Melatonin, Noncoding RNAs, Messenger RNA Stability and Epigenetics—Evidence, Hints, Gaps and Perspectives


Abstract
:1. Introduction
2. Melatonin Synthesizing Organs: Modulation of mRNA Stability and Demonstration of Cycling lncRNAs
3. Epigenetic Modulation of Melatonin Receptor Expression—Initial Findings
4. Circadian Oscillators and Epigenetics—A Role for Melatonin?

{kind=link}
{kind=link}
| Organism/Tissue or Cells | Main Findings | References | |
|---|---|---|---|
| Mouse/SCN | Circadian rhythms of miR-219 and miR-132; miR-219; knockdown lengthens circadian period; Cry1/Cry2 double knockout abolishes rhythms of pre-miR-219-1 and miR-219-1; CLOCK/BMAL1 over-expression stimulates pre-miR-219-1; miR-132 is induced by light (also by LAN) via ERK/MAPK, but acts as negative regulator of photic entrainment; miR-132 is presumably target of CBP (CREB binding protein) | [46,47] | |
| Rat/primary cortical neurons | miR-219 over-expression suppresses NMDA-induced Ca2+ influx | [46] | |
| Mouse/brain and P19 cells | miR-219 down-regulates NMDA signaling by targeting CamkII subunit γ mRNA | [48] | |
| Mouse/SCN | miR-132 targets mRNAs of proteins involved in chromatin remodeling (Mecp2, Ep300, Jarid1a) and translational control (Btg2, Paip2a); MeCP2 binds to Per1 and Per2 promoters; BTG2 and PAIP2A enhance decay of Per1 and Per2 mRNAs | [49] | |
| Mouse/retina | Circadian rhythms of 16 lincRNAs | [50] | |
| Mouse/retina | Circadian rhythms of 12 miRNAs, including those from miR-183-96-182 cluster | [47,51] | |
| Mouse/brain | Two overlapping imprinted ncRNAs from intergenic region Dlk1–Gtl2 that contains an ICR 1 are exclusively expressed at maternal chromosome, from both strands: Mico1 and Mico1os 2; both oscillate in a circadian fashion | [52] | |
| Mouse/hypothalamus, hindbrain, forebrain, cortex, hippocampus, cerebellum; neurons but not glia | Transcript of Prader–Willis locus SNORD116 is spliced into multiple snoRNAs and the lncRNA 116HG; 116HG forms subnuclear clouds that increase postnatally and are associated with large-scale chromatin decondensation; size of clouds smaller at ZT16 3 than at ZT6; in SNORD116−/− mice, expression of 6467 genes is altered at ZT6, of 3240 genes at ZT16; relative to WT, Clock, Cry1 and Per2 are up-regulated at ZT6, Cry1, Cry2 and Per1 down-regulated at ZT16 | [53] | |
| Mouse/serum | Circadian rhythms of miR-152 and miR-494; circulating miRNAs may influence oscillators via microvesicles | [54] | |
| Human/HEK293 cells | Bmal1 is targeted at 3'-UTR by miR-494 and miR-142-3p | [54] | |
| HTC116, HT29 and NIH3T3 cells | miR-192/194 cluster targets Per1, Per2, and Per3 and alters circadian rhythms | [55] | |
| Mouse/liver | Circadian rhythms of 85 miRNAs; several miRNA/mRNA target pairs identified, including core oscillator mRNAs; miR-181d and miR-191 are inversely correlated with Clock/Bmal1 and presumably involved in peripheral clocks; miR-328 and miR-383 positively correlated with Per1/Cry1 | [47,56] | |
| Mouse/liver | REV-ERBα drives miR-122 transcription; knockdown of miR-122 alters expression of hundreds of hepatic mRNAs | [57] | |
| Mouse/liver | miR-122 targets Noc mRNA; miR-122 knockdown increases the amplitude of the nocturnin rhythm | [58] | |
| Mouse/various organs | Noc mRNA is rhythmic in several brain regions, retina, heart, kidney, spleen, and liver | [59,60] | |
| Mouse/liver | Several hundred mRNAs exhibit circadian rhythms in poly(A) tail length, even in cases in which mRNA levels are not rhythmic | [61] | |
| Mouse/liver | CLOCK controls rhythmic transcription of Noc; Clock mutants exhibit dampened Noc rhythms | [62] | |
| Human/Huh7 hepatoma cells | Binding of CLOCK/BMAL1 to E-box in Noc promoter | [63] | |
| Mouse/liver | NOC stabilizes iNOS mRNA; NOC deficiency blunts the nocturnal peak of iNOS mRNA | [33] | |
| Mouse/liver | Circadian rhythms of 54 miRNAs, 16 lincRNAs and several antisense transcripts, including a Per2 antisense RNA (asPer2); rhythms in histone modifications: especially, H3K4me3, but also H3K4me1, H3K9ac, H3K27ac (at active enhancers), and H3K36me3 enriched in actively transcribed genes | [64] | |
| Mouse/embryo fibroblasts, liver | SIRT1, an accessory circadian oscillator protein, histone deacetylase and aging suppressor, promotes PER2 degradation by deacetylation, is required for high amplitudes of Per2, Cry1, Bmal1 and RORγ transcription rhythms, is recruited to the BMAL1/CLOCK complex and controls the expression of E-box-containing genes such as Per2, Cry1 and NAMPT via cycling NAD+ concentration | [65,66,67,68,69] | |
| Mouse/liver, lung, fibroblasts | PER proteins form complexes that include PSF 4, which recruits the scaffold SIN3A associated with a HDAC that rhythmically suppresses Per1 transcription by deacetylating histones at the promoter | [70] | |
| Mouse/liver, 3T3 cells | NONO 5 interacts with PER1 and modulates its activity | [71] | |
| Mouse/liver, brain areas incl. SCN | NONO associates with PER1 or PER2 at Rev-erbα and Dbp promoters; NONO couples the oscillator to cell cycle; NONO also interacts with ncRNAs | [72,73] | |
| Rat/GH4C1 cells | NONO and SFPQ 6 induce chromatin remodeling at prolactin promoter and couple Prl expression to circadian oscillator; NONO/SFPQ over-expression decreases promoter activity and disrupts circadian Prl rhythm | [74] | |
5. Light at Night—Epigenetic Changes during Chronodisruption and Melatonin Shutdown
6. Metabolic Disorders and Energy Expenditure, Melatonergic Counteractions and Epigenetic Regulation
7. Epigenetics of Inflammation and Oxidative Stress vs. Antioxidant Properties of Melatonin
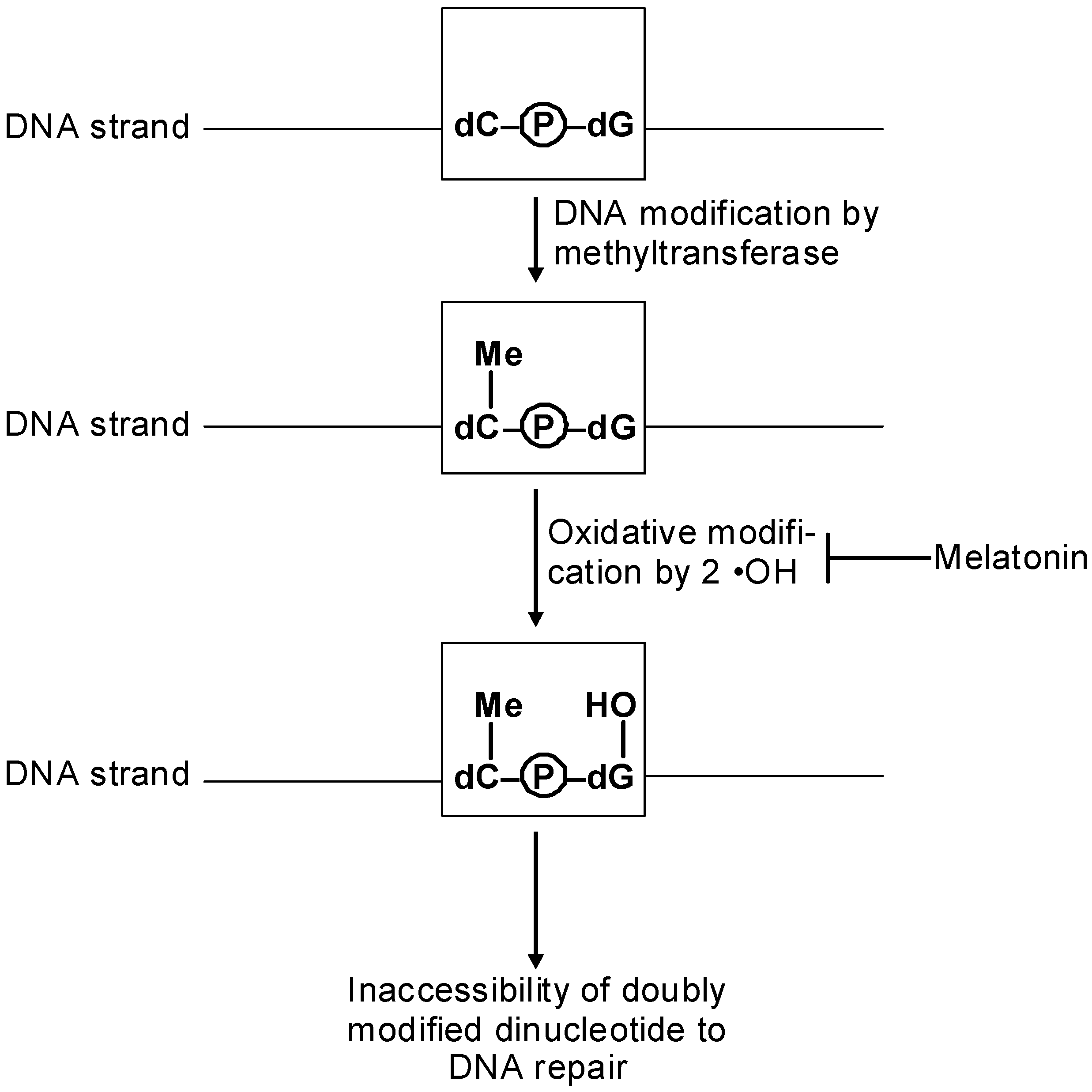
8. Melatonin, Cancer and Epigenetics
9. The Central Nervous System, Neurogenesis, Neuropsychiatric Disorders and Melatonin
10. Conclusions
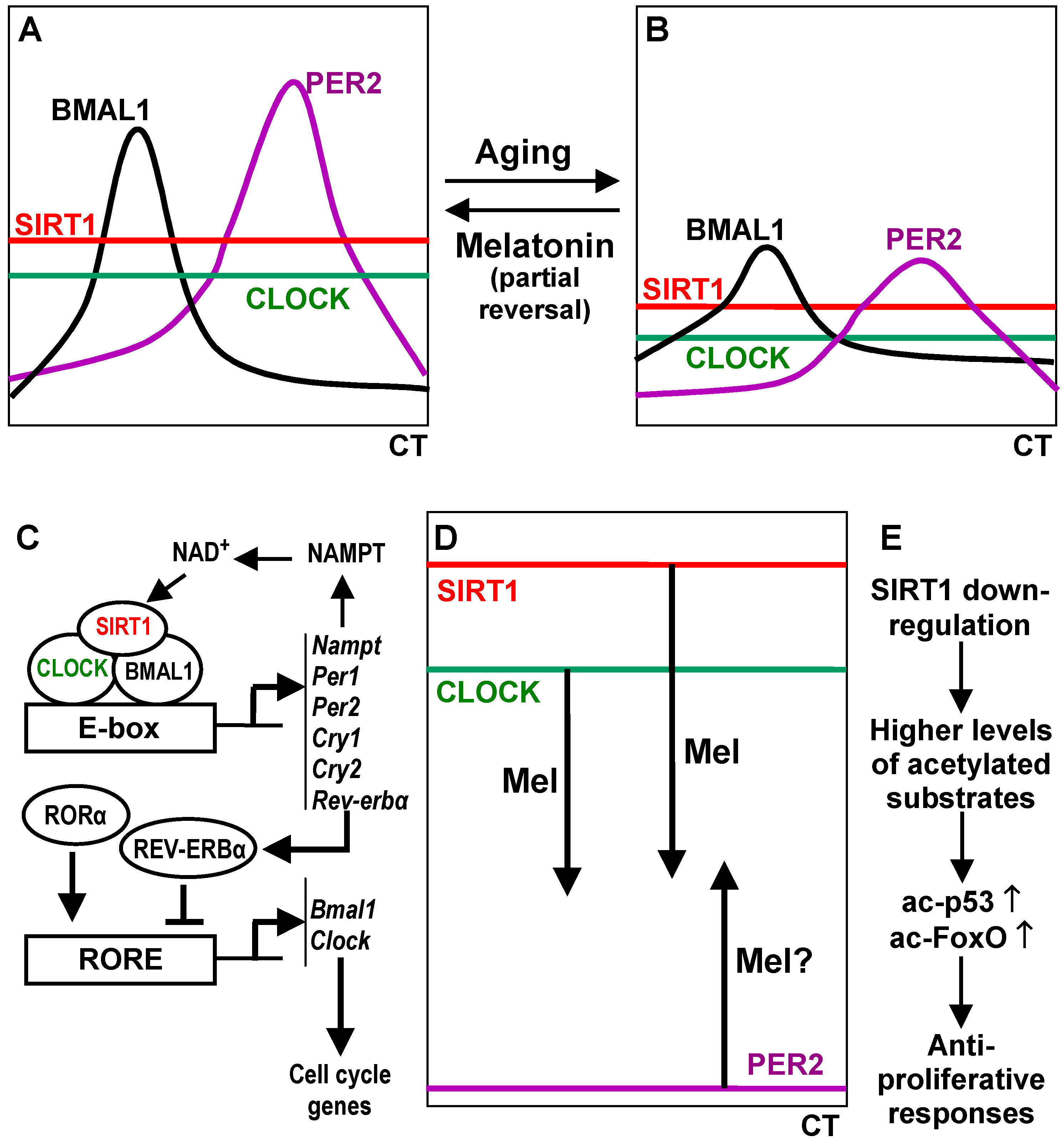
Conflicts of Interest
References
- Hardeland, R.; Cardinali, D.P.; Srinivasan, V.; Spence, D.W.; Brown, G.M.; Pandi-Perumal, S.R. Melatonin—A pleiotropic, orchestrating regulator molecule. Prog. Neurobiol. 2011, 93, 350–384. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J.; Tan, D.-X.; Mayo, J.C.; Sainz, R.M.; Leon, J.; Czarnocki, Z. Melatonin as an antioxidant: Biochemical mechanisms and pathophysiological implications in humans. Acta Biochim. Pol. 2003, 50, 1129–1146. [Google Scholar] [PubMed]
- Pandi-Perumal, S.R.; Srinivasan, V.; Maestroni, G.J.M.; Cardinali, D.P.; Poeggeler, B.; Hardeland, R. Melatonin: Nature’s most versatile biological signal? FEBS J. 2006, 273, 2813–2838. [Google Scholar] [CrossRef]
- Negi, G.; Kumar, A.; Sharma, S.S. Melatonin modulates neuroinflammation and oxidative stress in experimental diabetic neuropathy: Effects on NF-κB and Nrf2 cascades. J. Pineal Res. 2011, 50, 124–131. [Google Scholar] [PubMed]
- Korkmaz, A.; Rosales-Corral, S.; Reiter, R.J. Gene regulation by melatonin linked to epigenetic phenomena. Gene 2012, 503, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Carrillo-Vico, A.; Lardone, P.J.; Álvarez-Sánchez, N.; Rodríguez-Rodríguez, A.; Guerrero, J.M. Melatonin: Buffering the immune system. Int. J. Mol. Sci. 2013, 14, 8638–8683. [Google Scholar]
- Ehrlich, M.; Lacey, M. DNA methylation and differentiation: Silencing, upregulation and modulation of gene expression. Epigenomics 2013, 5, 553–568. [Google Scholar] [CrossRef] [PubMed]
- Mihan, K.N.; Chaillet, J.R. Cell and molecular biology of DNA methyltransferase 1. Int. Rev. Cell Mol. Biol. 2013, 306, 1–42. [Google Scholar] [PubMed]
- Kulis, M.; Queirós, A.C.; Beekman, R.; Martín-Subero, J.I. Intragenic DNA methylation in transcriptional regulation, normal differentiation and cancer. Biochim. Biophys. Acta 2013, 1829, 1161–1174. [Google Scholar] [CrossRef] [PubMed]
- Van Otterdijk, S.D.; Mathers, J.C.; Strathdee, G. Do age-related changes in DNA methylation play a role in the development of age-related diseases? Biochem. Soc. Trans. 2013, 41, 803–807. [Google Scholar]
- Zentner, G.E.; Henikoff, S. Regulation of nucleosome dynamics by histone modifications. Nat. Struct. Mol. Biol. 2013, 20, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Kaelin, W.G., Jr.; McKnight, S.L. Influence of metabolism on epigenetics and disease. Cell 2013, 153, 56–69. [Google Scholar] [CrossRef]
- Gut, P.; Verdin, E. The nexus of chromatin regulation and intermediary metabolism. Nature 2013, 502, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Jarome, T.J.; Lubin, F.D. Histone lysine methylation: A critical regulator of memory and behavior. Rev. Neurosci. 2013, 24, 375–387. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Qu, L. Non-coding RNAs and the acquisition of genomic imprinting in mammals. Sci. China C 2009, 52, 195–204. [Google Scholar] [CrossRef]
- Morozowa, N.; Zinovyev, A.; Nonne, N.; Pritchard, L.L.; Gorban, A.N.; Harel-Bellan, A. Kinetic signatures of microRNA modes of action. RNA 2012, 18, 1635–1655. [Google Scholar] [CrossRef] [PubMed]
- Tay, Y.; Rinn, J.; Pandolfi, P.P. The multilayered complexity of ceRNA crosstalk and competition. Nature 2014, 505, 344–352. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Liu, Y. Diverse small non-coding RNAs and RNA interference pathways. Methods Mol. Biol. 2011, 764, 169–182. [Google Scholar] [PubMed]
- Wilson, R.C.; Doudna, J.A. Molecular mechanisms of RNA interference. Annu. Rev. Biophys. 2013, 42, 217–239. [Google Scholar] [CrossRef] [PubMed]
- Ross, R.J.; Weiner, M.M.; Lin, H. PIWI proteins and PIWI-interacting RNAs in the soma. Nature 2014, 505, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Lowe, T. Small nucleolar RNAs and RNA-guided post-transcriptional modification. Essays Biochem. 2013, 54, 53–77. [Google Scholar] [CrossRef] [PubMed]
- Schoeftner, S.; Blasco, M.A. Chromatin regulation and non-coding RNAs at mammalian telomeres. Semin. Cell. Dev. Biol. 2010, 21, 186–193. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.T. Epigenetic regulation by long noncoding RNAs. Science 2012, 338, 1435–1439. [Google Scholar] [CrossRef] [PubMed]
- Marchese, F.P.; Huarte, M. Long non-coding RNAs and chromatin modifiers: Their place in the epigenetic code. Epigenetics 2013, 9, 21–26. [Google Scholar] [PubMed]
- Yang, L.; Froberg, J.E.; Lee, J.T. Long noncoding RNAs: Fresh perspectives into the RNA world. Trends Biochem. Sci. 2014, 39, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Hacisuleyman, E.; Goff, L.A.; Trapnell, C.; Williams, A.; Henao-Mejia, J.; Sun, L.; McClanahan, P.; Hendrickson, D.G.; Sauvagneau, M.; Kelley, D.R.; et al. Topological organization of multichromosomal regions by the long intergenic noncoding RNA Firre. Nat. Struct. Mol. Biol. 2014, 21, 198–206. [Google Scholar] [CrossRef]
- Li, X.; Wu, Z.; Mei, Q.; Li, X.; Guo, M.; Fu, X.; Han, W. Long non-coding RNA HOTAIR, a driver of malignancy, predicts negative prognosis and exhibits oncogenic activity in oesophageal squamous cell carcinoma. Br. J. Cancer 2013, 109, 2266–2278. [Google Scholar] [CrossRef] [PubMed]
- Romero, R.; Tarca, A.L.; Chaemsaithong, P.; Miranda, J.; Chaiworapongsa, T.; Jia, H.; Hassan, S.S.; Kalita, C.A.; Cai, J.; Yeo, L.; et al. Transcriptome interrogation of human myometrium identifies differentially expressed sense-antisense pairs of protein-coding and long non-coding RNA genes in spontaneous labor at term. J. Matern. Fetal Neonatal Med. 2014, 27, 1397–1408. [Google Scholar] [CrossRef]
- Wang, Y.; Pang, W.J.; Wei, N.; Xiong, Y.; Wu, W.J.; Zhao, C.Z.; Shen, Q.W.; Yang, G.S. Identification, stability and expression of Sirt1 antisense long non-coding RNA. Gene 2014, 539, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Green, C.B.; Douris, N.; Kojima, S.; Strayer, C.A.; Fogerty, J.; Lourim, D.; Keller, S.R.; Besharse, J.C. Loss of nocturnin, a circadian deadenylase, confers resistance to hepatic steatosis and diet-induced obesity. Proc. Natl. Acad. Sci. USA 2007, 104, 9888–9893. [Google Scholar] [CrossRef] [PubMed]
- Stubblefield, J.J.; Terrien, J.; Green, C.B. Nocturnin: At the crossroads of clocks and metabolism. Trends Endocrinol. Metab. 2012, 23, 326–333. [Google Scholar] [CrossRef] [PubMed]
- Godwin, A.R.; Kojima, S.; Green, C.B.; Wilusz, J. Kiss your tail goodbye: The role of PARN, Nocturnin, and Angel deadenylases in mRNA biology. Biochim. Biophys. Acta 2013, 1829, 571–579. [Google Scholar] [CrossRef] [PubMed]
- Niu, S.; Shingle, D.L.; Garbarino-Pico, E.; Kojima, S.; Gilbert, M.; Green, C.B. The circadian deadenylase Nocturnin is necessary for stabilization of the iNOS mRNA in mice. PLoS One 2011, 6, e26954. [Google Scholar] [CrossRef] [PubMed]
- Kloeden, P.E.; Rössler, R.; Rössler, O.E. Artificial life extension. The epigenetic approach. Ann. N. Y. Acad. Sci. 1994, 719, 474–482. [Google Scholar] [CrossRef]
- Korkmaz, A.; Topal, T.; Tan, D.-X.; Reiter, R.J. Role of melatonin in metabolic regulation. Rev. Endocr. Metab. Disord. 2009, 10, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Korkmaz, A.; Ma, S.; Topal, T.; Rosales-Corral, S.; Tan, D.-X.; Reiter, R.J. Glucose: A vital toxin and potential utility of melatonin in protecting against the diabetic state. Mol. Cell. Endocrinol. 2012, 349, 128–137. [Google Scholar] [CrossRef] [PubMed]
- Hardeland, R.; Madrid, J.A.; Tan, D.-X.; Reiter, R.J. Melatonin, the circadian multioscillator system and health: The need for detailed analyses of peripheral melatonin signaling. J. Pineal Res. 2012, 52, 139–166. [Google Scholar] [CrossRef] [PubMed]
- Hardeland, R. Chronobiology of melatonin beyond the feedback to the suprachiasmatic nucleus—Consequences to melatonin dysfunction. Int. J. Mol. Sci. 2013, 14, 5817–5841. [Google Scholar] [PubMed]
- Clokie, S.J.H.; Lau, P.; Kim, H.H.; Coon, S.L.; Klein, D.C. MicroRNAs in the pineal gland. miR-483 regulates melatonin synthesis by targeting arylalkylamine N-acetyltransferase. J. Biol. Chem. 2012, 287, 25312–23524. [Google Scholar]
- Ben-Moshe, Z.; Alon, S.; Mracek, P.; Faigenbloom, L.; Tovin, A.; Vatine, G.D.; Eisenberg, E.; Foulkes, N.S.; Gothilf, Y. The light-induced transcriptome of the zebrafish pineal gland reveals complex regulation of the circadian clockwork by light. Nucleic Acids Res. 2014, 42, 3750–3767. [Google Scholar] [CrossRef] [PubMed]
- Green, C.B.; Besharse, J.C. Identification of a novel vertebrate circadian clock-regulated gene encoding the protein nocturnin. Proc. Natl. Acad. Sci. USA 1996, 93, 14884–14888. [Google Scholar] [CrossRef] [PubMed]
- Green, C.B. Molecular control of Xenopus retinal circadian rhythms. J. Neuroendocrinol. 2003, 15, 350–354. [Google Scholar] [CrossRef] [PubMed]
- Coon, S.L.; Munson, P.J.; Cherukuri, P.F.; Sugden, D.; Rath, M.F.; Møller, M.; Clokie, S.J.H.; Fu, C.; Olanich, M.E.; Rangel, Z.; et al. Circadian changes in long noncoding RNAs in the pineal gland. Proc. Natl. Acad. Sci. USA 2012, 109, 13319–1324. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.; Rincón Castro, L.M.; Jawed, S.; Niles, L.P. Clinically relevant concentrations of valproic acid modulate melatonin MT1 receptor, HDAC and MeCP2 mRNA expression in C6 glioma cells. Eur. J. Pharmacol. 2008, 589, 45–48. [Google Scholar] [CrossRef] [PubMed]
- Esposito, T.; Magliocca, S.; Formicola, D.; Gianfrancesco, F. piR_015520 belongs to Piwi-associated RNAs regulates expression of the human melatonin receptor 1A gene. PLoS One 2011, 6, e22727. [Google Scholar] [CrossRef]
- Cheng, H.Y.M.; Papp, J.W.; Varlamova, O.; Dziema, H.; Russell, B.; Curfman, J.P.; Nakazawa, T.; Shimizu, K.; Okamura, H.; Impey, S.; et al. MicroRNA modulation of circadian clock-period and entrainment. Neuron 2007, 54, 813–829. [Google Scholar] [CrossRef]
- Mehta, N.; Cheng, H.Y.M. Micro-managing the circadian clock: The role of microRNAs in biological timekeeping. J. Mol. Biol. 2013, 425, 3609–3624. [Google Scholar] [CrossRef] [PubMed]
- Kocerha, J.; Faghihi, M.A.; Lopez-Toledano, M.A.; Huang, J.; Ramsey, A.J.; Caron, M.G.; Sales, N.; Willoughby, D.; Elmen, J.; Hansen, H.F.; et al. MicroRNA-219 modulates NMDA receptor-mediated neurobehavioral dysfunction. Proc. Natl. Acad. Sci. USA 2009, 106, 3507–3512. [Google Scholar] [CrossRef]
- Alvarez-Saavedra, M.; Antoun, G.; Yanagiya, A.; Oliva-Hernandez, R.; Cornejo-Palma, D.; Perez-Iratxeta, C.; Sonenberg, N.; Cheng, H.Y.M. MicroRNA-132 orchestrates chromatin remodeling and translational control of the circadian clock. Hum. Mol. Genet. 2011, 20, 371–351. [Google Scholar] [CrossRef]
- Mustafi, D.; Kevany, B.M.; Genoud, C.; Bai, X.; Palczewski, K. Photoreceptor phagocytosis is mediated by phosphoinositide signaling. FASEB J. 2013, 27, 4585–4595. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.B.; Witmer, P.D.; Lumayag, S.; Kovacs, B.; Valle, D. MicroRNA (miRNA) transcriptome of mouse retina and identification of a sensory organ-specific miRNA cluster. J. Biol. Chem. 2007, 282, 25053–25066. [Google Scholar] [PubMed]
- Labialle, S.; Croteau, S.; Bélanger, V.; McMurray, E.N.; Ruan, X.; Moussette, S.; Jonnaert, M.; Schmidt, J.V.; Cermakian, N.; Naumova, A.K. Novel imprinted transcripts from the Dlk1-Gtl2 intergenic region, Mico1 and Mico1os, show circadian oscillations. Epigenetics 2008, 3, 322–329. [Google Scholar] [CrossRef] [PubMed]
- Powell, W.T.; Coulson, R.L.; Crary, F.K.; Wong, S.S.; Ach, R.A.; Tsang, P.; Yamada, N.A.; Yasui, D.H.; LaSalle, J.M. A Prader–Willis locus lncRNA cloud modulates diurnal genes and energy expenditure. Hum. Mol. Genet. 2013, 22, 4318–4328. [Google Scholar] [CrossRef] [PubMed]
- Shende, V.R.; Goldrick, M.M.; Ramani, S.; Earnest, D.J. Expression and rhythmic modulation of circulating microRNAs targeting the clock gene Bmal1 in mice. PLoS One 2011, 6, e22586. [Google Scholar] [CrossRef] [PubMed]
- Nagel, R.; Clijsters, L.; Agami, R. the miRNA-192/194 cluster regulates the Period gene family and the circadian clock. FEBS J. 2009, 276, 5447–5455. [Google Scholar] [CrossRef] [PubMed]
- Na, Y.J.; Sung, J.H.; Lee, S.C.; Lee, Y.J.; Choi, Y.J.; Park, W.Y.; Shin, H.S.; Kim, J.H. Comprehensive analysis of microRNA–mRNA co-expression in circadian rhythm. Exp. Mol. Med. 2009, 41, 638–647. [Google Scholar] [CrossRef] [PubMed]
- Gatfield, D.; le Martelot, G.; Vejnar, C.E.; Gerlach, D.; Schaad, O.; Fleury-Olela, F.; Ruskeepää, A.L.; Oresic, M.; Esau, C.C.; Zdobnov, E.M.; et al. Integration of microRNA miR-122 in hepatic circadian gene expression. Genes Dev. 2009, 23, 1313–1326. [Google Scholar] [CrossRef]
- Kojima, S.; Gatfield, D.; Esau, C.C.; Green, C.B. MicroRNA-122 modulates the rhythmic expression profile of the circadian deadenylase Nocturnin in mouse liver. PLoS One 2010, 5, e11264. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Osterbur, D.L.; Megaw, P.L.; Tosini, G.; Fukuhara, C.; Green, C.B.; Besharse, J.C. Rhythmic expression of Nocturnin mRNA in multiple tissues of the mouse. BMC Dev. Biol. 2001, 1, 9. [Google Scholar] [CrossRef] [PubMed]
- Barbot, W.; Wasowicz, M.; Dupressoir, A.; Versaux-Botteri, C.; Heidmann, T. A murine gene with circadian expression revealed by transposon insertion: Self-sustained rhythmicity in the liver and the photoreceptors. Biochim. Biophys. Acta 2002, 1576, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Kojima, S.; Sher-Chen, E.L.; Green, C.B. Circadian control of mRNA polyadenylation dynamics regulates rhythmic protein expression. Genes Dev. 2012, 26, 2724–2736. [Google Scholar] [CrossRef] [PubMed]
- Oishi, K.; Miyazaki, K.; Kadota, K.; Kikuno, R.; Nagase, T.; Atsumi, G.; Ohkura, N.; Azama, T.; Mesaki, M.; Yukimasa, S.; et al. Genome-wide expression analysis of mouse liver reveals CLOCK-regulated circadian output genes. J. Biol. Chem. 2003, 278, 41519–41527. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Yue, J.; Zhang, Y.; Zhou, L.; Hao, W.; Yuan, J.; Qiang, B.; Ding, J.M.; Peng, X.; Cao, J.M. CLOCK/BMAL1 regulates human nocturnin transcription through binding to the E-box of nocturnin promoter. Mol. Cell. Biochem. 2008, 317, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Vollmers, C.; Schmitz, R.J.; Nathanson, J.; Yeo, G.; Ecker, J.R.; Panda, S. Circadian oscillations of protein-coding and regulatory RNAs in a highly dynamic mammalian liver epigenome. Cell Metab. 2012, 16, 833–845. [Google Scholar] [CrossRef] [PubMed]
- Nakahata, Y.; Kaluzova, M.; Grimaldi, B.; Sahar, S.; Hirayama, J.; Chen, D.; Guarente, L.P.; Sassone-Corsi, P. The NAD+-dependent deacetylase SIRT1 modulates CLOCK-mediated chromatin remodeling and circadian control. Cell 2008, 134, 329–340. [Google Scholar] [CrossRef]
- Grimaldi, B.; Nakahata, Y.; Kaluzova, M.; Masubuchi, S.; Sassone-Corsi, P. Chromatin remodeling, metabolism and circadian clocks: The interplay of CLOCK and SIRT1. Int. J. Biochem. Cell Biol. 2009, 41, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Nakahata, Y.; Sahar, S.; Astarita, G.; Kaluzova, M.; Sassone-Corsi, P. Circadian control of the NAD+ salvage pathway by CLOCK-SIRT1. Science 2009, 324, 654–657. [Google Scholar] [CrossRef] [PubMed]
- Bellet, M.M.; Orozco-Solis, R.; Sahar, S.; Eckel-Mahan, K.; Sassone-Corsi, P. The time of metabolism: NAD+, SIRT1, and the circadian clock. Cold Spring Harb. Symp. Quant. Biol. 2011, 76, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Sahar, S.; Sassone-Corsi, P. The epigenetic language of circadian clocks. Handb. Exp. Pharmacol. 2013, 217, 29–44. [Google Scholar] [PubMed]
- Duong, H.A.; Robles, M.S.; Knutti, D.; Weitz, C.J. A molecular mechanism for circadian clock negative feedback. Science 2011, 332, 1436–1439. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.A.; Ripperger, J.; Kadener, S.; Fleury-Olela, F.; Vilbois, F.; Rosbach, M.; Schibler, U. PERIOD1-associated proteins modulate the negative limb of the mammalian circadian oscillator. Science 2005, 308, 693–696. [Google Scholar] [CrossRef] [PubMed]
- Kowalska, E.; Ripperger, J.A.; Muheim, C.; Maier, B.; Kurihara, Y.; Fox, A.H.; Kramer, A.; Brown, S.A. Distinct roles of DBHS family members in the circadian transcriptional feedback loop. Mol. Cell. Biol. 2012, 32, 4585–4594. [Google Scholar] [PubMed]
- Kowalska, E.; Ripperger, J.A.; Hoegger, D.C.; Bruegger, P.; Buch, T.; Birchler, T.; Mueller, A.; Albrecht, U.; Contaldo, C.; Brown, S.A. NONO couples the circadian clock to the cell cycle. Proc. Natl. Acad. Sci. USA 2013, 110, 1592–1599. [Google Scholar] [CrossRef] [PubMed]
- Guillaumond, F.; Boyer, B.; Becquet, D.; Guillen, S.; Kuhn, L.; Garin, J.; Belghazi, M.; Bosler, O.; Franc, J.L.; François-Bellan, A.M. Chromatin remodeling as a mechanism for circadian prolactin transcription: Rhythmic NONO and SFPQ recruitment to HLTF. FASEB J. 2011, 25, 2740–2756. [Google Scholar] [CrossRef] [PubMed]
- Torres-Farfan, C.; Serón-Ferré, M.; Dinet, V.; Korf, H.W. Immunocytochemical demonstration of day/night changes of clock gene protein levels in the murine adrenal gland: Differences between melatonin-proficient (C3H) and melatonin-deficient (C57BL) mice. J. Pineal Res. 2006, 40, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Cuesta, J.; Tajes, M.; Jiménez, A.; Coto-Montes, A.; Camins, A.; Pallàs, M. Evaluation of potential pro-survival pathways regulated by melatonin in a murine senescence model. J. Pineal Res. 2008, 45, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.-M.; Wu, U.-I.; Lan, C.-T. Melatonin preserves longevity protein (sirtuin 1) expression in the hippocampus of total sleep-deprived rats. J. Pineal Res. 2009, 47, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Tajes, M.; Gutierrez-Cuesta, J.; Ortuño-Sahagun, D.; Camins, A.; Pallàs, M. Anti-aging properties of melatonin in an in vitro murine senescence model: Involvement of the sirtuin 1 pathway. J. Pineal Res. 2009, 47, 228–237. [Google Scholar] [CrossRef] [PubMed]
- Dubocovich, M.L.; Rivera-Bermudez, M.A.; Gerdin, M.J.; Masana, M.I. Molecular pharmacology, regulation and function of mammalian melatonin receptors. Front. Biosci. 2003, 8, d1093–d1108. [Google Scholar] [CrossRef] [PubMed]
- Dubocovich, M.L.; Markowska, M. Functional MT1 and MT2 melatonin receptors in mammals. Endocrine 2005, 27, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Goel, N.; Stunkard, A.J.; Rogers, N.L.; van Dongen, H.P.; Allison, K.C.; O’Reardon, J.P.; Ahima, R.S.; Cummings, D.E.; Heo, M.; Dinges, D.F. Circadian rhythm profiles in women with night eating syndrome. J. Biol. Rhythm. 2009, 24, 85–94. [Google Scholar] [CrossRef]
- Escames, G.; López, L.C.; Tapias, V.; Utrilla, P.; Reiter, R.J.; Hitos, A.B.; León, J.; Rodríguez, M.I.; Acuña-Castroviejo, D. Melatonin counteracts inducible mitochondrial nitric oxide synthase-dependent mitochondrial dysfunction in skeletal muscle of septic mice. J. Pineal Res. 2006, 40, 71–78. [Google Scholar] [CrossRef] [PubMed]
- López, L.C.; Escames, G.; Tapias, V.; Utrilla, P.; León, J.; Acuña-Castroviejo, D. Identification of an inducible nitric oxide synthase in diaphragm mitochondria from septic mice: Its relation with mitochondrial dysfunction and prevention by melatonin. Int. J. Biochem. Cell Biol. 2006, 38, 267–278. [Google Scholar] [CrossRef] [PubMed]
- Tapias, V.; Escames, G.; López, L.C.; López, A.; Camacho, E.; Carrión, M.D.; Entrena, A.; Gallo, M.A.; Espinosa, A.; Acuña-Castroviejo, D. Melatonin and its brain metabolite N1-acetyl-5-methoxykynuramine prevent mitochondrial nitric oxide synthase induction in parkinsonian mice. J. Neurosci. Res. 2009, 87, 3002–3010. [Google Scholar] [CrossRef] [PubMed]
- Kawai, M.; Green, C.B.; Lecka-Czernik, B.; Douris, N.; Gilbert, M.R.; Kojima, S.; Ackert-Bicknell, C.; Garg, N.; Horowitz, M.C.; Adamo, M.L.; et al. A circadian-regulated gene, Nocturnin, promotes adipogenesis by stimulating PPARγ nuclear translocation. Proc. Natl. Acad. Sci. USA 2010, 107, 10508–10513. [Google Scholar] [CrossRef] [PubMed]
- Lewy, A.J.; Wehr, T.A.; Goodwin, F.K.; Newsome, D.A.; Markey, S.P. Light suppresses melatonin secretion in humans. Science 1980, 210, 1267–1269. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J. The melatonin rhythm: Both a clock and a calendar. Experientia 1993, 49, 654–664. [Google Scholar] [CrossRef] [PubMed]
- Klein, D.C. Arylalkylamine N-acetyltransferase: “The timezyme”. J. Biol. Chem. 2007, 282, 4233–4237. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J.; Tan, D.-X.; Korkmaz, A.; Erren, T.C.; Piekarski, C.; Tamura, H.; Manchester, L.C. Light at night, chronodisruption, melatonin suppression, and cancer risk: A review. Crit. Rev. Oncog. 2007, 13, 303–328. [Google Scholar] [CrossRef] [PubMed]
- Erren, T.C.; Reiter, R.J. Light hygiene: Time to make preventive use of insights—old and new—into the nexus of the drug light, melatonin, clocks, chronodisruption and public health. Med. Hyptheses 2009, 73, 537–541. [Google Scholar] [CrossRef]
- Stevens, R.G. Working against our endogenous circadian clock: Breast cancer and electric lighting in the modern world. Mutat. Res. 2009, 680, 106–108. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J.; Tan, D.-X.; Erren, T.C.; Fuentes-Broto, L.; Paredes, S.D. Light-mediated perturbations of circadian timing and cancer risk: A mechanistic analysis. Integr. Cancer Ther. 2009, 8, 354–360. [Google Scholar] [CrossRef] [PubMed]
- Anisimov, V.N.; Vinogradova, I.A.; Panchenko, A.V.; Popovich, I.G.; Zabezhinski, M.A. Light-at-night-induced circadian disruption, cancer and aging. Curr. Aging Sci. 2012, 5, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J.; Tan, D.-X.; Korkmaz, A.; Ma, S. Obesity and metabolic syndrome: Association with chronodisruption, sleep deprivation, and melatonin suppression. Ann. Med. 2012, 44, 564–577. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Stevens, R.G.; Hoffman, A.E.; Tjonneland, A.; Vogel, U.B.; Zheng, T.; Hansen, J. Epigenetic impact of long-term shiftwork: Pilot evidence from circadian genes and whole-genome methylation analysis. Chronobiol. Int. 2011, 28, 852–861. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, D.I.; Hansen, J.; Fu, A.; Stevens, R.G.; Tjonneland, A.; Vogel, U.B.; Zheng, T.; Zhu, Y. Methylation alterations at imprinted genes detected among long-term shiftworkers. Environ. Mol. Mutagen. 2013, 54, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Schwimmer, H.; Metzer, A.; Pilosof, Y.; Szyf, M.; Machnes, Z.M.; Fares, F.; Harel, O.; Haim, A. Light at night and melatonin have opposite effects on breast cancer tumors in mice assessed by growth rates and global DNA methylation. Chronobiol. Int. 2014, 31, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Peschke, E. Melatonin, endocrine pancreas and diabetes. J. Pineal Res. 2008, 44, 26–40. [Google Scholar] [PubMed]
- Shieh, J.-M.; Wu, H.-T.; Cheng, K.-C.; Cheng, J.-T. Melatonin ameliorates high fat diet-induced diabetes and stimulates glycogen synthesis via a PKCζ-Akt-GSK3β pathway in hepatic cells. J. Pineal Res. 2009, 47, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Peschke, E.; Mühlbauer, E. New evidence for a role of melatonin in glucose regulation. Best Pract. Res. Clin. Endocrinol. Metab. 2010, 24, 829–841. [Google Scholar] [CrossRef] [PubMed]
- Hardeland, R. Melatonin and the theories of aging: A critical appraisal of melatonin’s role in antiaging mechanisms. J. Pineal Res. 2013, 55, 325–356. [Google Scholar] [PubMed]
- Corbalán-Tutau, D.; Madrid, J.A.; Nicolás, F.; Garaulet, M. Daily profile of two circadian markers “melatonin and cortisol” and associations with metabolic syndrome components. Physiol. Behav. 2014, 123, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S.; Inoue, I.; Nakajima, Y.; Seo, M.; Nakano, T.; Yang, F.; Kimagai, M.; Komoda, T.; Awata, T.; Ikeda, M.; et al. A promoter in the novel exon of hPPARγ directs the circadian expression of PPARγ. J. Atheroscler. Thromb. 2010, 17, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Su, P.; Xu, C.; Chen, C.; Liang, A.; Du, K.; Peng, Y.; Huang, D. Melatonin inhibits adipogenesis and enhances osteogenesis of human mesenchymal stem cells by suppressing PPARγ expression and enhancing Runx2 expression. J. Pineal Res. 2010, 49, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Vale, M.I.C.; Peres, S.B.; Vernochet, C.; Farmer, S.R.; Lima, F.B. Adipocyte differentiation is inhibited by melatonin through the regulation of C/EBPβ transcriptional activity. J. Pineal Res. 2009, 47, 221–227. [Google Scholar] [CrossRef] [PubMed]
- González, A.; Alvarez-García, V.; Martínez-Campa, C.; Alonso-González, C.; Cos, S. Melatonin promotes differentiation of 3T3-L1 fibroblasts. J. Pineal Res. 2012, 52, 12–20. [Google Scholar] [CrossRef] [PubMed]
- De Luxán-Delgado, B.; Caballero, B.; Potes, Y.; Rubio-González, A.; Rodríguez, I.; Gutiérrez-Rodríguez, J.; Solano, J.J.; Coto-Montes, A. Melatonin administration decreases adipogenesis in the liver of ob/ob mice through autophagy modulation. J. Pineal Res. 2014, 56, 126–133. [Google Scholar] [CrossRef]
- Tan, D.-X.; Manchester, L.C.; Fuentes-Broto, L.; Paredes, S.D.; Reiter, R.J. Significance and application of melatonin in the regulation of brown adipose tissue metabolism: Relation to human obesity. Obes. Rev. 2011, 12, 167–188. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Aranda, A.; Fernández-Vázquez, G.; Campos, D.; Tassi, M.; Velasco-Perez, L.; Tan, D.-X.; Reiter, R.J.; Agil, A. Melatonin induces browning of inguinal adipose tissue in Zucker diabetic fatty rats. J. Pineal Res. 2013, 55, 416–423. [Google Scholar] [PubMed]
- Vinogradova, I.; Anisimov, V. Melatonin prevents the development of the metabolic syndrome in male rats exposed to different light/dark regimes. Biogerontology 2013, 14, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Douris, N.; Kojima, S.; Pan, X.; Lerch-Gaggl, A.F.; Duong, S.Q.; Hussain, M.M.; Green, C.B. Nocturnin regulates circadian trafficking of dietary lipid in intestinal enterocytes. Curr. Biol. 2011, 21, 1347–1355. [Google Scholar] [CrossRef] [PubMed]
- Hussain, M.M.; Pan, X. Clock regulation of dietary lipid absorption. Curr. Opin. Clin. Nutr. Metab. Care 2012, 15, 336–341. [Google Scholar] [CrossRef] [PubMed]
- Douris, N.; Green, C.B. NOC out the fat: A short review of the circadian deadenylase Nocturnin. Ann. Med. 2008, 40, 622–626. [Google Scholar] [CrossRef] [PubMed]
- Kawai, M.; Rosen, C.J. PPARγ: A circadian transcription factor in adipogenesis and osteogenesis. Nat. Rev. Endocrinol. 2010, 6, 629–636. [Google Scholar] [CrossRef] [PubMed]
- Lecka-Czernik, B.; Rosen, C.J.; Kawai, M. Skeletal aging and the adipocyte program. Cell Cycle 2010, 9, 3648–3654. [Google Scholar] [CrossRef] [PubMed]
- Guntur, A.R.; Kawai, M.; Le, P.; Bouxsein, M.L.; Bornstein, S.; Green, C.B.; Rosen, C.J. An essential role for the circadian-regulated gene Nocturnin in osteogenesis: The importance of local timekeeping in skeletal homeostasis. Ann. N. Y. Acad. Sci. 2011, 1237, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Kawai, M.; Delany, A.M.; Green, C.B.; Adamo, M.L.; Rosen, C.J. Nocturnin suppresses igf1 expression in bone by targeting the 3'-untranslated region of igf1 mRNA. Endocrinology 2010, 151, 4861–4870. [Google Scholar] [CrossRef] [PubMed]
- Sethi, S.; Radio, N.M.; Kotlarczyk, M.P.; Chen, C.T.; Wei, Y.H.; Jockers, R.; Witt-Enderby, P.A. Determination of the minimal melatonin exposure required to induce osteoblast differentiation from human mesenchymal stem cells and these effects on downstream signaling pathways. J. Pineal Res. 2010, 49, 222–238. [Google Scholar] [CrossRef] [PubMed]
- Park, K.-H.; Kang, J.W.; Lee, E.M.; Kim, J.S.; Rhee, Y.H.; Kim, M.; Jeong, S.J.; Park, Y.G.; Kim, S.H. Melatonin promotes osteoblastic differentiation through the BMP/ERK/Wnt signaling pathways. J. Pineal Res. 2011, 51, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, J.; Ling, Y.; Chen, C.; Liang, A.; Peng, Y.; Chang, H.; Su, P.; Huang, D. Sustained release of melatonin from poly (lactic-co-glycolic acid) (PLGA) microspheres to induce osteogenesis of human mesenchymal stem cells in vitro. J. Pineal Res. 2013, 54, 24–32. [Google Scholar] [PubMed]
- Aravamudhan, A.; Ramos, D.M.; Nip, J.; Subramanian, A.; James, R.; Harmon, M.D.; Yu, X.; Kumbar, S.G. Osteoinductive small molecules: Growth factor alternatives for bone tissue engineering. Curr. Pharm. Des. 2013, 19, 3420–3428. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.-H.; Wang, S.-G.; Liu, S.-X.; Li, J.-P.; Zhang, Y.-X.; Sun, Z.-Y.; Fan, Q.-M.; Tian, J.-W. PPARγ forms a bridge between DNA methylation and histone acetylation at the C/EBPα gene promoter to regulate the balance between osteogenesis and adipogenesis of bone marrow stromal cells. FEBS J. 2013, 280, 5801–5814. [Google Scholar] [CrossRef] [PubMed]
- Hemming, S.; Cakouros, D.; Isenmann, S.; Cooper, L.; Menicanin, D.; Zannettino, A.; Gronthos, S. EZH2 and KDM6A act as an epigenetic switch to regulate mesenchymal stem cell lineage specification. Stem Cells 2014, 32, 802–815. [Google Scholar] [CrossRef] [PubMed]
- Garbarino-Pico, E.; Green, C.B. Posttranscriptional regulation of mammalian circadian clock output. Cold Spring Harb. Symp. Quant. Biol. 2007, 72, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Hee, S.-W.; Tsai, S.-H.; Chang, Y.-C.; Chang, C.-J.; Yu, I.-S.; Lee, P.-C.; Lee, W.-J.; Chang, Y.-C.E.; Chuang, L.-M. The role of nocturnin in early adipogenesis and modulation of sytemic insulin resistance in human. Obesity 2012, 20, 1558–1565. [Google Scholar] [CrossRef] [PubMed]
- Garbarino-Pico, E.; Niu, S.; Rollag, M.D.; Strayer, C.A.; Besharse, J.C.; Green, C.B. Immediate early response of the circadian polyA ribonuclease nocturnin to two extracellular stimuli. RNA 2007, 13, 745–755. [Google Scholar] [CrossRef] [PubMed]
- Massiera, F.; Barbry, P.; Guesnet, P.; Joly, A.; Luquet, S.; Moreilhon-Brest, C.; Mohsen-Kanson, T.; Amri, E.-Z.; Ailhaud, G. A Western-like fat diet is sufficient to induce a gradual enhancement in fat mass over generations. J. Lipid Res. 2010, 51, 2352–2361. [Google Scholar] [CrossRef] [PubMed]
- Hardeland, R. Antioxidative protection by melatonin—Multiplicity of mechanisms from radical detoxification to radical avoidance. Endocrine 2005, 27, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Hardeland, R. Melatonin and its metabolites as anti-nitrosating and anti-nitrating agents. J. Exp. Integr. Med. 2011, 1, 67–81. [Google Scholar] [CrossRef]
- Arese, M.; Magnifico, M.C.; Mastronicola, D.; Altieri, F.; Grillo, C.; Blanck, T.J.J.; Sarti, P. Nanomolar melatonin enhances nNOS expression and controls HaCaT-cells bioenergetics. IUBMB Life 2012, 64, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Sarti, P.; Magnifico, M.C.; Altieri, F.; Mastronicola, D.; Arese, M. New evidence for cross talk between melatonin and mitochondria mediated by a circadian-compatible interaction with nitric oxide. Int. J. Mol. Sci. 2013, 14, 11259–11276. [Google Scholar] [CrossRef] [PubMed]
- Cencioni, C.; Spallotta, F.; Martelli, F.; Valente, S.; Mai, A.; Zeiher, A.M.; Gaetano, C. Oxidative stress and epigenetic regulation in ageing and age-related diseases. Int. J. Mol. Sci. 2013, 14, 17643–17663. [Google Scholar] [PubMed]
- Ito, T.; Yagi, S.; Yamakuchi, M. MicroRNA-34a regulation of endothelial senescence. Biochem. Biophys. Res. Commun. 2010, 398, 735–740. [Google Scholar] [CrossRef] [PubMed]
- Boon, R.A.; Iekushi, K.; Lechner, S.; Seeger, T.; Fischer, A.; Heydt, S.; Kaluza, D.; Treguer, K.; Carmona, G.; Bonauer, A.; et al. MicroRNA-34a regulates cardiac ageing and function. Nature 2013, 35, 107–119. [Google Scholar]
- Reiter, R.J.; Tan, D.-X.; Paredes, S.D.; Fuentes-Broto, L. Beneficial effects of melatonin in cardiovascular disease. Ann. Med. 2010, 42, 276–285. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Rodriguez, A. Melatonin in cardiovascular disease. Expert Opin. Investig. Drugs 2012, 21, 1593–1596. [Google Scholar] [PubMed]
- Lochner, A.; Huisamen, B.; Nduhirabandi, F. Cardioprotective effect of melatonin against ischemia/reperfusion damage. Front. Biosci. 2013, 5, 305–315. [Google Scholar] [CrossRef]
- Magenta, A.; Cencioni, C.; Fasanaro, P.; Zaccagnini, G.; Greco, S.; Sarra-Ferraris, G.; Antonini, A.; Martelli, F.; Capogrossi, M.C. miR-200c is up-regulated by oxidative stress and induces endothelial cell apoptosis and senescence via ZEB1 inhibition. Cell Death Differ. 2011, 18, 1628–1639. [Google Scholar] [CrossRef] [PubMed]
- Cabrer, J.; Burkhardt, S.; Tan, D.-X.; Manchester, L.C.; Karbownik, M.; Reiter, R.J. Autoxidation and toxicant-induced oxidation of lipid and DNA in monkey liver: Reduction of molecular damage by melatonin. Pharmacol. Toxicol. 2001, 89, 225–230. [Google Scholar] [CrossRef] [PubMed]
- López-Burillo, S.; Tan, D.-X.; Mayo, J.C.; Sainz, R.M.; Manchester, L.C.; Reiter, R.J. Melatonin, xanthurenic acid, resveratrol, EGCG, vitamin C and α-lipoic acid differentially reduce oxidative DNA damage induced by Fenton reagents: A study of their individual and synergistic actions. J. Pineal Res. 2003, 34, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Korkmaz, A.; Topal, T.; Oter, S.; Tan, D.-X.; Reiter, R.J. Hyperglycemia-related pathophysiologic mechanisms and potential beneficial actions of melatonin. Mini-Rev. Med. Chem. 2008, 8, 1144–1153. [Google Scholar] [CrossRef] [PubMed]
- Korkmaz, A.; Kunak, Z.I.; Paredes, S.D.; Yaren, H.; Tan, D.-X.; Reiter, R.J. The use of melatonin to combat mustard toxicity. Neuroendocrinol. Lett. 2008, 29, 614–619. [Google Scholar] [PubMed]
- Natarajan, M.; Sadeghi, K.; Reiter, R.J.; Meltz, M.L. The neurohormone melatonin inhibits cytokine, mitogen, and ionizing radiation induced NF-κB. Biochem. Mol. Biol. Int. 1995, 37, 1063–1070. [Google Scholar] [PubMed]
- Deng, W.G.; Tang, S.T.; Tseng, H.P.; Wu, K.K. Melatonin suppresses macrophage cyclooxygenase-2 and inducible nitric oxide synthase expression by inhibiting p52 acetylation and binding. Blood 2006, 108, 518–524. [Google Scholar] [CrossRef] [PubMed]
- Chuang, J.I.; Natarajan, M.; Meltz, M.L.; Reiter, R.J. Effect of melatonin on NF-κB DNA-binding activity in the rat spleen. Cell Biol. Int. 1996, 20, 687–692. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.D.; Lee, J.S. Interplay between epigenetics and genetics in cancer. Genomics Inf. 2013, 11, 164–173. [Google Scholar] [CrossRef]
- Tam, W.L.; Weinberg, R.A. The epigenetics of endothelial-mesenchymal plasticity in cancer. Nat. Med. 2013, 19, 1438–1449. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.M.; Kwon, M.J.; Shin, Y.K. Over-expression of cancer-associated genes via epigenetic derepression mechanisms in gynecologic cancer. Front. Oncol. 2014, 4, 12. [Google Scholar] [CrossRef] [PubMed]
- Kanai, Y.; Arai, E. Multilayer-omics analyses of human cancers: Exploration of biomarkers and drug targets based in the activities of the International Human Epigenome Consortium. Front. Genet. 2014, 5, 24. [Google Scholar] [PubMed]
- Huarte, M.; Rinn, J.L. Large non-coding RNAs: Missing links to cancer? Hum. Mol. Genet. 2010, 19, R152–R161. [Google Scholar]
- Hung, T.; Chang, H.Y. Long noncoding RNA in genome regulation: Prospects and mechanisms. RNA Biol. 2010, 7, 583–585. [Google Scholar] [CrossRef]
- Ye, C.; Li, L. 5-Hydroxymethylcytosine: A new insight into epigenetics in cancer. Cancer Biol. Ther. 2014, 15, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.C.; Ling, Z.Q. The role of TET family proteins and 5-hydroxymethylcytosine in human tumors. Histol. Histopathol. 2014, 29, 991–997. [Google Scholar] [PubMed]
- Lam, L.L.; Emberly, E.; Fraser, H.B.; Neumann, S.M.; Chen, E.; Miller, G.E.; Kobor, M.S. Factors underlying variable DNA methylation in human community cohort. Proc. Natl. Acad. Sci. USA 2012, 109, 17253–17260. [Google Scholar] [CrossRef] [PubMed]
- Stankiewicz, A.M.; Swiergiel, A.H.; Lisowski, P. Epigenetics of stress adaptations in the brain. Brain Res. Bull. 2013, 98, 76–92. [Google Scholar] [CrossRef]
- Klengel, T.; Pape, J.; Binder, E.B.; Mehta, D. The role of DNA methylation in stress-related psychiatric disorders. Neuropharmacology 2014, 80, 115–132. [Google Scholar] [CrossRef] [PubMed]
- Vojta, A.; Zoldož, V. Adaptation or malignant transformation: The two faces of epigenetically mediated responses to stress. Biomed. Res. Int. 2013, 2013, 954060. [Google Scholar] [CrossRef] [PubMed]
- Bob, P.; Fedor-Freybergh, P. Melatonin, consciousness, and traumatic stress. J. Pineal Res. 2008, 44, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Haridas, S.; Kumar, M.; Manda, K. Melatonin ameliorates chronic mild stress induced behavioral dysfunctions in mice. Physiol. Behav. 2013, 119, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Muller, M.D.; Sauder, C.L.; Ray, C.A. Melatonin attenuates the skin sympathetic nerve response to mental stress. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H1382–H1386. [Google Scholar] [CrossRef]
- Ruksee, N.; Tongjaroenbuangam, W.; Mahanam, T.; Govitrapong, P. Melatonin pretreatment prevented the effect of dexamethasone negative alterations on behavior and hippocampal neurogenesis in the mouse brain. J. Steroid Biochem. Mol. Biol. 2014, 143C, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Bosch-Presegué, L.; Vaquero, A. Sirtuins in stress response: Guardians of the genome. Oncogene 2014, 33, 3764–3775. [Google Scholar] [CrossRef] [PubMed]
- Bosch-Presegué, L.; Vaquero, A. The dual role of sirtuins in cancer. Genes Cancer 2011, 2, 648–662. [Google Scholar] [CrossRef] [PubMed]
- Asher, G.; Gatfield, D.; Stratmann, M.; Reinke, H.; Dibner, C.; Kreppel, F.; Mostoslavsky, R.; Alt, F.W.; Schibler, U. SIRT1 regulates circadian clock gene expression through PER2 deacetylation. Cell 2008, 134, 317–328. [Google Scholar] [CrossRef] [PubMed]
- Joshi, P.; Quach, O.L.; Giguere, S.S.; Cristea, I.M. A functional proteomics perspective of DBC1 as a regulator of transcription. J. Prot. Bioinform. 2013, (Suppl. 2), 002. [Google Scholar]
- Jung-Hynes, B.; Nihal, M.; Zhong, W.; Ahmad, N. Role of histone deacetylase SIRT1 in prostate cancer. A target for prostate cancer management via its inhibition? J. Biol. Chem. 2009, 284, 3823–3832. [Google Scholar]
- Jung-Hynes, B.; Ahmad, N. SIRT1 controls circadian clock circuitry and promotes cell survival: A connection with age-related neoplasms. FASEB J. 2009, 23, 2803–2809. [Google Scholar] [CrossRef]
- Jung-Hynes, B.; Reiter, R.J.; Ahmad, N. Sirtuins, melatonin and circadian rhythms: Building a bridge between aging and cancer. J. Pineal Res. 2010, 48, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Jung-Hynes, B.; Schmit, T.L.; Reagan-Shaw, S.R.; Siddiqui, I.A.; Mukhtar, H.; Ahmad, N. Melatonin, a novel Sirt1 inhibitor, imparts antiproliferative effects against prostate cancer in vitro in culture and in vivo in TRAMP model. J. Pineal Res. 2011, 50, 140–149. [Google Scholar] [PubMed]
- Hill, S.M.; Frasch, T.; Xiang, S.; Yuan, L.; Duplessis, T.; Mao, L. Molecular mechanisms of melatonin anticancer effects. Integr. Cancer Ther. 2009, 8, 337–346. [Google Scholar] [CrossRef]
- Lee, S.E.; Kim, S.J.; Youn, J.-P.; Hwang, S.Y.; Park, C.-S.; Park, Y.S. MicroRNA and gene expression analysis of melatonin-exposed breast cancer cell lines indicating involvement of the anticancer effect. J. Pineal Res. 2011, 51, 345–352. [Google Scholar] [PubMed]
- Lee, S.E.; Kim, S.J.; Youn, J.-P.; Yu, S.Y.; Yang, H.; Jeong, S.I.; Hwang, S.Y.; Park, C.-S.; Park, Y.S. Genome-wide profiling in melatonin-exposed human breast cancer cell lines identifies differentially methylated genes involved in the anticancer effect of melatonin. J. Pineal Res. 2013, 54, 80–88. [Google Scholar] [PubMed]
- Sweatt, J.D. The emerging field of neuroepigenetics. Neuron 2013, 80, 624–632. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.Q.; Jordan, I.K.; Kunyak, V.V. Epigenetics components of aging in the central nervous system. Neurotherapeutics 2013, 10, 647–663. [Google Scholar] [CrossRef] [PubMed]
- Guzman-Karlsson, M.C.; Meadows, J.P.; Gavin, C.F.; Hablitz, J.J.; Sweatt, J.D. Transcriptional and epigenetic regulation of Hebbian and non-Hebbian plasticity. Neuropharmacology 2014, 80, 3–17. [Google Scholar] [CrossRef]
- Rudenko, A.; Tsai, L.H. Epigenetic modifications in the nervous system and their impact upon cognitive impairments. Neuropharmacology 2014, 80, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Saab, B.J.; Mansuy, I.M. Neuroepigenetics of memory formation and impairment: The role of microRNAs. Neuropharmacology 2014, 80, 61–69. [Google Scholar] [CrossRef]
- Detanico, B.C.; Piato, A.L.; Freitas, J.J.; Lhullier, F.-L.; Hidalgo, M.P.; Caumo, W.; Elisabetsky, E. Antidepressant-like effects of melatonin in the mouse chronic mild stress model. Eur. J. Pharmacol. 2009, 607, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, V.; Pandi-Perumal, S.R.; Trakht, I.; Spence, D.W.; Hardeland, R.; Poeggeler, B.; Cardinali, D.P. Pathophysiology of depression: Role of sleep and the melatonergic system. Psychiatr. Res. 2009, 165, 201–214. [Google Scholar] [CrossRef]
- Maldonado, M.D.; Reiter, R.J.; Pérez-San-Gregorio, M.A. Melatonin as a potential therapeutic agent in psychiatric illness. Hum. Psychopharmacol. 2009, 24, 391–400. [Google Scholar] [CrossRef]
- Hardeland, R. Melatonin in aging and disease—Multiple consequences of reduced secretion, options and limits of treatment. Aging Dis. 2012, 3, 194–225. [Google Scholar] [PubMed]
- Morley-Fletcher, S.; Mairesse, J.; Soumier, A.; Banasr, M.; Fagioli, F.; Gabriel, C.; Mocaer, E.; Daszuta, A.; McEwen, B.; Nicoletti, F.; et al. Chronic agomelatine treatment corrects behavioral, cellular, and biochemical abnormalities induced by prenatal stress in rats. Psychopharmacology 2011, 217, 301–313. [Google Scholar] [PubMed]
- Irmak, M.K.; Sizlan, A. Essential hypertension seems to result from melatonin-induced epigenetic modifications in area postrema. Med. Hypotheses 2006, 66, 1000–1007. [Google Scholar] [CrossRef] [PubMed]
- Irmak, M.K.; Topal, T.; Oter, S. Melatonin seems to be a mediator that transfers the environmental stimuli to oocytes for inheritance of adaptive changes through epigenetic inheritance system. Med. Hypotheses 2005, 64, 1138–1143. [Google Scholar]
- Niles, L.P.; Pan, Y.; Kang, S.; Lacoul, A. Melatonin induces histone hyperacetylation in the rat brain. Neurosci. Lett. 2013, 541, 49–53. [Google Scholar] [PubMed]
- Sharma, R.; Ottenhof, T.; Rzeczkowska, P.A.; Niles, L.P. Epigenetic targets for melatonin: Induction of histone H3 hyperacetylation and gene expression in C17.2 neural stem cells. J. Pineal Res. 2008, 45, 277–264. [Google Scholar] [CrossRef] [PubMed]
- Sarlak, G.; Jenwitheesuk, A.; Chetsawang, B.; Govitrapong, P. Effects of melatonin on nervous system aging: Neurogenesis and neurodegeneration. J. Pharmacol. Sci. 2013, 123, 9–24. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Dai, E.; Zhang, Y.; Chen, X.; Liu, X.; Wang, S.; Wang, L.; Jiang, W. Constructing and characterizing a bioactive small molecule and microRNA association network for Alzheimer’s disease. J. R. Soc. Interface 2013, 11, 20131057. [Google Scholar] [CrossRef] [PubMed]
- Bacalini, M.G.; Friso, S.; Olivieri, F.; Pirazzini, C.; Guiliani, C.; Capri, M.; Santoro, A.; Franceschi, C.; Garagnani, P. Present and future of anti-ageing epigenetic diets. Mech. Ageing Dev. 2014, 136, 101–115. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, Z.-H.; Wu, Y.-Y.; Tang, H.; Tan, L.; Wang, X.; Gao, X.-Y.; Xiong, Y.-S.; Liu, D.; Wang, J.-Z.; et al. Melatonin attenuates scopolamine-induced memory/synaptic disorder by rescuing EPACs/miR-124/Egr1 pathway. Mol. Neurobiol. 2013, 47, 373–381. [Google Scholar] [CrossRef]
- Wilkinson, B.; Campbell, D.B. Contribution of long non-coding RNAs to autism spectrum disorders risk. Int. Rev. Neurobiol. 2013, 113, 35–59. [Google Scholar] [PubMed]
- Lanfumey, L.; Mongeau, R.; Hamon, M. Biological rhythms and melatonin in mood disorders and their treatments. Pharmacol. Ther. 2013, 138, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Dodd, S.; Maes, M.; Anderson, G.; Dean, O.M.; Moylan, S.; Berk, M. Putative neuroprotective agents in neuropsychiatric disorders. Prog. Neuropsychopharmacol. Biol. Psychiatry 2013, 42, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Vogel-Ciernia, A.; Wood, M.A. Neuron-specific chromatin remodeling: A missing link in epigenetic mechanisms underlying synaptic plasticity, memory, and intellectual disability disorders. Neuropharmacology 2014, 80, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Millan, M.J. On “polypharmacy” and multi-target agents, complementary strategies for improving the treatment of depression: A comparative appraisal. Int. J. Neuropsychopharmacol. 2014, 17, 1009–1037. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, A.; Malchow, B.; Hasan, A.; Falkai, P. The impact of environmental factors in severe psychiatric disorders. Front. Neurosci. 2014, 8, 19. [Google Scholar] [CrossRef] [PubMed]
- Orozco-Solis, R.; Sassone-Corsi, P. Epigenetic control and the circadian clock: Linking metabolism to neuronal responses. Neuroscience 2014, 264C, 76–87. [Google Scholar] [CrossRef] [PubMed]
- Akula, N.; Barb, J.; Jiang, X.; Wendland, J.R.; Choi, K.H.; Sen, S.K.; Hou, L.; Chen, D.T.W.; Laje, G.; Johnson, K.; et al. RNA-sequencing of the brain transcriptome implicates dysregulation of neuroplasticity, circadian rhythms and GTPase binding in bipolar disorder. Mol. Psychiatry 2014. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hardeland, R. Melatonin, Noncoding RNAs, Messenger RNA Stability and Epigenetics—Evidence, Hints, Gaps and Perspectives. Int. J. Mol. Sci. 2014, 15, 18221-18252. https://doi.org/10.3390/ijms151018221
Hardeland R. Melatonin, Noncoding RNAs, Messenger RNA Stability and Epigenetics—Evidence, Hints, Gaps and Perspectives. International Journal of Molecular Sciences. 2014; 15(10):18221-18252. https://doi.org/10.3390/ijms151018221
Chicago/Turabian StyleHardeland, Rüdiger. 2014. "Melatonin, Noncoding RNAs, Messenger RNA Stability and Epigenetics—Evidence, Hints, Gaps and Perspectives" International Journal of Molecular Sciences 15, no. 10: 18221-18252. https://doi.org/10.3390/ijms151018221
APA StyleHardeland, R. (2014). Melatonin, Noncoding RNAs, Messenger RNA Stability and Epigenetics—Evidence, Hints, Gaps and Perspectives. International Journal of Molecular Sciences, 15(10), 18221-18252. https://doi.org/10.3390/ijms151018221