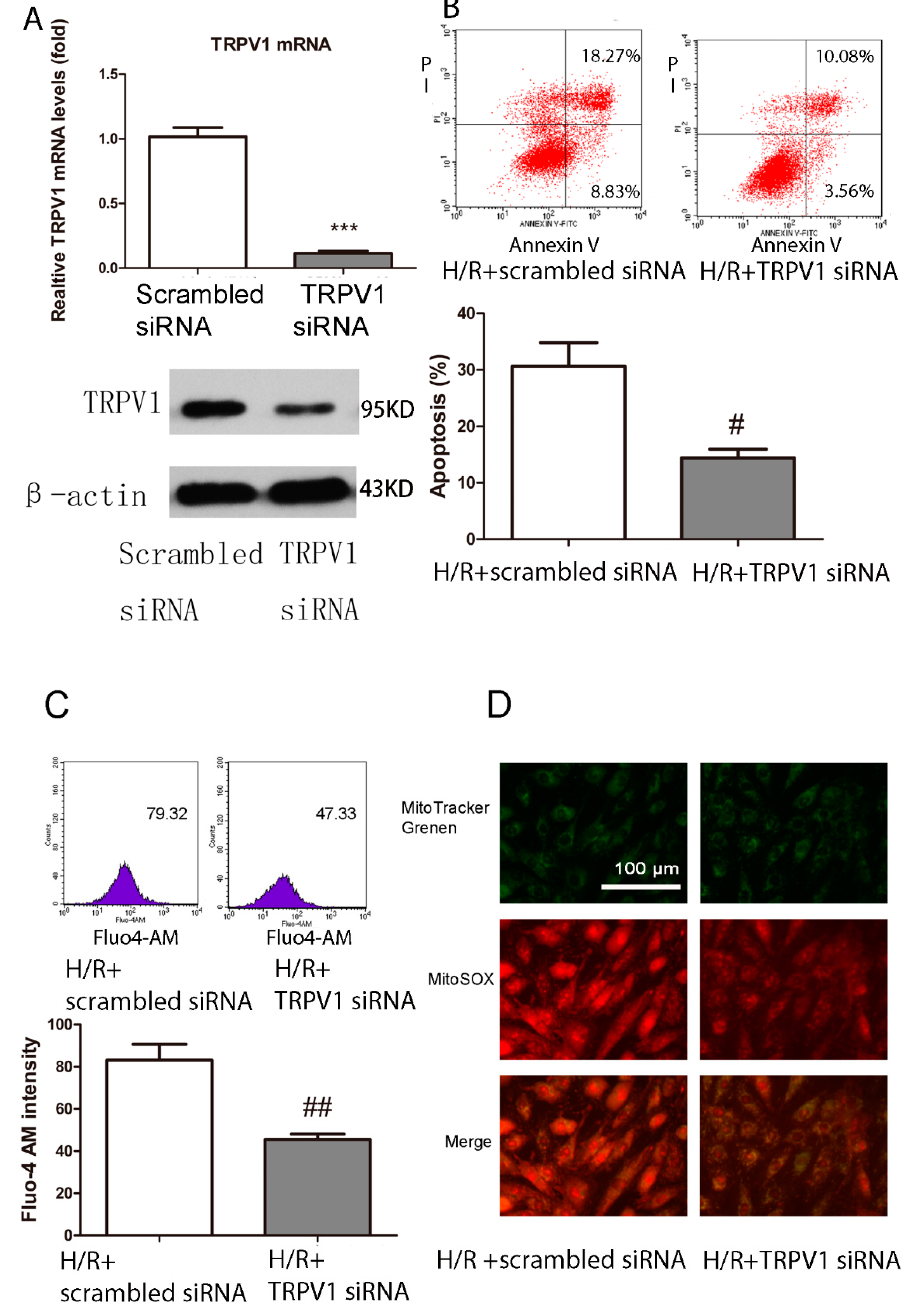
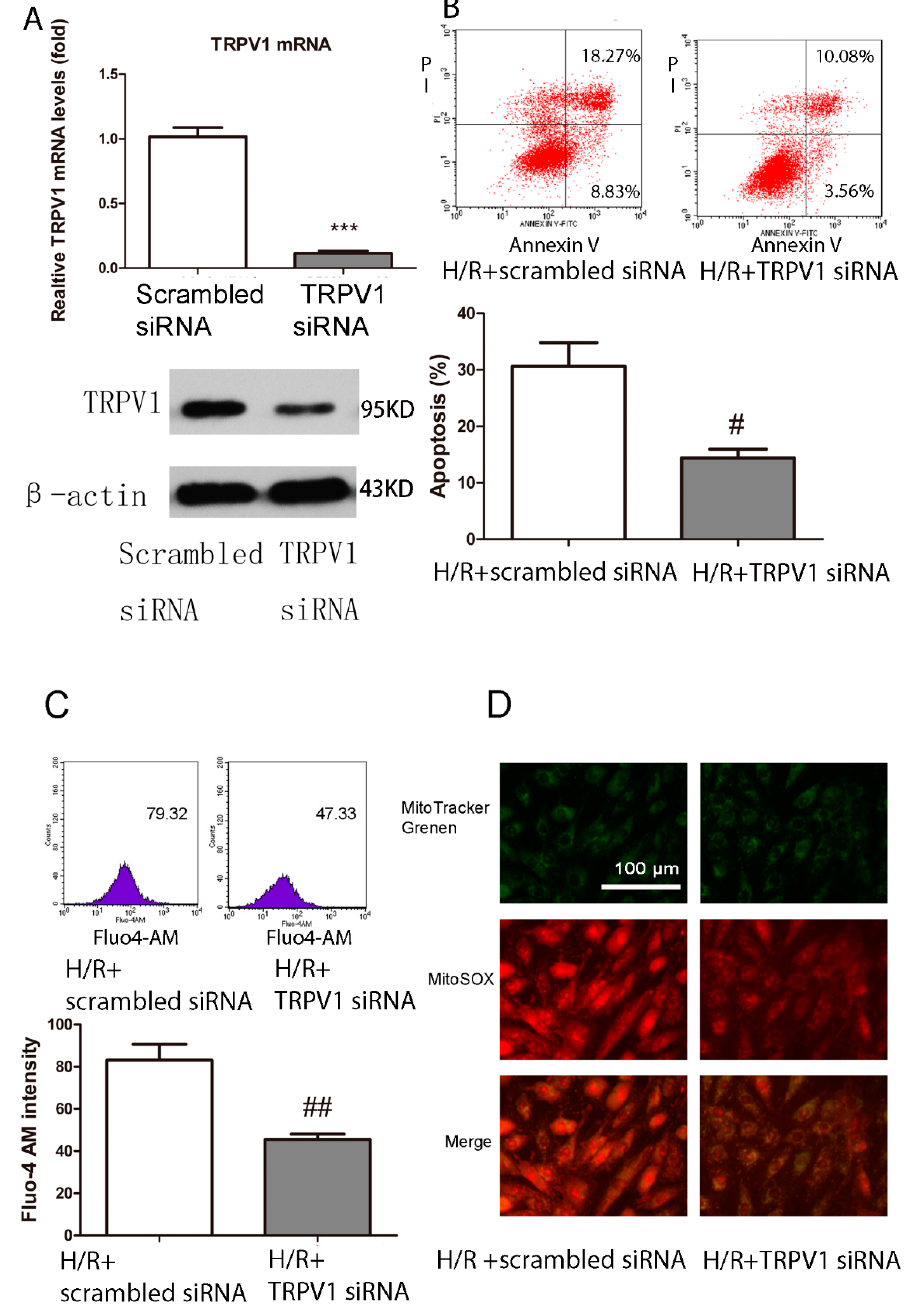
2.1.6. Knockdown of TRPV1 by siRNA Inhibits Apoptosis and Improves Mitochondrial Function
2.1.7. TRPV1 Activation Reduces Cell Viability and Increases Mitochondrial Superoxide Production in Primary Cardiomyocytes during H/R
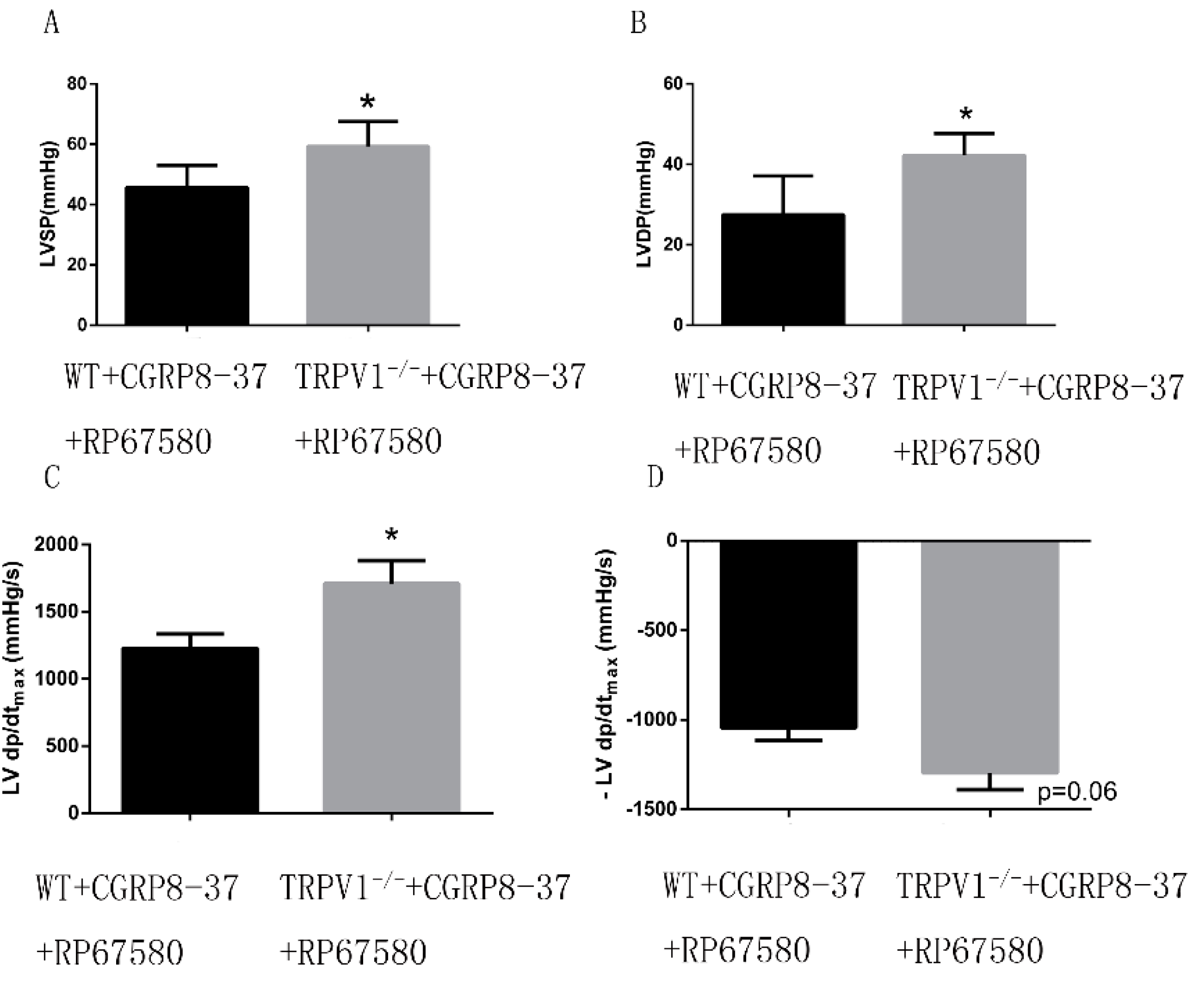
2.1.8. Improved Cardiac Function in ex Vivo TRPV1−/− Hearts in the Presence of both CGRP8–37 and RP67580
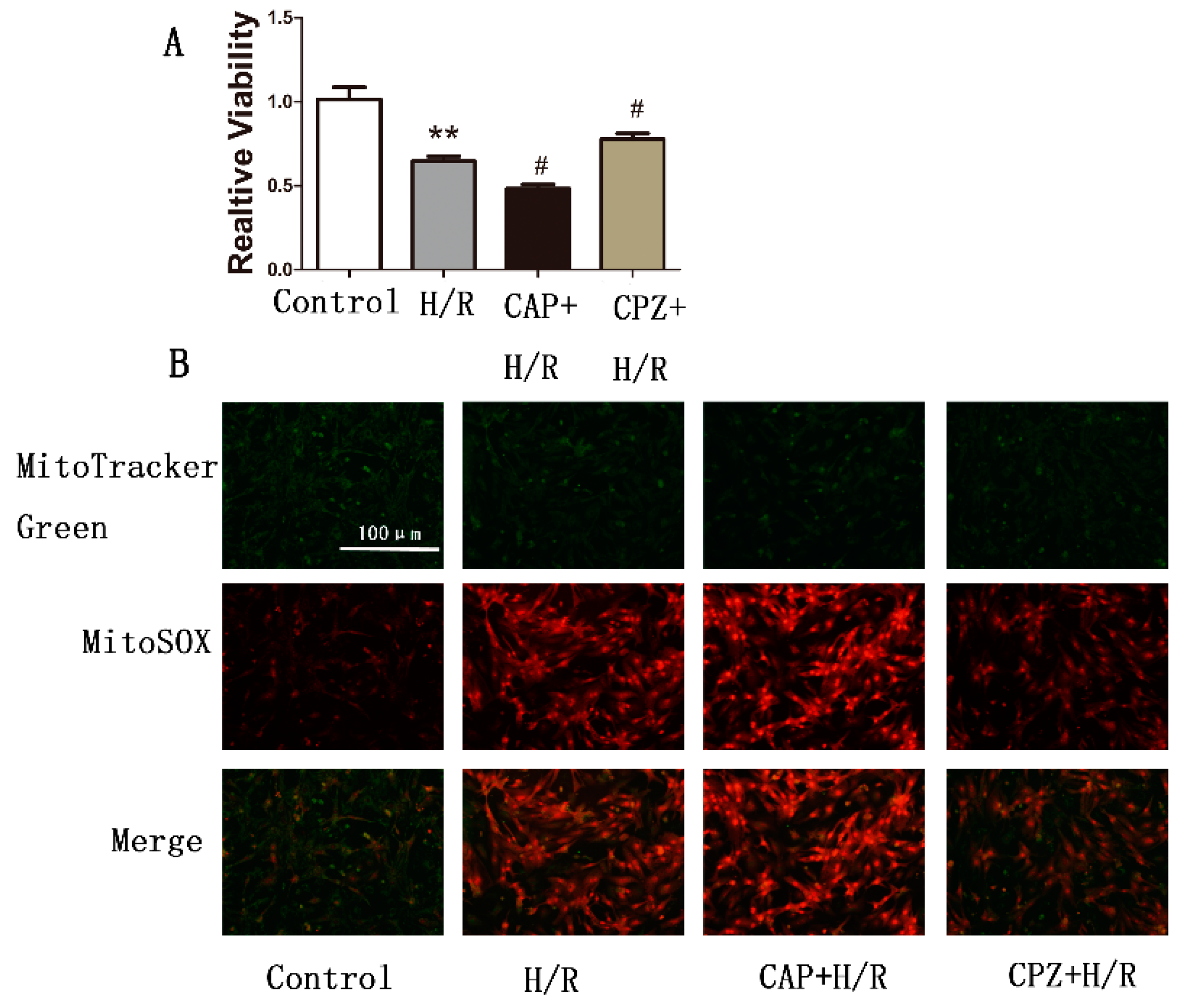
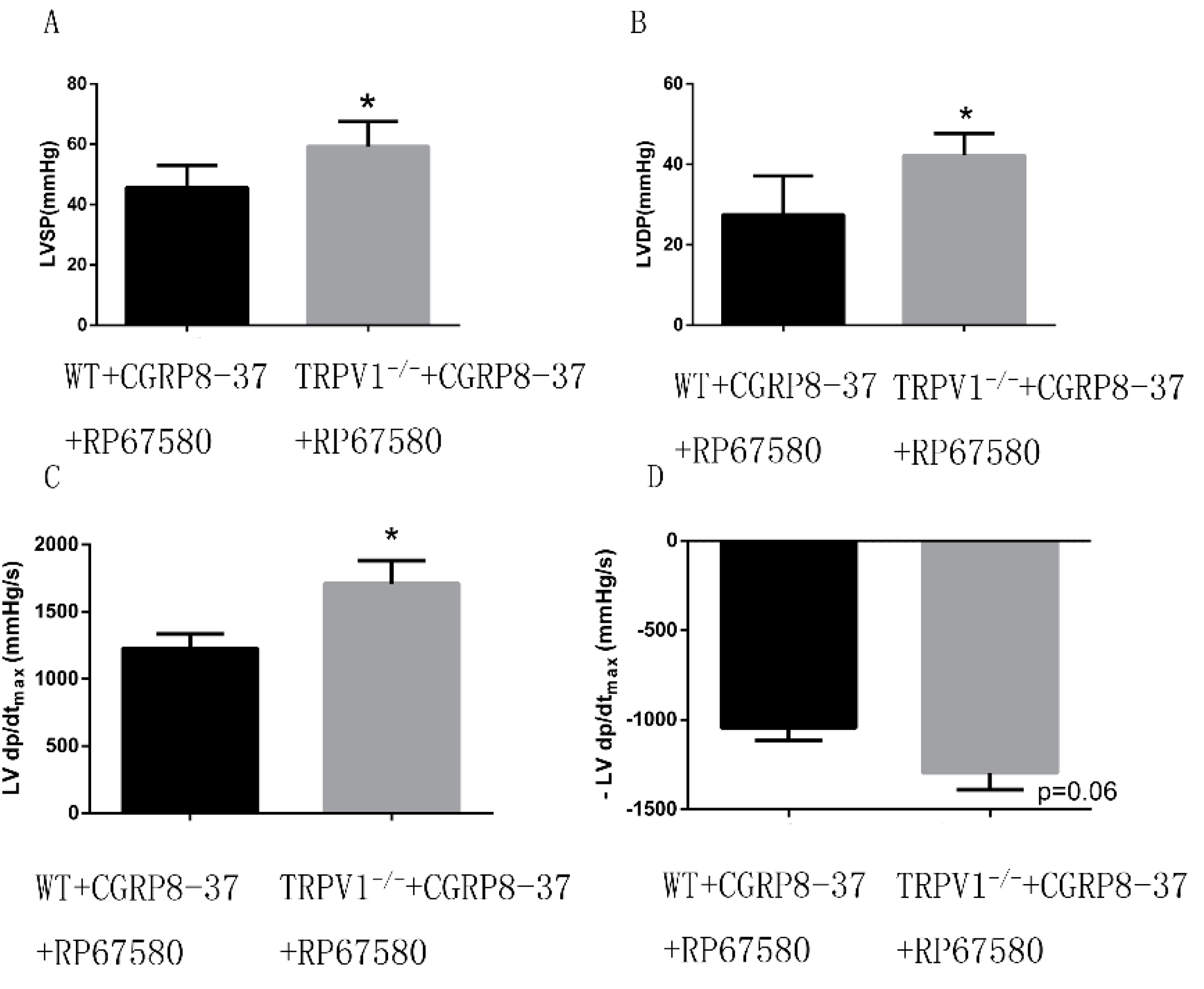
2.2. Discussion
3. Experimental Section
3.1. Reagents
3.2. Cell Culture
3.3. Hypoxia/Reoxygenation
3.4. Cell Viability
3.5. Cell Apoptosis
3.6. RT-PCR
3.7. Western Blot Analysis
3.8. Immunofluorescence Analysis
3.9. Intracellular Calcium Level Measurement
3.10. Determination of Mitochondrial Superoxide Production
3.11. Assessment of Mitochondrial Membrane Potential
3.12. siRNA Transfection
3.13. Animal Preparation
3.14. Langendorff Heart Preparation and Measurements of Cardiac Function
3.15. Statistical Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Caterina, M.J.; Schumacher, M.A.; Tominaga, M.; Rosen, T.A.; Levine, J.D.; Julius, D. The capsaicin receptor: A heat-activated ion channel in the pain pathway. Nature 1997, 389, 816–824. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; He, E.Y.; Scott, G.I.; Ren, J. α,β-Unsaturated aldehyde pollutant acrolein suppresses cardiomyocyte contractile function: Role of TRPV1 and oxidative stress. Environ. Toxicol. 2013. [Google Scholar] [CrossRef]
- Pei, Z.; Zhuang, Z.; Sang, H.; Wu, Z.; Meng, R.; He, E.Y.; Scott, G.I.; Maris, J.R.; Li, R.; Ren, J. α,β-Unsaturated aldehyde crotonaldehyde triggers cardiomyocyte contractile dysfunction: Role of TRPV1 and mitochondrial function. Pharmacol. Res. 2014, 82, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Wang, L.; Han, J.; Song, J.; Yao, L.; Shao, L.; Sun, Z.; Zheng, L. Decreased expression of transient receptor potential vanilloid 1 impaires the postischemic recovery of diabetic mouse hearts. Circ. J. 2009, 73, 1127–1132. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, D.H. TRPV1 gene knockout impairs postischemic recovery in isolated perfused heart in mice. Circulation 2005, 112, 3617–3623. [Google Scholar] [CrossRef] [PubMed]
- Hoover, D.B. Effects of capsaicin on release of substance P-like immunoreactivity and physiological parameters in isolated perfused guinea-pig heart. Eur. J. Pharmacol. 1987, 141, 489–492. [Google Scholar] [CrossRef] [PubMed]
- Manzini, S.; Perretti, F.; de Benedetti, L.; Pradelles, P.; Maggi, C.A.; Geppetti, P. A comparison of bradykinin- and capsaicin-induced myocardial and coronary effects in isolated perfused heart of guinea-pig: Involvement of substance P and calcitonin gene-related peptide release. Br. J. Pharmacol. 1989, 97, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Ustinova, E.E.; Bergren, D.; Schultz, H.D. Neuropeptide depletion impairs postischemic recovery of the isolated rat heart: Role of substance P. Cardiovasc. Res. 1995, 30, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.H.; Zhou, S.X.; Li, R.C.; Zheng, L.R.; Zhu, J.H.; Hu, S.J.; Sun, Y.L. Serum levels of calcitonin gene-related peptide and substance P are decreased in patients with diabetes mellitus and coronary artery disease. J. Int. Med. Res. 2012, 40, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Sappington, R.M.; Sidorova, T.; Long, D.J.; Calkins, D.J. TRPV1, contribution to retinal ganglion cell apoptosis and increased intracellular Ca2+ with exposure to hydrostatic pressure. Investig. Ophthalmol. Vis. Sci. 2009, 50, 717–728. [Google Scholar] [CrossRef]
- Hu, F.; Sun, W.W.; Zhao, X.T.; Cui, Z.J.; Yang, W.X. TRPV1 mediates cell death in rat synovial fibroblasts through calcium entry-dependent ROS production and mitochondrial depolarization. Biochem. Biophys. Res. Commun. 2008, 369, 989–993. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.J.; Kee, K.H.; Suh, C.H.; Lim, S.C.; Oh, S.H. Capsaicin-induced apoptosis is regulated by endoplasmic reticulum stress- and calpain-mediated mitochondrial cell death pathways. Toxicology 2009, 264, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Naziroglu, M.; Cig, B.; Ozgul, C. Neuroprotection induced by N-acetylcysteine against cytosolic glutathione depletion-induced Ca2+ influx in dorsal root ganglion neurons of mice: Role of TRPV1 channels. Neuroscience 2013, 242, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Chien, C.S.; Ma, K.H.; Lee, H.S.; Liu, P.S.; Li, Y.H.; Huang, Y.S.; Chueh, S.H. Dual effect of capsaicin on cell death in human osteosarcoma G292 cells. Eur. J. Pharmacol. 2013, 718, 350–360. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.H.; Lu, W.C.; Wang, C.W.; Chan, Y.C.; Chen, M.K. Capsaicin induces cell cycle arrest and apoptosis in human KB cancer cells. BMC Complement. Altern. Med. 2013, 13, 46. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Kim, J.Y.; Lee, S.M.; Jun, C.H.; Cho, S.B.; Park, C.H.; Joo, Y.E.; Kim, H.S.; Choi, S.K.; Rew, J.S. Capsaicin induces apoptosis and modulates MAPK signaling in human gastric cancer cells. Mol. Med. Rep. 2014, 9, 499–502. [Google Scholar] [PubMed]
- Amantini, C.; Mosca, M.; Nabissi, M.; Lucciarini, R.; Caprodossi, S.; Arcella, A.; Giangaspero, F.; Santoni, G. Capsaicin-induced apoptosis of glioma cells is mediated by TRPV1 vanilloid receptor and requires p38 MAPK activation. J. Neurochem. 2007, 102, 977–990. [Google Scholar] [CrossRef] [PubMed]
- Kamakura, T.; Ishida, Y.; Nakamura, Y.; Yamada, T.; Kitahara, T.; Takimoto, Y.; Horii, A.; Uno, A.; Imai, T.; Okazaki, S.; et al. Functional expression of TRPV1 and TRPA1 in rat vestibular ganglia. Neurosci. Lett. 2013, 552, 92–97. [Google Scholar] [CrossRef] [PubMed]
- Amantini, C.; Mosca, M.; Lucciarini, R.; Perfumi, M.; Morrone, S.; Piccoli, M.; Morrone, S.; Piccoli, M.; Santoni, G. Distinct thymocyte subsets express the vanilloid receptor VR1 that mediates capsaicin-induced apoptotic cell death. Cell Death Differ. 2004, 11, 1342–1356. [Google Scholar] [CrossRef] [PubMed]
- Agopyan, N.; Head, J.; Yu, S.; Simon, S.A. TRPV1 receptors mediate particulate matter-induced apoptosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 286, L563–L572. [Google Scholar] [CrossRef] [PubMed]
- Reilly, C.A.; Johansen, M.E.; Lanza, D.L.; Lee, J.; Lim, J.O.; Yost, G.S. Calcium-dependent and independent mechanisms of capsaicin receptor (TRPV1)-mediated cytokine production and cell death in human bronchial epithelial cells. J. Biochem. Mol. Toxicol. 2005, 19, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Stout, A.K.; Raphael, H.M.; Kanterewicz, B.I.; Klann, E.; Reynolds, I.J. Glutamate-induced neuron death requires mitochondrial calcium uptake. Nat. Neurosci. 1998, 1, 366–373. [Google Scholar] [CrossRef] [PubMed]
- Verkhratsky, A.; Toescu, E.C. Endoplasmic reticulum Ca2+ homeostasis and neuronal death. J. Cell. Mol. Med. 2003, 7, 351–361. [Google Scholar]
- Hajnoczky, G.; Davies, E.; Madesh, M. Calcium signaling and apoptosis. Biochem. Biophys. Res. Commun. 2003, 304, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Cadenas, E.; Boveris, A. Enhancement of hydrogen peroxide formation by protophores and ionophores in antimycin-supplemented mitochondria. Biochem. J. 1980, 188, 31–37. [Google Scholar] [PubMed]
- Sousa, S.C.; Maciel, E.N.; Vercesi, A.E.; Castilho, R.F. Ca2+-induced oxidative stress in brain mitochondria treated with the respiratory chain inhibitor rotenone. FEBS Lett. 2003, 543, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Cleeter, M.W.; Cooper, J.M.; Darley-Usmar, V.M.; Moncada, S.; Schapira, A.H. Reversible inhibition of cytochrome c oxidase, the terminal enzyme of the mitochondrial respiratory chain, by nitric oxide. Implications for neurodegenerative diseases. FEBS Lett. 1994, 345, 50–54. [Google Scholar] [CrossRef] [PubMed]
- Viola, H.M.; Arthur, P.G.; Hool, L.C. Transient exposure to hydrogen peroxide causes an increase in mitochondria-derived superoxide as a result of sustained alteration in L-type Ca2+ channel function in the absence of apoptosis in ventricular myocytes. Circ. Res. 2007, 100, 1036–1044. [Google Scholar] [CrossRef]
- Zhang, R.; Humphreys, I.; Sahu, R.P.; Shi, Y.; Srivastava, S.K. In vitro and in vivo induction of apoptosis by capsaicin in pancreatic cancer cells is mediated through ROS generation and mitochondrial death pathway. Apoptosis 2008, 13, 1465–1478. [Google Scholar] [CrossRef] [PubMed]
- Pramanik, K.C.; Boreddy, S.R.; Srivastava, S.K. Role of mitochondrial electron transport chain complexes in capsaicin mediated oxidative stress leading to apoptosis in pancreatic cancer cells. PLoS One 2011, 6, e20151. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.M.; Pyo, J.O.; Kim, G.Y.; Yu, R.; Han, I.S.; Ju, S.A.; Kim, W.H.; Kim, B.S. Capsaicin induces apoptosis by generating reactive oxygen species and disrupting mitochondrial transmembrane potential in human colon cancer cell lines. Cell Mol. Biol. Lett. 2009, 14, 497–510. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.S.; Nam, D.H.; Kim, J.A. Induction of apoptosis by capsaicin in A172 human glioblastoma cells. Cancer Lett. 2000, 161, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.S.; Kwon, E.J.; Jin, D.Q.; Park, S.H.; Kang, Y.S.; Huh, K.; Kim, J.A. Redox status-dependent regulation of cyclooxygenases mediates the capsaicin-induced apoptosis in human neuroblastoma cells. J. Environ. Pathol. Toxicol. Oncol. 2002, 21, 113–120. [Google Scholar] [PubMed]
- Baek, Y.M.; Hwang, H.J.; Kim, S.W.; Hwang, H.S.; Lee, S.H.; Kim, J.A.; Yun, J.W. A comparative proteomic analysis for capsaicin-induced apoptosis between human hepatocarcinoma (HepG2) and human neuroblastoma (SK-N-SH) cells. Proteomics 2008, 8, 4748–4767. [Google Scholar] [CrossRef] [PubMed]
- Peterson, Y.K.; Cameron, R.B.; Wills, L.P.; Trager, R.E.; Lindsey, C.C.; Beeson, C.C.; Schnellmann, R.G. β2-Adrenoceptor agonists in the regulation of mitochondrial biogenesis. Bioorganic Med. Chem. Lett. 2013, 23, 5376–5381. [Google Scholar]
- Attardi, G.; Schatz, G. Biogenesis of mitochondria. Annu. Rev. Cell Biol. 1988, 4, 289–333. [Google Scholar] [CrossRef]
- Rehman, H.; Krishnasamy, Y.; Haque, K.; Thurman, R.G.; Lemasters, J.J.; Schnellmann, R.G.; Zhong, Z. Green tea polyphenols stimulate mitochondrial biogenesis and improve renal function after chronic cyclosporin a treatment in rats. PLoS One 2013, 8, e65029. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.J.; Xiao, Z.S.; Peng, C.F.; Deng, H.W. Calcitonin gene-related peptide-induced preconditioning protects against ischemia-reperfusion injury in isolated rat hearts. Eur. J. Pharmacol. 1996, 311, 163–167. [Google Scholar] [CrossRef] [PubMed]
- Kaygisiz, Z.; Erksap, N.; Uyar, R.; Kabadere, S.; Kabadere, T.E.; Dernek, S. The effect of adrenomedullin, amylin fragment 8–37 and calcitonin gene-related peptide on contractile force, heart rate and coronary perfusion pressure in isolated rat hearts. Acta Physiol. Hung. 2003, 90, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Amantini, C.; Ballarini, P.; Caprodossi, S.; Nabissi, M.; Morelli, M.B.; Lucciarini, R.; Cardarelli, M.A.; Mammana, G.; Santoni, G. Triggering of transient receptor potential vanilloid type 1 (TRPV1) by capsaicin induces Fas/CD95-mediated apoptosis of urothelial cancer cells in an ATM-dependent manner. Carcinogenesis 2009, 30, 1320–1329. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.R.; Kim, S.U.; Oh, U.; Jin, B.K. Transient receptor potential vanilloid subtype 1 mediates microglial cell death in vivo and in vitro via Ca2+-mediated mitochondrial damage and cytochrome c release. J. Immunol. 2006, 177, 4322–4329. [Google Scholar] [CrossRef] [PubMed]
- Kamga, P.C.; Mo, L.; Quesnelle, K.; Dagda, R.K.; Murillo, D.; Geary, L.; Corey, C.; Portella, R.; Zharikov, S.; St Croix, C.; et al. Nitrite activates protein kinase A in normoxia to mediate mitochondrial fusion and tolerance to ischaemia/reperfusion. Cardiovasc. Res. 2014, 101, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Funk, J.A.; Schnellmann, R.G. Accelerated recovery of renal mitochondrial and tubule homeostasis with SIRT1/PGC-1α activation following ischemia-reperfusion injury. Toxicol. Appl. Pharmacol. 2013, 273, 345–354. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhang, K.; Yan, S.; Li, A.; Hu, X.; Zhang, L.; Liu, C. Enhancement of apamin-sensitive medium afterhyperpolarization current by anandamide and its role in excitability control in cultured hippocampal neurons. Neuropharmacology 2011, 60, 901–909. [Google Scholar] [CrossRef] [PubMed]
- Drebot, I.; Boldyriev, O.I.; Dosenko, V.; Kostiuk, P.H. Expression of genes encoding vanilloid receptors type 1 and type 2 in the cultured hippocampal neurons. Fiziol. Zh. 2008, 54, 3–9. [Google Scholar] [PubMed]
- Xu, S.; Ning, W.; Xu, Z.; Zhou, S.; Chiang, H.; Luo, J. Chronic exposure to GSM 1800-MHz microwaves reduces excitatory synaptic activity in cultured hippocampal neurons. Neurosci. Lett. 2006, 398, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Cingolani, E.; Ramirez, C.G.; Kizana, E.; Murata, M.; Cho, H.C.; Marban, E. Gene therapy to inhibit the calcium channel β subunit: Physiological consequences and pathophysiological effects in models of cardiac hypertrophy. Circ. Res. 2007, 101, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Funk, J.A.; Odejinmi, S.; Schnellmann, R.G. SRT1720 induces mitochondrial biogenesis and rescues mitochondrial function after oxidant injury in renal proximal tubule cells. J. Pharmacol. Exp. Ther. 2010, 333, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Rasbach, K.A.; Schnellmann, R.G. Signaling of mitochondrial biogenesis following oxidant injury. J. Biol. Chem. 2007, 282, 2355–2362. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, Z.; Han, J.; Zhao, W.; Zhang, Y.; Wang, S.; Ye, L.; Liu, T.; Zheng, L. TRPV1 Activation Exacerbates Hypoxia/Reoxygenation-Induced Apoptosis in H9C2 Cells via Calcium Overload and Mitochondrial Dysfunction. Int. J. Mol. Sci. 2014, 15, 18362-18380. https://doi.org/10.3390/ijms151018362
Sun Z, Han J, Zhao W, Zhang Y, Wang S, Ye L, Liu T, Zheng L. TRPV1 Activation Exacerbates Hypoxia/Reoxygenation-Induced Apoptosis in H9C2 Cells via Calcium Overload and Mitochondrial Dysfunction. International Journal of Molecular Sciences. 2014; 15(10):18362-18380. https://doi.org/10.3390/ijms151018362
Chicago/Turabian StyleSun, Zewei, Jie Han, Wenting Zhao, Yuanyuan Zhang, Shuai Wang, Lifang Ye, Tingting Liu, and Liangrong Zheng. 2014. "TRPV1 Activation Exacerbates Hypoxia/Reoxygenation-Induced Apoptosis in H9C2 Cells via Calcium Overload and Mitochondrial Dysfunction" International Journal of Molecular Sciences 15, no. 10: 18362-18380. https://doi.org/10.3390/ijms151018362
APA StyleSun, Z., Han, J., Zhao, W., Zhang, Y., Wang, S., Ye, L., Liu, T., & Zheng, L. (2014). TRPV1 Activation Exacerbates Hypoxia/Reoxygenation-Induced Apoptosis in H9C2 Cells via Calcium Overload and Mitochondrial Dysfunction. International Journal of Molecular Sciences, 15(10), 18362-18380. https://doi.org/10.3390/ijms151018362