The Cellular and Molecular Carcinogenic Effects of Radon Exposure: A Review


Abstract
:
1. Introduction
2. Genetic Effects
2.1. Environmental Investigations at the TP53 Locus
2.1.1. Hotspot Mutations of the TP53 Gene
2.1.2. Investigations of TP53 Mutations in Uranium Miners
2.1.3. Investigations of TP53 Mutations in Non-Mining Individuals
2.2. TP53 Laboratory Investigations
2.3. Environmental Investigations at the HPRT locus
2.3.1. Investigations of HPRT Mutations in Uranium Miners
2.3.2. Investigation of HPRT and BCL-2 Mutations in Non Mining Individuals
2.4. HPRT Laboratory Investigations
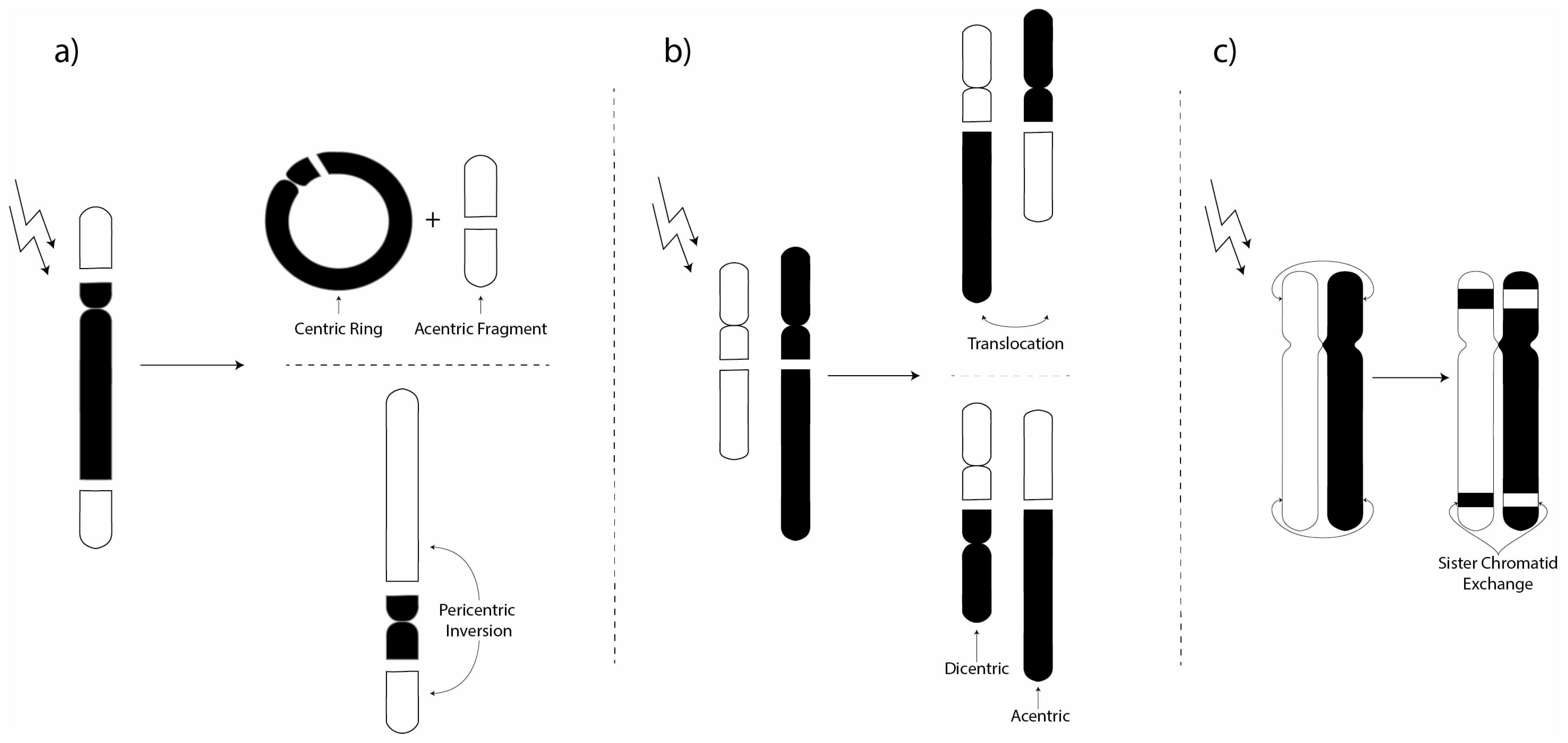
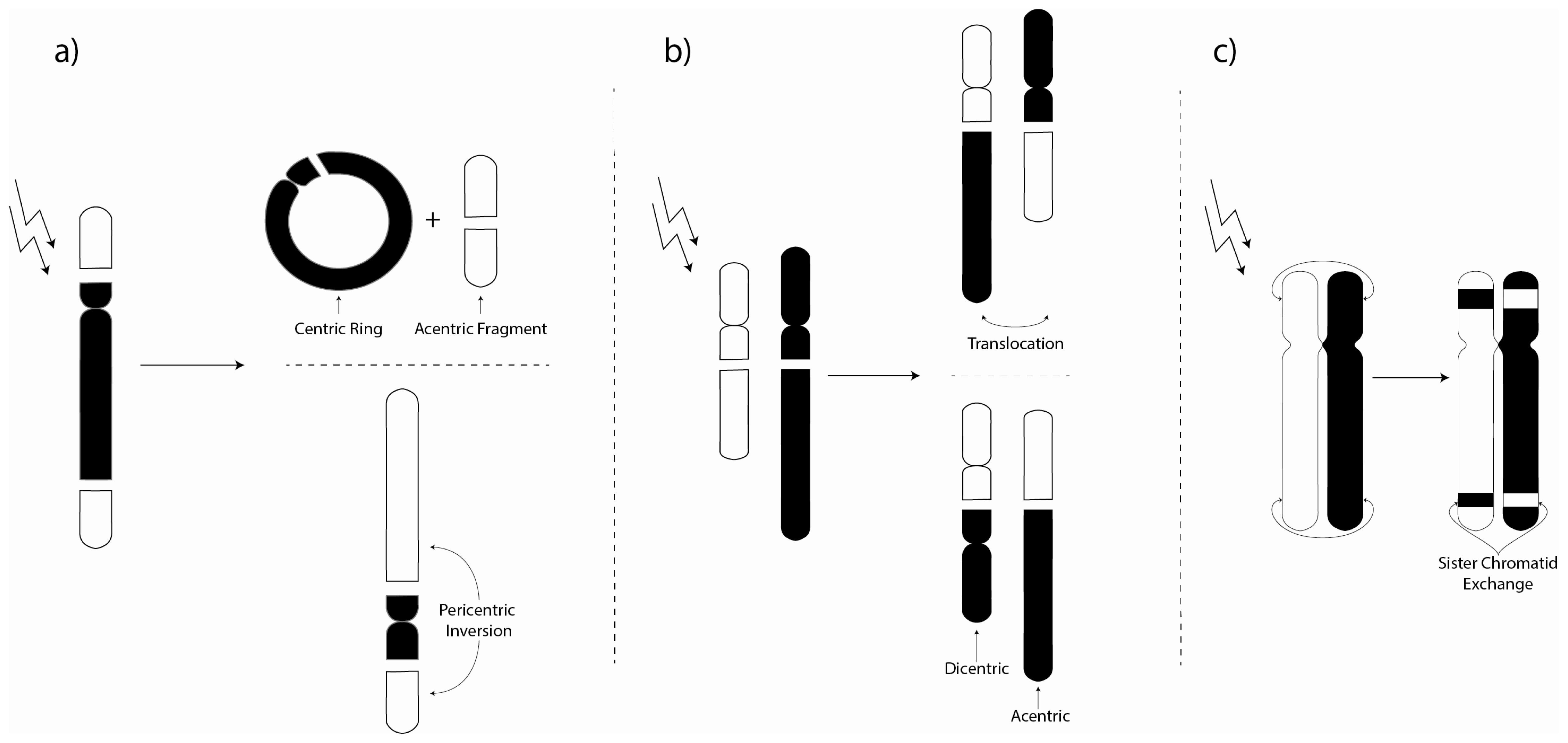
3. Chromosome Aberrations
3.1. Environmental Investigations of Chromosome Aberrations
3.1.1. Investigations of Chromosome Aberrations in Uranium Miners
3.1.2. Investigations of Chromosome Aberrations in Non Mining Individuals
3.2. Laboratory Chromosome Aberration Investigations
3.3. Biomarker Chromosome Aberration Ratio Studies
3.4. Experimental Investigations of Micronuclei
3.5. Genomic Studies
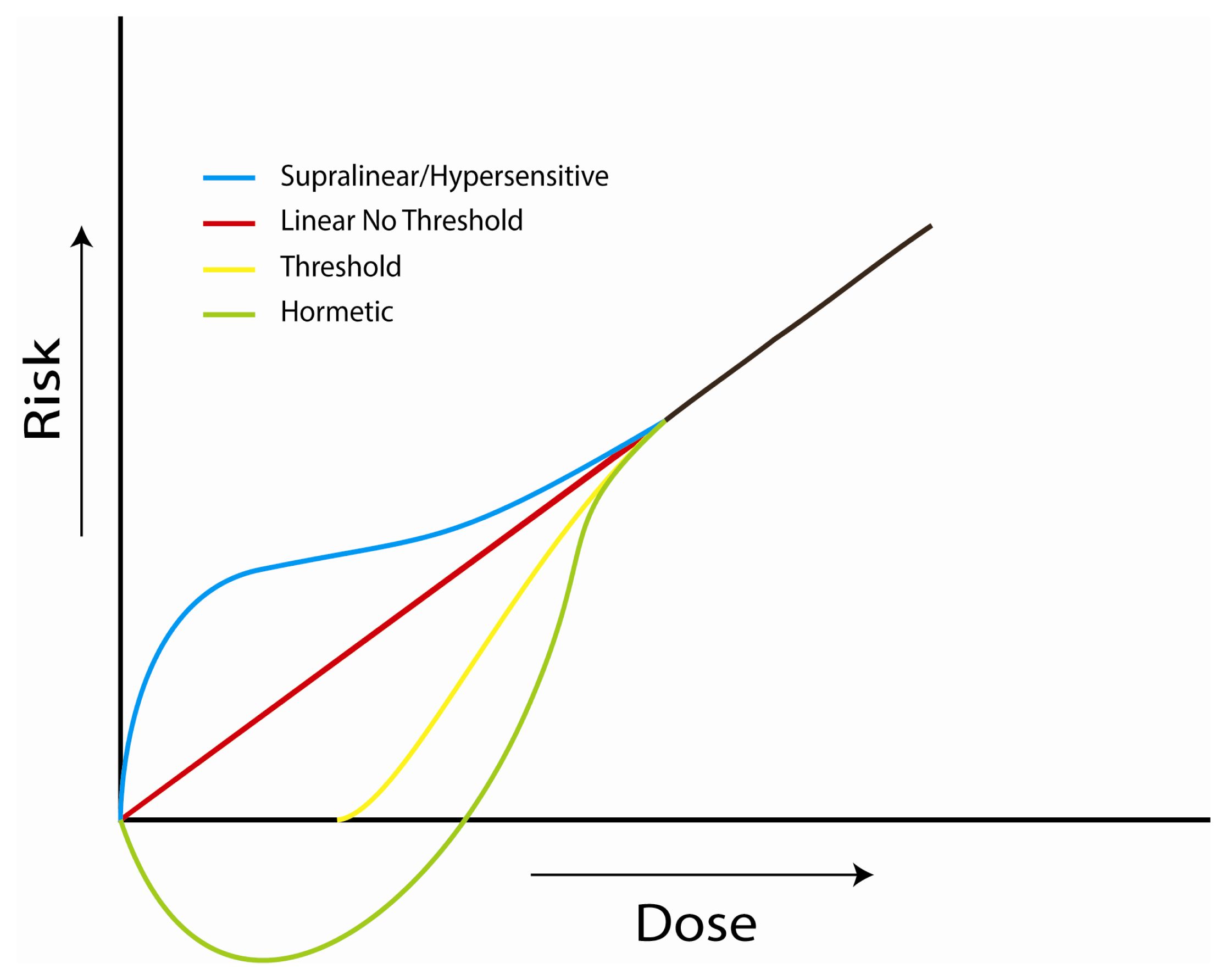
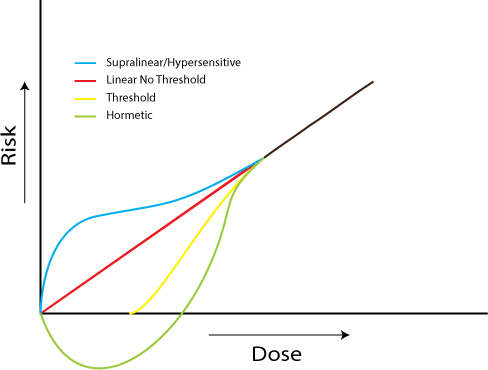
4. Implications at Low Doses
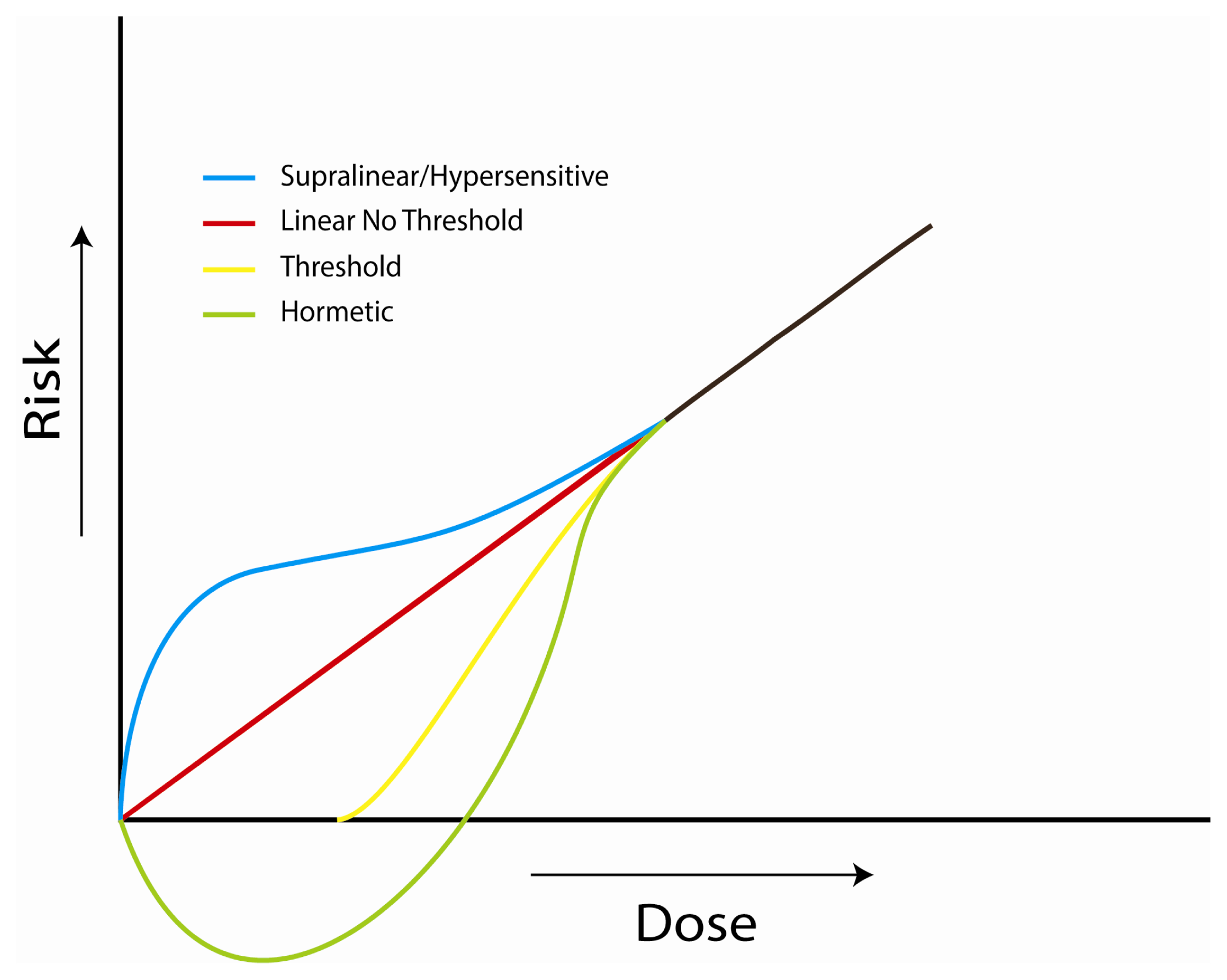
4.1. Investigations of the Linear, No-Threshold Dose-Response Hypothesis
4.2. Investigations of Bystander Effects
4.3. Investigations of Adaptive Responses
5. Discussion
6. Conclusions
Acknowledgements
Conflict of Interest
References
- United Nations Scientific Committee on the Effects of Atomic Radiation (UNSCEAR), Report of the United Nations Scientific Committee on the Effects of Atomic Radiation to the General Assembly; United Nations: New York, NY, USA, 2000.
- United Nations Scientific Committee on the Effects of Atomic Radiation (UNSCEAR), Biological Mechanisms of Radiation Actions at Low Doses. A white paper to guide the Scientific Committee’s future programme of work; United Nations: New York, NY, USA, 2012.
- Sachs, H.; Hernandez, T.; Ring, J. Regional geology and radon variability in buildings. Environ. Int 1982, 8, 97–103. [Google Scholar]
- Lubin, J. Lung cancer risk from residential radon: Meta-analysis of eight epidemiologic studies. J. Natl. Cancer 1997, 89, 49–57. [Google Scholar]
- Pavia, M.; Bianco, A.; Pileggi, C.; Angelillo, I.F. Meta-analysis of residential exposure to radon gas and lung cancer. Bull. World Health Organ 2003, 81, 732–738. [Google Scholar]
- Sethi, T.; El-Ghamry, M.; Kloecker, G. Radon and lung cancer. Clin. Adv. Hematol. Oncol 2012, 10, 157–164. [Google Scholar]
- Darby, S.; Hill, D.; Auvinen, A.; Barros-Dios, J.M.; Baysson, H.; Bochicchio, F.; Deo, H.; Falk, R.; Forastiere, F.; Hakama, M.; et al. Radon in homes and risk of lung cancer: Collaborative analysis of individual data from 13 European case-control studies. Br. Med. J. 2005, 330. [Google Scholar] [CrossRef]
- Committee on Health Risks of Exposure to Radon, National Reseaerch Council, Health Effects of Exposure to Radon; The National Academies Press: Washington, DC, USA, 1999.
- Krewski, D.; Lubin, J.H.; Zielinski, J.M.; Alavanja, M.; Catalan, V.S.; Field, R.W.; Klotz, J.B.; Letourneau, E.G.; Lynch, C.F.; Lyon, J.I. Residential radon and risk of lung cancer: A combined analysis of 7 North American case-control studies. Epidemiology 2005, 16, 137. [Google Scholar]
- Al-Zoughool, M.; Krewski, D. Health effects of radon: A review of the literature. Int. J. Radiat. Biol 2009, 85, 57–69. [Google Scholar]
- Committee to Assess Health Risks from Exposure to Low Levels of Ionizing Radiation, National Research Council, Health Risks from Exposure to Low Levels of Ionizing Radiation: BEIR VII Phase 2; The National Academies Press: Washington, DC, USA, 2006.
- Morawska, L.; Phillips, C. Attachment of radon progeny to cigarette smoke aerosol. Aerosol Sci. Technol 1992, 17, 149–158. [Google Scholar]
- Biermann, A.H.; Sawyer, S.R. Attachment of radon progeny to cigarette-smoke aerosols. Lawrence Livermore Natl. Lab. 1995. [Google Scholar] [CrossRef]
- McLaughlin, J.; O’Byrne, F. The role of daughter product plateout in passive radon detection. Radiat. Prot. Dosim 1984, 7, 115–119. [Google Scholar]
- Porstendörfer, J. Behaviour of radon daughter products in indoor air. Radiat. Prot. Dosim 1984, 7, 107–113. [Google Scholar]
- Neman, R.; Hadler, N.J.C.; Iunes, P.J.; Paulo, S.R. On indoor radon daughters’ plate-out on material surfaces. Radiat. Meas 2005, 39, 653–655. [Google Scholar]
- Richardson, R.; Eatough, J.; Henshaw, D. Dose to red bone marrow from natural radon and thoron exposure. Br. J. Radiol 1991, 64, 608–624. [Google Scholar]
- Steinhausler, F.; Hofmann, W.; Lettner, H. Thoron exposure of man: A negligible issue? Radiat. Prot. Dosim 1994, 56, 127–131. [Google Scholar]
- Tokonami, S. Why is Rn-220 (thoron) measurement important? Radiat. Prot. Dosim 2010, 141, 335–339. [Google Scholar]
- Akiba, S.; Tokonami, S.; Bochicchio, F.; McLaughlin, J.; Tommasino, L.; Harley, N. Thoron: Its metrology, health effects and implications for radon epidemiology: A summary of roundtable discussions. Radiat. Prot. Dosim 2010, 141, 477–481. [Google Scholar]
- Eatough, J.; Henshaw, D. Radon and thoron associated dose to the basal layer of the skin. Phys. Med. Biol 1992, 37, 955–967. [Google Scholar]
- Reeves, D.; Stuck, R. Clinical and experimental results with thorotrast. Medicine 1938, 17, 37–74. [Google Scholar]
- Looney, W. An investigation of the late clinical findings following thorotrast (thorium dioxide) administration. J. Occup. Environ. Med 1960, 83, 163–185. [Google Scholar]
- Ishikawa, Y.; Mori, T.; Kato, Y.; Tsuchiya, E.; Machinami, R.; Sugano, H.; Kitagawa, T. Lung cancers associated with thorotrast exposure: High incidence of small-cell carcinoma and implications for estimation of radon risk. Int. J. Cancer 1992, 52, 570–574. [Google Scholar]
- Spiethoff, A.; Wesch, H.; Wegener, K.; Klimisch, H. The effects of Thorotrast and quartz on the induction of lung tumors in rats. Health Phys 1992, 63, 101–110. [Google Scholar]
- Hofmann, W.; Hornik, S. Lung dosimetry for thorotrast patients: Implications for inhalation of radon progeny. Radiat. Res 1999, 152, S93–S96. [Google Scholar]
- Dos Santos Silva, I.; Malveiro, F.; Jones, M.; Swerdlow, A. Mortality after radiological investigation with radioactive Thorotrast: A follow-up study of up to fifty years in Portugal. Radiat. Res 2003, 159, 521–534. [Google Scholar]
- Wang, L.; Liu, D.; Shimizu, T.; Fukumoto, M. Mechanisms of liver carcinogenesis by chronic exposure to alpha-particles form internally deposited Thorotrast. Int. Congr. Ser 2005, 1276, 192–194. [Google Scholar]
- Kendall, G.M.; Smith, T.J. Doses to organs and tissues from radon and its decay products. J. Radiol. Prot 2002, 22, 389. [Google Scholar]
- Axelson, O. Cancer risks from exposure to radon in homes. Environ. Health Perspect 1995, 103, 37–43. [Google Scholar]
- Deshpande, A.; Goodwin, E.; Bailey, S.; Marrone, B.; Lehnert, B. Alpha-particle-induced sister chromatid exchange in normal human lung fibroblasts: Evidence for an extranuclear target. Radiat. Res 1996, 145, 260–267. [Google Scholar]
- Lehnert, B.; Goodwin, E. Extracellular factor(s) following exposure to α particles can cause sister chromatid exchanges in normal human cells. Cancer Res 1997, 57, 2164–2171. [Google Scholar]
- Madas, B.G.; Balásházy, I. Mutation induction by inhaled radon progeny modeled at the tissue level. Radiat. Environ. Biophys 2011, 50, 553–570. [Google Scholar]
- Balásházy, I.; Farkas, A.; Madas, B.G.; Hofmann, W. Non-linear relationship of cell hit and transformation probabilities in a low dose of inhaled radon progenies. J. Radiol. Prot 2009, 29, 147–162. [Google Scholar]
- Farkas, A.; Hofmann, W.; Balásházy, I.; Szoke, I.; Madas, B.G.; Moustafa, M. Effect of site-specific bronchial radon progeny deposition on the spatial and temporal distributions of cellular responses. Radiat. Environ. Biophys 2011, 50, 281–297. [Google Scholar]
- Konishi, E.; Yoshizawa, Y. Estimation of depth of basal cell layer of skin for radiation protection. Radiat. Prot. Dosim 1985, 11, 29–33. [Google Scholar]
- Eatough, J.P. Alpha-particle dosimetry for the basal layer of the skin and the radon progeny 218-Po and 214-Po. Phys. Med. Biol 1997, 42, 1899–1911. [Google Scholar]
- Charles, M. Radon exposure of the skin: I. Biological effects. J. Radiol. Prot 2007, 27, 231–252. [Google Scholar]
- Etherington, D.; Pheby, D.; Bray, F. An ecological study of cancer incidence and radon levels in South West England. Eur. J. Cancer 1996, 32A, 1189–1197. [Google Scholar]
- Wheeler, B.; Allen, J.; Depledge, M.; Curnow, A. Radon and skin cancer in Southwest England: An ecologic study. Epidemiology 2012, 23, 44–52. [Google Scholar]
- Henshaw, D.; Eatough, J.; Richardson, R. Radon as a causative factor in induction of myeloid leukaemia and other cancers. Lancet 1990, 335, 1008–1012. [Google Scholar]
- Kadhim, M.A.; Lorimore, S.A.; Goodhead, D.T.; Wright, E.G.; Hepburn, M.D.; Buckle, V.J. Alpha-particle-induced chromosomal instability in human bone marrow cells. Lancet 1994, 344, 987–988. [Google Scholar]
- Wilkinson, G. Gastric cancer in New Mexico counties with significant deposits of uranium. Arch. Environ. Health 1985, 40, 307–312. [Google Scholar]
- Kjellberg, S.; Wiseman, J. The relationship of radon to gastrointestinal malignancies. Am. Surg 1995, 61, 822–825. [Google Scholar]
- Darby, S.C.; Whitley, E.; Howe, G.R.; Hutchings, S.J.; Kusiak, R.A.; Lubin, J.H.; Morrison, H.I.; Tirmarche, M.; Tomasek, L.; Radford, E.; et al. Radon and cancers other than lung-cancer in underground miners: A collaborative analysis of 11 studies. J. Natl. Cancer Inst 1995, 87, 378–384. [Google Scholar]
- Auvinen, A.; Salonen, L.; Pekkanen, J.; Pukkala, E.; Ilus, T.; Kurttio, P. Radon and other natural radionuclides in drinking water and risk of stomach cancer: A case-cohort study in Finland. Int. J. Cancer 2005, 114, 109–113. [Google Scholar]
- Chen, D.J.; Strniste, G.F.; Tokita, N. The genotoxicity of alpha particles in human embryonic skin fibroblasts. Radiat. Res 1984, 100, 321–327. [Google Scholar]
- Narayanan, P.K.; Goodwin, E.H.; Lehnert, B.E. Alpha particles initiate biological production of superoxide anions and hydrogen peroxide in human cells. Cancer Res 1997, 57, 3963–3971. [Google Scholar]
- Fleischer, R.; Meyer, N.; Hadley, S.; MacDonald, J.; Cavallo, A. Personal radon dosimetry from eyeglass lenses. Radiat. Prot. Dosim 2001, 97, 251–258. [Google Scholar]
- Bigu, J.; Raz, R. Passive radon/thoron personal dosimeter using an electrostatic collector and a diffused-junction detector. Rev. Sci. Instrum 1985, 56, 146. [Google Scholar]
- Eatough, J.; Worley, A.; Moss, G. Personal monitoring of 218Po and 214Po radionuclide deposition onto individuals under normal environmental exposure conditions. Phys. Med. Biol 1999, 44, 2227–2239. [Google Scholar]
- Erickson, B. The therapeutic use of radon: A biomedical treatment in Europe; an “alternative” remedy in the United States. Dose-Response 2007, 5, 48–62. [Google Scholar]
- Steinhäusler, F. Radon spas: Source term, doses and risk assessment. Radiat. Prot. Dosim 1988, 24, 257–259. [Google Scholar]
- Franke, A.; Reiner, L.; Pratzel, H.; Franke, T.; Resch, K. Long-term efficacy of radon spa therapy in rheumatoid arthritis-a randomized, sham-controlled study and follow-up. Rheumatology 2000, 39, 894–902. [Google Scholar]
- U.S. Environmental Protection Agency Office of Radiation and Indoor Air, EPA assessment of risks from radon in homes; United States Environmental Protection Agency: Washington, DC, USA, 2003.
- Wazer, D.E.; Chu, Q.; Liu, X.L.; Gao, Q.; Safaii, H.; Band, V. Loss of p53 protein during radiation transformation of primary human mammary epithelial cells. Mol. Cell. Biol 1994, 14, 2468–2478. [Google Scholar]
- Vähäkangas, K.H.; Bennett, W.P.; Castrén, K.; Welsh, J.A.; Khan, M.A.; Blömeke, B.; Alavanja, M.C.; Harris, C.C. p53 and K-ras mutations in lung cancers from former and never-smoking women. Cancer Res. 2001, 61, 4350–4356. [Google Scholar]
- Hainaut, P.; Pfeifer, G.P. Patterns of p53 G→T transversions in lung cancers reflect the primary mutagenic signature of DNA-damage by tobacco smoke. Carcinogenesis 2001, 22, 367–374. [Google Scholar]
- Toh, C.; Lim, W. Lung cancer in never-smokers. J. Clin. Pathol 2007, 60, 337–340. [Google Scholar]
- Vähäkangas, K.H.; Samet, J.M.; Metcalf, R.A.; Welsh, J.A.; Bennett, W.P.; Lane, D.P.; Harris, C.C. Mutations of p53 and ras genes in radon-associated lung cancer from uranium miners. Lancet 1992, 339, 576–580. [Google Scholar]
- Hollstein, M.; Sidransky, D.; Vogelstein, B.; Harris, C.C. p53 mutations in human cancers. Science 1991, 253, 49–53. [Google Scholar]
- Taylor, J.; Watson, M.; Michels, R.; Saccomanno, G.; Anderson, M. p53 mutation hotspot in radon-associated lung cancer. Lancet 1994, 343, 86–87. [Google Scholar]
- McDonald, J.W.; Taylor, J.A.; Watson, M.A.; Saccomanno, G.; Devereux, T.R. p53 and K-ras in radon-associated lung adenocarcinoma. Cancer Epidemiol. Biomarkers Prev. 1995, 4, 791–793. [Google Scholar]
- Wesch, H.; Wiethege, T.; Spiethoff, A.; Wegener, K.; Müller, K.M.; Mehlhorn, J. German uranium miner study: Historical background and available histopathological material. Radiat. Res 1999, 152, S48–S51. [Google Scholar]
- Hollstein, M.; Bartsch, H.; Wesch, H.; Kure, E.; Mustonen, R.; Muhlbauer, K.; Spiethoff, A.; Wegener, K.; Wiethege, T.; Muller, K. p53 gene mutation analysis in tumors of patients exposed to alpha-particles. Carcinogenesis 1997, 18, 511–516. [Google Scholar]
- Wiethege, T.; Wesch, H.; Wegener, K.; Muller, K.; Mehlhorn, J.; Spiethoff, A.; Schomig, D.; Hollstein, M.; Bartsch, H. German uranium miner study: Pathological and molecular genetic findings. J. Radiat. Res 1999, 152, S52–S55. [Google Scholar]
- Yang, Q.; Wesch, H.; Mueller, K.M.; Bartsch, H.; Wegener, K.; Hollstein, M. Analysis of radon-associated squamous cell carcinomas of the lung for a p53 gene hotspot mutation. Br. J. Cancer 2000, 82, 763–766. [Google Scholar]
- Popp, W.; Vahrenholz, C.; Schuster, H.; Wiesner, B.; Bauer, P.; Täuscher, F.; Plogmann, H.; Morgenroth, K.; Konietzko, N.; Norpoth, K. P53 mutations and codon 213 polymorphism of p53 in lung cancers of former uranium miners. J. Cancer Res. Clin. Oncol 1999, 125, 309–312. [Google Scholar]
- Hussain, S.P.; Kennedy, C.H.; Amstad, P.; Lui, H.; Lechner, J.F.; Harris, C.C. Radon and lung carcinogenesis: Mutability of p53 codons 249 and 250 to 238Pu alpha-particles in human bronchial epithelial cells. Carcinogenesis 1997, 18, 121–125. [Google Scholar]
- Yngveson, A.; Williams, C.; Hjerpe, A.; Söderkvist, P.; Pershagen, G. p53 mutations in lung cancer associated with residential radon exposure. Cancer Epidemiol 1999, 8, 433–438. [Google Scholar]
- Ruano-Ravina, A.; Pérez-Becerra, R.; Fraga, M.; Kelsey, K.; Barros-Dios, J. Analysis of the relationship between p53 immunohistochemical expression and risk factors for lung cancer, with special emphasis on residential radon exposure. Ann. Oncol 2008, 19, 109–114. [Google Scholar]
- Lo, Y.; Darby, S.; Noakes, L.; Whitley, E.; Silcocks, P.; Fleming, K.; Bell, J. Screening for codon 249 p53 mutation in lung cancer associated with domestic radon exposure. Lancet 1995, 345, 60. [Google Scholar]
- Venitt, S.; Biggs, P.J. Radon, mycotoxins, p53, and uranium mining. Lancet 1994, 343, 795. [Google Scholar]
- Alavanja, M.; Brownson, R.; Lubin, J.; Berger, E.; Chang, J.; Boice, J.D. Residential radon exposure and lung cancer among nonsmoking women. J. Natl. Cancer Inst 1994, 86, 1829–1837. [Google Scholar]
- Johnson, N.; Carpenter, T.; Jaramillo, R.; Liberati, T. DNA damage-inducible genes as biomarkers for exposures to environmental agents. Environ. Health Perspect 1997, 105, 913–918. [Google Scholar]
- Li, H.; Gu, Y.; Miki, J.; Hukku, B.; McLeod, D.G.; Hei, T.K.; Rhim, J.S. Malignant transformation of human benign prostate epithelial cells by high linear energy transfer alpha-particles. Int. J. Oncol 2007, 31, 537–544. [Google Scholar]
- Gu, Y.; Li, H.; Miki, J.; Kim, K.-H.; Furusato, B.; Sesterhenn, I.A.; Chu, W.-S.; McLeod, D.G.; Srivastava, S.; Ewing, C.M.; et al. Phenotypic characterization of telomerase-immortalized primary non-malignant and malignant tumor-derived human prostate epithelial cell lines. Exp. Cell Res 2006, 312, 831–843. [Google Scholar]
- Belinsky, S.A.; Swafford, D.S.; Finch, G.L.; Mitchell, C.E.; Kelly, G.; Hahn, F.F.; Anderson, M.W.; Nikula, K.J. Alterations in the K-ras and p53 genes in rat lung tumors. Environ. Health Perspect 1997, 105, 901–906. [Google Scholar]
- Bastide, K.; Guilly, M.-N.; Bernaudin, J.-F.; Joubert, C.; Lectard, B.; Levalois, C.; Malfoy, B.; Chevillard, S. Molecular analysis of the Ink4a/Rb1-Arf/Tp53 pathways in radon-induced rat lung tumors. Lung Cancer 2009, 63, 348–353. [Google Scholar]
- Stout, J.T.; Caskey, C.T. HPRT: Gene structure, expression, and mutation. Ann. Rev. Genet 1985, 19, 127–148. [Google Scholar]
- Lesch, M.; Nyhan, W.L. A familial disorder of uric acid metabolism and central nervous system function. Am. J. Med 1964, 36, 561–570. [Google Scholar]
- Edwards, A.; Voss, H.; Rice, P.; Civitello, A.; Stegemann, J.; Schwager, C.; Zimmermann, J.; Erfle, H.; Caskey, C.; Ansorge, W. Automated DNA sequencing of the human HPRT locus. Genomics 1990, 6, 593–608. [Google Scholar]
- Cariello, N.F.; Craft, T.R.; Vrieling, H.; van Zeeland, A.A.; Adams, T.; Skopek, T.R. Human HPRT mutant database: Software for data entry and retrieval. Environ. Mol. Mutagenes 1992, 20, 81–83. [Google Scholar]
- Cariello, N. Database and software for the analysis of mutations at the human hprt gene. Nucleic Acids Res 1994, 22, 3547–3548. [Google Scholar]
- Cariello, N.; Douglas, G.; Gorelick, N.; Hart, D.; Wilson, J.; Soussi, T. Databases and software for the analysis of mutations in the human p53 gene, human hprt gene and both the lacI and lacZ gene in transgenic rodents. Nucleic Acids Res 1998, 26, 198–199. [Google Scholar]
- Hakoda, M.; Hirai, Y.; Kyoizumi, S.; Akiyama, M. Molecular analyses of in vivo hprt mutant T cells from atomic bomb survivors. Environ. Mol. Mutagenes 1989, 13, 25–33. [Google Scholar]
- Hakoda, M.; Akiyama, M.; Kyoizumi, S.; Awa, A.; Yamakido, M.; Otake, M. Increased somatic cell mutant frequency in atomic bomb survivors. Mutat. Res 1988, 201, 39–48. [Google Scholar]
- Zwingmann, I.; Welle, I.; Engelen, J.; Schilderman, P.; de Jong, J.; Kleinjans, J. Analysis of oxidative DNA damage and HPRT mutant frequencies in cancer patients before and after radiotherapy. Mutat. Res 1999, 431, 361–369. [Google Scholar]
- Jinnah, H.; de Gregorio, L.; Harris, J.; Nyhan, W.; O’Neill, J. The spectrum of inherited mutations causing HPRT deficiency: 75 new cases and a review of 196 previously reported cases. Mutat. Res 2000, 463, 309–326. [Google Scholar]
- Shanahan, E.M.; Peterson, D.; Roxby, D.; Quintana, J.; Morely, A.A.; Woodward, A. Mutation rates at the glycophorin A and HPRT loci in uranium miners exposed to radon progeny. Occup. Environ. Med 1996, 53, 439–444. [Google Scholar]
- Woodward, A.; Roder, D.; McMichael, A.J.; Crouch, P.; Mylvaganam, A. Radon daughter exposures at the Radium Hill uranium mine and lung cancer rates among former workers, 1952–87. Cancer Causes Control 1991, 2, 213–220. [Google Scholar]
- Bridges, B.; Cole, J.; Arlett, C.; Green, M.; Waugh, A.; Beare, D.; Henshaw, D.; Last, R. Possible association between mutant frequency in peripheral lymphocytes and domestic radon concentrations. Lancet 1991, 337, 1187–1189. [Google Scholar]
- Yunis, J.J.; Oken, M.M.; Kaplan, M.E.; Ensrud, K.M.; Howe, R.R.; Theologides, A. Distinctive chromosomal abnormalities in histologic subtypes of non-Hodgkin’s lymphoma. N. Engl. J. Med 1982, 307, 1231–1236. [Google Scholar]
- Gribben, J.; Freedman, A.; Woo, S.; Blake, K.; Shu, R.; Freeman, G.; Longtine, J.; Pinkus, G.; Nadler, L. All advanced stage non-Hodgkin’s lymphomas with a polymerase chain reaction amplifiable breakpoint of bcl-2 have residual cells containing the bcl-2 rearrangement. Blood 1991, 78, 3275–3280. [Google Scholar]
- Cory, S.; Huang, D.; Adams, J. The Bcl-2 family: Roles in cell survival and oncogenesis. Oncogene 2003, 22, 8590–8607. [Google Scholar]
- Cole, J.; Green, M.; Bridges, B.; Waugh, A.; Beare, D.; Henshaw, D.; Last, R.; Liu, Y.; Cortopassi, G. Lack of evidence for an association between the frequency of mutants or translocations in circulating lymphocytes and exposure to radon gas in the home. Radiat. Res 1996, 145, 61–69. [Google Scholar]
- Liu, Y.; Cortopassi, G.; Steingrimsdottir, H.; Waugh, A.P.; Beare, D.M.; Green, M.H.; Robinson, D.R.; Cole, J. Correlated mutagenesis of bcl2 and hprt loci in blood lymphocytes. Environ. Mol. Mutagenes 1997, 29, 36–45. [Google Scholar]
- Liu, Y.; Hernandez, A.; Shibata, D.; Cortopassi, G. BCL2 translocation frequency rises with age in humans. Proc. Natl. Acad. Sci. USA 1994, 91, 8910–8914. [Google Scholar]
- Curry, J.; Karnaoukhova, L.; Guenette, G.; Glickman, B. Influence of sex, smoking and age on human hprt mutation frequencies and spectra. Genetics 1999, 152, 1065–1077. [Google Scholar]
- Bell, D.; Liu, Y.; Cortopassi, G. Occurrence of bcl-2 oncogene translocation with increased frequency in the peripheral blood of heavy smokers. J. Natl. Cancer Inst 1995, 87, 223–224. [Google Scholar]
- Albering, H.J.; Hageman, G.J.; Kleinjans, J.C.; Engelen, J.J.; Koulishcer, L.; Herens, C. Indoor radon exposure and cytogenetic damage. Lancet 1992, 340, 739. [Google Scholar]
- Albering, H.; Engelen, J.; Koulischer, L.; Welle, I.; Kleinjans, J. Indoor radon, an extrapulmonary genetic risk? Lancet 1994, 344, 750–751. [Google Scholar]
- Ruttenber, A.; Harrison, L.; Baron, A.; McClure, D.; Glanz, J.; Quillin, R.; O’Neill, J.; Sullivan, L.; Campbell, J.; Nicklas, J. hprt mutant frequencies, nonpulmonary malignancies, and domestic radon exposure: “Postmortem” analysis of an interesting hypothesis. Environ. Mol. Mutagenes 2001, 37, 7–16. [Google Scholar]
- Zwingmann, I.; Welle, I.; van Herwijnen, M.; Engelen, J.; Schilderman, P.; Smid, T.; Kleinjans, J. Oxidative DNA damage and cytogenetic effects in flight engineers exposed to cosmic radiation. Environ. Mol. Mutagenes 1998, 32, 121–129. [Google Scholar]
- Seifert, A.; Demers, C.; Dubeau, H.; Messing, K. HPRT-mutant frequency and lymphocyte characteristics of workers exposed to ionizing radiation on a sporadic basis: A comparison of two exposure indicators, job title and dose. Mutat. Res 1993, 319, 61–70. [Google Scholar]
- McDiarmid, M.; Albertini, R.; Tucker, J.; Vacek, P.; Carter, E.; Bakhmutsky, M.; Oliver, M.; Engelhardt, S.; Squibb, K. Measures of genotoxicity in Gulf war I veterans exposed to depleted uranium. Environ. Mol. Mutagenes 2011, 52, 569–581. [Google Scholar]
- Xie, H.; LaCerte, C.; Thompson, W.; Wise, J. Depleted uranium induces neoplastic transformation in human lung epithelial cells. Chem. Res. Toxicol 2010, 23, 373–378. [Google Scholar]
- Wise, S.S.; Thompson, W.D.; Aboueissa, A.-M.; Mason, M.D.; Wise, J.P. Particulate depleted uranium is cytotoxic and clastogenic to human lung cells. Chem. Res. Toxicol 2007, 20, 815–820. [Google Scholar]
- Cui, F.; Fan, S.; Hu, M.; Nie, J.; Li, H.; Tong, J. Micronuclei rate and hypoxanthine phosphoribosyl transferase mutation in radon-exposed rats. Prog. Nat. Sci 2008, 18, 1305–1308. [Google Scholar]
- Jostes, R.F.; Fleck, E.W.; Morgan, T.L.; Stiegler, G.L.; Cross, F.T. Southern blot and polymerase chain reaction exon analyses of HPRT-mutations induced by radon and radon progeny. Radiat. Res 1994, 137, 371–379. [Google Scholar]
- Jostes, R.; Hui, T.; James, A.; Cross, F.; Schwartz, J.; Rotmensch, J.; Atcher, R.; Evans, H.; Mencl, J.; Bakale, G.; et al. In vitro exposure of mammalian cells to radon: Dosimetric considerations. Radiat. Res 1991, 127, 211–219. [Google Scholar]
- Kiefer, J.; Schreiber, A.; Gutermuth, F.; Koch, S.; Schmidt, P. Mutation induction by different types of radiation at the Hprt locus. Mutat. Res 1999, 431, 429–448. [Google Scholar]
- Bao, C.Y.; Ma, A.; Evans, H.H.; Horng, M.F.; Mencl, J.; Hui, T.E.; Sedwick, W.D. Molecular analysis of hypoxanthine phosphoribosyltransferase gene deletions induced by alpha- and X-radiation in human lymphoblastoid cells. Mutat. Res 1994, 326, 1–15. [Google Scholar]
- Bakale, G.; Rao, P.S.; Mencl, J.; Adams, R.B.; Evans, H.H. A radon generator/delivery system. Radiat. Res 1993, 133, 277–281. [Google Scholar]
- Chaudhry, M.; Jiang, Q.; Ricanati, M.; Horng, M.; Evans, H. Characterization of multilocus lesions in human cells exposed to X radiation and radon. Radiat. Res 1996, 145, 31–38. [Google Scholar]
- Amundson, S.; Chen, D.; Okinaka, R. Alpha particle mutagenesis of human lymphoblastoid cell lines. Int. J. Radiat. Biol 1996, 70, 219–226. [Google Scholar]
- Hsie, A.; Porter, R.; Xu, Z.; Yu, Y.; Sun, J.; Meltz, M.; Schwartz, J. Molecular markers of ionizing radiation-induced gene mutations in mammalian cells. Environ. Health Perspect 1996, 104, 675–678. [Google Scholar]
- Schwartz, J.; Hsie, A. Genetic and cytogenetic markers of exposure to high-linear energy transfer radiation. Radiat. Res 1997, 148, S87–S92. [Google Scholar]
- Albertini, R.; Clark, L.; Nicklas, J.; O’Neill, J.; Hui, T.; Jostes, R. Radiation quality affects the efficiency of induction and the molecular spectrum of HPRT mutations in human T cells. Radiat. Res 1997, 148, S76–S86. [Google Scholar]
- O’Neill, P.; Nicklas, J.; Hirsch, B.; Jostes, R.; Hunter, T.; Sullivan, L.; Albertini, R. In vitro studies of the genotoxicity of ionizing radiation in human G(0) T lymphocytes. Environ. Mol. Mutagenes 2005, 46, 207–220. [Google Scholar]
- Schwartz, J.L.; Jordan, R.; Sun, J.; Ma, H.; Hsieb, A. Dose-dependent changes in the spectrum of mutations induced by ionizing radiation. Radiat. Res 2000, 153, 312–317. [Google Scholar]
- Nagasawa, H.; Little, J. Unexpected sensitivity to the induction of mutations by very low doses of alpha-particle radiation: Evidence for a bystander effect. Radiat. Res 1999, 152, 552–557. [Google Scholar]
- Huo, L.; Nagasawa, H.; Little, J. HPRT mutants induced in bystander cells by very low fluences of alpha particles result primarily from point mutations. Radiat. Res 2001, 156, 521–525. [Google Scholar]
- Nagasawa, H.; Huo, L.; Little, J.B. Increased bystander mutagenic effect in DNA double-strand break repair-deficient mammalian cells. Int. J. Radiat. Biol 2003, 79, 35–41. [Google Scholar]
- Da Cruz, A.; Curry, J.; Curado, M.; Glickman, B. Monitoring hprt mutant frequency over time in T-lymphocytes of people accidentally exposed to high doses of ionizing radiation. Environ. Mol. Mutagenes 1996, 27, 165–175. [Google Scholar]
- Mitelman, F. Catalog of chromosome aberrations in cancer, 4th edition, in two parts. Am. J. Hum. Genet 1992, 50, 1354–1355. [Google Scholar]
- Solomon, E.; Borrow, J.; Goddard, A.D. Chromosome aberrations and cancer. Science 1991, 254, 1153–1160. [Google Scholar]
- Albertson, D.G.; Collins, C.; McCormick, F.; Gray, J.W. Chromosome aberrations in solid tumors. Nat. Genet 2003, 34, 369–376. [Google Scholar]
- Bender, M.; Griggs, H.; Bedford, J. Mechanisms of chromosomal aberration production III. Chemicals and ionizing radiation. Mutat. Res 1974, 23, 197–212. [Google Scholar]
- Sorsa, M.; Wilbourn, J.; Vainio, H. Human cytogenetic damage as a predictor of cancer risk. IARC Sci. Publ. 1992, 543–554. [Google Scholar]
- Norppa, H. Cytogenetic biomarkers and genetic polymorphisms. Toxicol. Lett 2004, 149, 309–334. [Google Scholar]
- Tucker, J.D.; Auletta, A.; Cimino, M.C.; Dearfield, K.L.; Jacobson-Kram, D.; Tice, R.R.; Carrano, A.V. Sister-chromatid exchange: Second report of the Gene-Tox program. Mutat. Res 1993, 297, 101–180. [Google Scholar]
- Anderson, B.E.; Zeiger, E.; Shelby, M.D.; Resnick, M.A.; Gulati, D.K.; Ivett, J.L.; Loveday, K.S. Chromosome aberration and sister chromatid exchange test results with 42 chemicals. Environ. Mol. Mutagenes 1990, 16, 55–137. [Google Scholar]
- Galloway, S.M.; Armstrong, M.J.; Reuben, C.; Colman, S.; Brown, B.; Cannon, C.; Bloom, A.D.; Nakamura, F.; Ahmed, M.; Duk, S. Chromosome aberrations and sister chromatid exchanges in Chinese hamster ovary cells: Evaluations of 108 chemicals. Environ. Mol. Mutagenes 1987, 10, 1–35. [Google Scholar]
- Wald, N.; Koizumi, A.; Pan, S. A pilot study of the relationship between chromosome aberrations and occupational external and internal radiation exposure. Hum. Radiat. Cytogenet. 1968, 183–193. [Google Scholar]
- Boyd, J.; Brown, W.; Vennart, J.; Woodcock, G. Chromosome studies on women formerly employed as luminous-dial painters. Br. Med. J 1966, 1, 377–382. [Google Scholar]
- Sax, K. Chromosome aberrations induced by X-rays. Genetics 1938, 23, 494–516. [Google Scholar]
- Brown, J.; McNeill, J. Aberrations in leukocyte chromosomes of personnel occupationally exposed to low levels of radiation. Radiat. Res 1969, 40, 534–543. [Google Scholar]
- Ishihara, T.; Kumatori, T. Chromosome studies on japanese exposed to radiation resulting from nuclear bomb explosions. Hum. Radiat. Cytogenet. 1968, 144–166. [Google Scholar]
- Awa, A.; Honda, T.; Sofuni, T.; Neriishi, S. Chromosome-aberration frequency in cultured blood-cells in relation to radiation dose of A-bomb survivors. Lancet 1971, 298, 903–905. [Google Scholar]
- Müller, I.; Geinitz, H.; Braselmann, H.; Baumgartner, A.; Fasan, A.; Thamm, R.; Molls, M.; Meineke, V.; Zitzelsberger, H. Time-course of radiation-induced chromosomal aberrations in tumor patients after radiotherapy. Int. J. Radiat. Oncol. Biol. Phys 2005, 63, 1214–1220. [Google Scholar]
- Jones, L.A.; Scott, D.; Cowan, R.; Roberts, S.A. Abnormal radiosensitivity of lymphocytes from breast cancer patients with excessive normal tissue damage after radiotherapy: Chromosome aberrations after low dose-rate irradiation. Int. J. Radiat. Biol 1995, 67, 519–528. [Google Scholar]
- Barrios, L.; Miró, R.; Caballín, M.R.; Fuster, C.; Guedea, F.; Subias, A.; Egozcue, J. Cytogenetic effects of radiotherapy. Breakpoint distribution in induced chromosome aberrations. Cancer Genet. Cytogenet 1989, 41, 61–70. [Google Scholar]
- AlSuhaibani, E.S. Chromosomal aberration analysis among underground water well workers in Saudi Arabia. Radiat. Prot. Dosim 2011, 144, 651–654. [Google Scholar]
- Liu, Q.; Cao, J.; Wang, Z.; Bai, Y.; Lü, Y.; Huang, Q.; Zhao, W.; Li, J.; Jiang, L.; Tang, W.; et al. Dose estimation by chromosome aberration analysis and micronucleus assays in victims accidentally exposed to 60Co radiation. Br. J. Radiol 2009, 82, 1027–1032. [Google Scholar]
- Liu, Q.; Cao, J.; Liu, Y.; Lü, Y.; Qin, B.; Jiang, B.; Jiang, L.; Fu, B.; Zhao, F.; Jiang, E.; et al. Follow-up study by chromosome aberration analysis and micronucleus assays in victims accidentally exposed to 60Co radiation. Health Phys 2010, 98, 885–888. [Google Scholar]
- Heimers, A.; Brede, H.J.; Giesen, U.; Hoffmann, W. Chromosome aberration analysis and the influence of mitotic delay after simulated partial-body exposure with high doses of sparsely and densely ionising radiation. Radiat. Environ. Biophys 2006, 45, 45–54. [Google Scholar]
- Savage, J. Classification and relationships of induced chromosomal structual changes. J. Med. Genet 1976, 12, 103–122. [Google Scholar]
- Shadley, J.; Whitlock, J.; Rotmensch, J.; Atcher, R.; Tang, J.; Schwartz, J. The effects of radon daughter alpha-particle irradiation in K1 and xrs-5 CHO cell lines. Mutat. Res 1991, 248, 73–83. [Google Scholar]
- Wolff, S.; Jostes, R.; Cross, F.T.; Hui, T.E.; Afzal, V.; Wiencke, J.K. Adaptive response of human lymphocytes for the repair of radon-induced chromosomal damage. Mutat. Res 1991, 250, 299–306. [Google Scholar]
- Kadhim, M.; Macdonald, D.; Goodhead, D.; Lorimore, S.; Marsden, S.; Wright, E. Transmission of chromosomal instability after plutonium alpha-particle irradiation. Nature 1992, 355, 738–740. [Google Scholar]
- Nagasawa, H.; Robertson, J.; Little, J. Induction of chromosomal aberrations and sister chromatid exchanges by alpha particles in density-inhibited cultures of mouse 10T1/2 and 3T3 cells. Int. J. Radiat. Biol 1990, 57, 35–44. [Google Scholar]
- Nagasawa, H.; Little, J.B. Induction of sister chromatid exchanges by extremely low doses of α-particles. Cancer Res 1992, 52, 6394–6396. [Google Scholar]
- Iyer, R.; Lehnert, B.E.; Svensson, R. Factors underlying the cell growth-related bystander responses to α particles. Cancer Res 2000, 60, 1290–1298. [Google Scholar]
- Azzam, E.; de Toledo, S.; Gooding, T.; Little, J. Intercellular communication is involved in the bystander regulation of gene expression in human cells exposed to very low fluences of alpha particles. Radiat. Res 1998, 150, 497–504. [Google Scholar]
- Pohl-Rüling, J.; Lettner, H.; Hofmann, W.; Eckl, P.; Haas, O.; Obe, G.; Grell-Büchtmann, I.; van Buul, P.; Schroeder-Kurth, T.; Atzmüller, C.; et al. Chromosomal aberrations of blood lymphocytes induced in vitro by radon-222 daughter alpha irradiation. Mutat. Res 2000, 449, 7–19. [Google Scholar]
- Hamza, V.Z.; Mohankumar, M.N. Cytogenetic damage in human blood lymphocytes exposed in vitro to radon. Mutat. Res 2009, 661, 1–9. [Google Scholar]
- Meenakshi, C.; Mohankumar, M. Radon-induced chromosome damage in blood lymphocytes of smokers. Res. J. Environ. Toxicol 2012, 6, 51–58. [Google Scholar]
- Costa-Ribeiro, C.; Barcinski, M.A.; Figueiredo, N.; Franca, E.P.; Lobão, N. Radiobiological aspects and radiation levels associated with the milling of monazite sand. Health Phys 1975, 28, 225–231. [Google Scholar]
- Livingston, G.K.; Falk, R.B.; Schmid, E. Effect of occupational radiation exposures on chromosome aberration workers rates in former plutonium workers. Radiat. Res 2006, 166, 89–97. [Google Scholar]
- Smerhovsky, Z.; Landa, K.; Rössner, P.; Brabec, M.; Zudova, Z.; Hola, N.; Pokorna, Z.; Mareckova, J.; Hurychova, D. Risk of cancer in an occupationally exposed cohort with increased level of chromosomal aberrations. Environ. Health Perspect 2001, 109, 41–45. [Google Scholar]
- Smerhovsky, Z.; Landa, K.; Rössner, P.; Juzova, D.; Brabec, M.; Zudova, Z.; Hola, N.; Zarska, H.; Nevsimalova, E. Increased risk of cancer in radon-exposed miners with elevated frequency of chromosomal aberrations. Mutat. Res 2002, 514, 165–176. [Google Scholar]
- Brandom, W.F.; Saccomanno, G.; Archer, V.E.; Archer, P.G.; Coors, M.E. Chromosome aberrations in uranium miners occupationally exposed to 222 Radon. Radiat. Res 1972, 52, 204–215. [Google Scholar]
- Brandom, W.F.; Saccomanno, G.; Archer, V.E.; Archer, P.G.; Bloom, A.D. Chromosome aberrations as a biological dose-response indicator of radiation exposure in uranium miners. Radiat. Res 1978, 76, 159–171. [Google Scholar]
- Bilban, M. Influence of the work environment in a Pb-Zn mine on the incidence of cytogenetic damage in miners. Am. J. Ind. Med 1998, 34, 455–463. [Google Scholar]
- Bilban, M.; Jakopin, C. Incidence of cytogenetic damage in lead-zinc mine workers exposed to radon. Mutagenesis 2005, 20, 187–191. [Google Scholar]
- Popp, W.; Plappert, U.; Müller, W.U.; Rehn, B.; Schneider, J.; Braun, A.; Bauer, P.C.; Vahrenholz, C.; Presek, P.; Brauksiepe, A.; et al. Biomarkers of genetic damage and inflammation in blood and bronchoalveolar lavage fluid among former German uranium miners: A pilot study. Radiat. Environ. Biophys 2000, 39, 275–282. [Google Scholar]
- Sram, R.; Binková, B. Monitoring genotoxic exposure in uranium miners. Environ. Health Perspect 1993, 99, 303–305. [Google Scholar]
- Zölzer, F.; Hon, Z.; Skalická, Z.F.; Havránková, R.; Navrátil, L.; Rosina, J.; Škopek, J. Micronuclei in lymphocytes from currently active uranium miners. Radiat. Environ. Biophys 2012, 51, 277–282. [Google Scholar]
- Zaire, R.; Notter, M.; Riedel, W.; Thiel, E. Unexpected rates of chromosomal instabilities and alterations of hormone levels in Namibian uranium miners. Radiat. Res 1997, 147, 579–584. [Google Scholar]
- Mészáros, G.; Bognár, G.; Köteles, G.J. Long-term persistence of chromosome aberrations in uranium miners. J. Occup. Health 2004, 46, 310–315. [Google Scholar]
- Tawn, E.; Whitehouse, C.; Tarone, R. FISH chromosome aberration analysis on retired radiation workers from the Sellafield nuclear facility. Radiat. Res 2004, 162, 249–256. [Google Scholar]
- Barcinski, M.; do Céu Abreu, M.; de Almeida, J.; Naya, J.; Fonseca, L.; Castro, L. Cytogenetic investigation in a Brazilian population living in an area of high natural radioactivity. Am. J. Hum. Genet 1975, 27, 802–806. [Google Scholar]
- Pohl-Rüling, J.; Fischer, P. The dose-effect relationship of chromosome aberrations to α and γ irradiation in a population subjected to an increased burden of natural radioactivity. Radiat. Res 1979, 80, 61–81. [Google Scholar]
- Chen, D.; Wei, L. Chromosome aberration, cancer mortality and hormetic phenomena among inhabitants in areas of high background radiation in china. J. Radiat. Res 1991, 32, 46–53. [Google Scholar]
- Bauchinger, M.; Schmid, E.; Braselmann, H.; Kulka, U. Chromosome aberrations in peripheral lymphocytes from occupants of houses with elevated indoor radon concentrations. Mutat. Res 1994, 310, 135–142. [Google Scholar]
- Lindholm, C.; Mäkeläinen, I.; Paile, W.; Koivistoinen, A.; Salomaa, S. Domestic radon exposure and the frequency of stable or unstable chromosomal aberrations in lymphocytes. Int. J. Radiat. Biol 1999, 75, 921–928. [Google Scholar]
- Boffetta, P.; van der Hel, O.; Norppa, H.; Fabianova, E.; Fucic, A.; Gundy, S.; Lazutka, J.; Cebulska-Wasilewska, A.; Puskailerova, D.; Znaor, A.; et al. Chromosomal aberrations and cancer risk: results of a cohort study from Central Europe. Am. J. Epidemiol 2007, 165, 36–43. [Google Scholar]
- Bilban, M.; Vaupoti, J. Chromosome aberrations study of pupils in high radon level elementary school. Health Phys 2001, 80, 157–163. [Google Scholar]
- Minina, V.I.; Druzhinin, V.G.; Golovina, T.A.; Larin, S.A.; Mun, S.A.; Akhmat’ianova, V.R.; Bakanova, M.L.; Glushkov, A.N. Spread of carcinogenic and mutagenic effects in the population of Gornaia Shoria. Gig. Sanit. 2011, 35–38. [Google Scholar]
- Druzhinin, V.G.; Akhmat’ianova, V.R.; Golovina, T.A.; Volkov, A.N.; Minina, V.I.; Larionov, A.V.; Makeeva, E.A. Genome sensitivity and genotoxic effects features in children-teenagers affected by radon radiation in living and educational environment. Radiats. Biol. Radioecol 2009, 49, 568–573. [Google Scholar]
- Minina, V.; Druzhinin, V.; Lunina, A.; Larionov, A.; Golovina, T.; Glushkov, A. Association of DNA repair gene polymorphism with chromosomal aberrations frequency in human lymphocytes. Russ. J. Genet 2012, 2, 171–176. [Google Scholar]
- Bilban, M.; Bilban-Jakopin, C.; Vrhovec, S. Incidence of chromosomal aberrations and micronuclei in cave tour guides. Neoplasma 2001, 48, 278–284. [Google Scholar]
- Lehnert, B.; Goodwin, E. A new mechanism for DNA alterations induced by alpha particles such as those emitted by radon and radon progeny. Environ. Health Perspect 1997, 105, 1095–1101. [Google Scholar]
- Hamza, V.Z.; Kumar, P.R.V.; Jeevanram, R.K.; Santanam, R.; Danalaksmi, B.; Mohankumar, M.N. A simple method to irradiate blood cells in vitro with radon gas. Radiat. Prot. Dosim 2008, 130, 343–350. [Google Scholar]
- Bender, M.; Gooch, P. Persistent chromosome aberrations in irradiated human subjects. II. Three and one-half year investigation. Radiat. Res 1963, 18, 389–396. [Google Scholar]
- Awa, A. Persistent chromosome aberrations in the somatic cells of A-bomb survivors, Hiroshima and Nagasaki. Radiat. Res 1991, 32, 265–274. [Google Scholar]
- Bender, M.; Gooch, P. Persistent chromosome aberrations in irradiated human subjects. Radiat. Res 1962, 16, 44–53. [Google Scholar]
- Brenner, D.; Sachs, R. Chromosomal “fingerprints” of prior exposure to densely ionizing radiation. Radiat. Res 1994, 140, 134–142. [Google Scholar]
- Brenner, D.; Okladnikova, N.; Hande, P.; Burak, L.; Geard, C.; Azizova, T. Biomarkers specific to densely-ionising (high LET) radiations. Radiat. Prot. Dosim 2001, 97, 69–73. [Google Scholar]
- Anderson, R.M.; Stevens, D.L.; Sumption, N.D.; Townsend, K.M.S.; Goodhead, D.T.; Hill, M.A. Effect of linear energy transfer (LET) on the complexity of alpha-particle-induced chromosome aberrations in human CD34+ cells. Radiat. Res 2007, 167, 541–550. [Google Scholar]
- Tawn, E.; Whitehouse, C. Chromosome intra- and inter-changes determined by G-banding in radiation workers with in vivo exposure to plutonium. J. Radiol. Prot 2005, 25, 83–88. [Google Scholar]
- Fenech, M.; Morley, A. Measurement of micronuclei in lymphocytes. Mutat. Res 1985, 147, 29–36. [Google Scholar]
- Heddle, J.A.; Carrano, A.V. The DNA content of micronuclei induced in mouse bone marrow by gamma-irradiation: Evidence that micronuclei arise from acentric chromosomal fragments. Mutat. Res 1977, 44, 63–69. [Google Scholar]
- Countryman, P.; Heddle, J. The production of micronuclei from chromosome aberrations in irradiated cultures of human lymphocytes. Mutat. Res 1976, 41, 321–331. [Google Scholar]
- Bonassi, S.; El-Zein, R.; Bolognesi, C.; Fenech, M. Micronuclei frequency in peripheral blood lymphocytes and cancer risk: Evidence from human studies. Mutagenesis 2011, 26, 93–100. [Google Scholar]
- Khan, M.A.; Cross, F.T.; Jostes, R.; Hui, E.; Morris, J.E.; Brooks, A.L. Micronuclei induced by radon and its progeny in deep-lung fibroblasts of rats in vivo and in vitro. Radiat. Res 1994, 139, 53–59. [Google Scholar]
- Taya, A.; Morgan, A.; Baker, S.; Humphreys, J.; Bisson, M.; Collier, C. Changes in the rat lung after exposure to radon and its progeny: Effects on incorporation of bromodeoxyuridine in epithelial cells and on the incidence of nuclear aberrations in alveolar macrophages. Radiat. Res 1994, 139, 170–177. [Google Scholar]
- Nelson, J.; Brooks, A.; Metting, N.; Khan, M.; Buschbom, R.; Duncan, A.; Miick, R.; Braby, L. Clastogenic effects of defined numbers of 3.2 MeV alpha particles on individual CHO-K1 cells. Radiat. Res 1996, 145, 568–574. [Google Scholar]
- Hei, T.K.; Wu, L.J.; Liu, S.X.; Vannais, D.; Waldren, C.A.; Randers-Pehrson, G. Mutagenic effects of a single and an exact number of alpha particles in mammalian cells. Proc. Natl. Acad. Sci. USA 1997, 94, 3765–3770. [Google Scholar]
- Kennedy, C.H.; Mitchell, C.E.; Fukushima, N.H.; Neft, R.E.; Lechner, J.F. Induction of genomic instability in normal human bronchial epithelial cells by 238Pu alpha-particles. Carcinogenesis 1996, 17, 1671–1676. [Google Scholar]
- Tobias, E.S.; Hurlstone, A.F.; MacKenzie, E.; McFarlane, R.; Black, D.M. The TES gene at 7q31.1 is methylated in tumours and encodes a novel growth-suppressing LIM domain protein. Oncogene 2001, 20, 2844–2853. [Google Scholar]
- Chêne, L.; Giroud, C.; Desgrandchamps, F.; Boccon-Gibod, L.; Cussenot, O.; Berthon, P.; Latil, A. Extensive analysis of the 7q31 region in human prostate tumors supports TES as the best candidate tumor suppressor gene. Int. J. Cancer 2004, 111, 798–804. [Google Scholar]
- Zenklusen, J.C.; Thompson, J.C.; Klein-szanto, A.J.P.; Conti, C.J. Frequent loss of heterozygosity in human primary squamous cell and colon carcinomas at 7q31.1: Evidence for a broad range tumor suppressor gene. Cancer Res 1995, 55, 1347–1350. [Google Scholar]
- Ruas, M.; Peters, G. The p16 INK4a/CDKN2A tumor suppressor and its relatives. Biochim. Biophys. Acta 1998, 1378, F115–F177. [Google Scholar]
- Belchior, A.; Gil, O.M.; Almeida, P.; Vaz, P. Evaluation of the cytotoxicity and the genotoxicity induced by α radiation in an A549 cell line. Radiat. Meas 2011, 46, 958–961. [Google Scholar]
- Chaudhry, M.A. Analysis of gene expression in normal and cancer cells exposed to gamma-radiation. J. Biomed. Biotechnol 2008, 2008, 541678. [Google Scholar]
- Chaudhry, M.; Omaruddin, R.; Kreger, B.; de Toledo, S.; Azzam, E. Micro RNA responses to chronic or acute exposures to low dose ionizing radiation. Mol. Biol. Rep 2012, 39, 7549–7558. [Google Scholar]
- Chaudhry, M.; Omaruddin, R. Differential regulation of MicroRNA expression in irradiated and bystander cells. Mol. Biol 2012, 46, 569–578. [Google Scholar]
- Chaudhry, M.; Omaruddin, R. Transcriptional changes of mitochondrial genes in irradiated cells proficient or deficient in p53. J. Genet 2012, 91, 105–110. [Google Scholar]
- Amundson, S.; do, K.; Shahab, S.; Bittner, M.; Meltzer, P.; Trent, J.; Fornace, A. Identification of potential mRNA biomarkers in peripheral blood lymphocytes for human exposure to ionizing radiation. Radiat. Res 2000, 154, 342–346. [Google Scholar]
- Scott, B.R.; Walker, D.M.; Tesfaigzi, Y.; Schöllnberger, H.; Walker, V. Mechanistic basis for nonlinear dose-response relationships for low-dose radiation-induced stochastic effects. Nonlinearity Biol. Toxicol. Med 2003, 1, 93–122. [Google Scholar]
- Feinendegen, L.E. Evidence for beneficial low level radiation effects and radiation hormesis. Br. J. Radiol 2005, 78, 3–7. [Google Scholar]
- Baskar, R. Emerging role of radiation induced bystander effects: Cell communications and carcinogenesis. Genome Integr 2010, 1, 13. [Google Scholar]
- Zhou, H.; Suzuki, M.; Randers-Pehrson, G.; Vannais, D.; Chen, G.; Trosko, J.E.; Waldren, C.A.; Hei, T.K. Radiation risk to low fluences of alpha particles may be greater than we thought. Proc. Natl. Acad. Sci. USA 2001, 98, 14410–14415. [Google Scholar]
- Little, M.P.; Wakeford, R. The bystander effect in experimental systems and compatibility with radon-induced lung cancer in humans. J. Radiol. Prot 2002, 22, A27–A31. [Google Scholar]
- Little, M.; Wakeford, R.; Tawn, E.; Bouffler, S.; de Gonzalez, A. Risks associated with low doses and low dose rates of ionizing radiation: Why linearity may be (almost) the best we can do. Radiology 2009, 251, 6–12. [Google Scholar]
- Sawant, S.G.; Zheng, W.; Hopkins, K.M.; Randers-Pehrson, G.; Lieberman, H.B.; Hall, E.J. The radiation-induced bystander effect for clonogenic survival. Radiat. Res 2002, 157, 361–364. [Google Scholar]
- Goldberg, Z.; Lehnert, B.E. Radiation-induced effects in unirradiated cells: A review and implications in cancer. Int. J. Oncol 2002, 21, 337–349. [Google Scholar]
- Wu, L.J.; Randers-Pehrson, G.; Xu, A.; Waldren, C.A.; Geard, C.R.; Yu, Z.; Hei, T.K. Targeted cytoplasmic irradiation with alpha particles induces mutations in mammalian cells. Proc. Natl. Acad. Sci. USA 1999, 96, 4959–4964. [Google Scholar]
- Prise, K.; O’Sullivan, J. Radiation-induced bystander signalling in cancer therapy. Nature Rev. Cancer 2009, 9, 351–360. [Google Scholar]
- Belyakov, O.V.; Mitchell, S.A.; Parikh, D.; Randers-Pehrson, G.; Marino, S.A.; Amundson, S.A.; Geard, C.R.; Brenner, D.J. Biological effects in unirradiated human tissue induced by radiation damage up to 1 mm away. Proc. Natl. Acad. Sci. USA 2005, 102, 14203–14208. [Google Scholar]
- Chen, S.; Zhao, Y.; Han, W.; Chiu, S.K.; Zhu, L.; Wu, L.; Yu, K.N. Rescue effects in radiobiology: Unirradiated bystander cells assist irradiated cells through intercellular signal feedback. Mutat. Res 2011, 706, 59–64. [Google Scholar]
- Zhou, H.; Ivanov, V.N.; Gillespie, J.; Geard, C.R.; Amundson, S.A.; Brenner, D.J.; Yu, Z.; Lieberman, H.B.; Hei, T.K. Mechanism of radiation-induced bystander effect: Role of the cyclooxygenase-2 signaling pathway. Proc. Natl. Acad. Sci. USA 2005, 102, 14641–14646. [Google Scholar]
- Nagasawa, H.; Cremesti, A.; Kolesnick, R.; Fuks, Z.; Little, J. Involvement of membrane signaling in the bystander effect in irradiated cells. Cancer Res 2002, 62, 2531–2534. [Google Scholar]
- Yu, H. Typical cell signaling response to ionizing radiation: DNA damage and extranuclear damage. Chin. J. Cancer Res 2012, 24, 83–89. [Google Scholar]
- Shao, C.; Folkard, M.; Michael, B.D.; Prise, K.M. Targeted cytoplasmic irradiation induces bystander responses. Proc. Natl. Acad. Sci. USA 2004, 101, 13495–13500. [Google Scholar]
- Burdak-Rothkamm, S.; Short, S.C.; Folkard, M.; Rothkamm, K.; Prise, K.M. ATR-dependent radiation-induced gamma H2AX foci in bystander primary human astrocytes and glioma cells. Oncogene 2007, 26, 993–1002. [Google Scholar]
- Schaue, D.; Kachikwu, E.L.; McBride, W.H. Cytokines in radiobiological responses: A review. Radiat. Res 2012, 178, 505–523. [Google Scholar]
- Hall, E.J. The bystander effect. Health Phys 2003, 85, 31–35. [Google Scholar]
- Morgan, W. Is there a common mechanism underlying genomic instability, bystander effects and other nontargeted effects of exposure to ionizing radiation? Oncogene 2003, 22, 7094–7099. [Google Scholar]
- Morgan, W. Non-targeted and delayed effects of exposure to ionizing radiation: I. Radiation induced genomic instability and bystander effects in vitro. Radiat. Res 2003, 159, 567–580. [Google Scholar]
- Little, J.B. Radiation carcinogenesis. Carcinogenesis 2000, 21, 397–404. [Google Scholar]
- Iyer, R.; Lehnert, B. Low dose, low-LET ionizing radiation-induced radioadaptation and associated early responses in unirradiated cells. Mutat. Res 2002, 503, 1–9. [Google Scholar]
- Matsumoto, H.; Hayashi, S.; Hatashita, M.; Ohnishi, K.; Shioura, H.; Ohtsubo, T.; Kitai, R.; Ohnishi, T.; Kano, E. Induction of radioresistance by a nitric oxide-mediated bystander effect. Radiat. Res 2001, 155, 387–396. [Google Scholar]
- Barcellos-Hoff, M.; Brooks, A. Extracellular signaling through the microenvironment: A hypothesis relating carcinogenesis, bystander effects, and genomic instability. Radiat. Res 2001, 156, 618–627. [Google Scholar]
- Wolff, S. The adaptive response in radiobiology: Evolving insights and implications. Environ. Health Perspect 1998, 106, 277–283. [Google Scholar]
- Bonner, W. Low-dose radiation: Thresholds, bystander effects, and adaptive responses. Proc. Natl. Acad. Sci 2003, 100, 4973–4975. [Google Scholar]
- Ghiassi-nejad, M.; Mortazavi, S.M.J.; Cameron, J.R.; Niroomand-rad, A.; Karam, P. A Very high background radiation areas of Ramsar, Iran: Preliminary biological studies. Health phys. 2002, 82, 87–93. [Google Scholar]
- Mothersill, C.; Seymour, C.B. Radiation-induced bystander effects and the DNA paradigm: An “out of field” perspective. Mutat. Res 2006, 597, 5–10. [Google Scholar]
- Forastiere, F.; Sperati, A.; Cherubini, G.; Miceli, M.; Biggeri, A.; Axelson, O. Adult myeloid leukaemia, geology, and domestic exposure to radon and gamma radiation: A case control study in central Italy. Occup. Environ. Med 1998, 55, 106–110. [Google Scholar]
- Rechavi, G.; Berkowicz, M.; Rosner, E.; Neuman, Y.; Ben-Bassat, I.; Ramot, B. Chromosomal aberrations suggestive of mutagen-related leukemia after 21 years of “therapeutic” radon exposure. Cancer Genet. Cytogenet 1990, 48, 125–130. [Google Scholar]
- Eatough, J.; Henshaw, D. Radon dose to the skin and the possible induction of skin cancers. Radiat. Prot. Dosim 1991, 39, 33. [Google Scholar]
- Attar, M.; Kondolousy, Y.; Khansari, N. Effect of high dose natural ionizing radiation on the immune system of the exposed residents of Ramsar Town, Iran. Iran J. Allergy Asthma 2007, 6, 73–78. [Google Scholar]
- Chauhan, V.; Howland, M.; Kutzner, B.; McNamee, J.P.; Bellier, P.V.; Wilkins, R.C. Biological effects of alpha particle radiation exposure on human monocytic cells. Int. J. Hyg. Environ. Health 2012, 215, 339–344. [Google Scholar]
- Jostes, R.F. Genetic, cytogenetic, and carcinogenic effects of radon: a review. Mutat. Res 1996, 340, 125–139. [Google Scholar]
- Leonard, B.E.; Thompson, R.E.; Beecher, G.C. Human lung cancer risks from radon-Part II—Influence from combined adaptive response and bystander effects—A microdose analysis. Dose-Response 2011, 9, 502–553. [Google Scholar]
- Hall, E.J.; Hei, T.K. Genomic instability and bystander effects induced by high-LET radiation. Oncogene 2003, 22, 7034–7042. [Google Scholar]
- Schöllnberger, H.; Mitchel, R.; Redpath, J.; Crawford-Brown, D.; Hofmann, W. Detrimental and protective bystander effects: A model approach. Radiat. Res. Soc 2009, 168, 614–626. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Isotope | Emission energy (MeV) | Decay type |
|---|---|---|
| Radon-222 | 5.49 | α |
| Polonium-218 | 6.00 | α |
| Lead-214 | 0.67, 0.73 | β |
| 0.35, 0.30, 0.24 | γ | |
| Bismuth-214 | 1.54, 1.51, 3.27 | β |
| 0.61, 1.76, 1.12 | γ | |
| Polonium-214 | 7.69 | α |
| Lead-210 | 0.06, 0.02 | β |
| 0.05 | γ | |
| Bismuth-210 | 1.16 | β |
| 0.27, 0.30 | γ | |
| Polonium-210 | 5.30 | α |
| Lead-206 | Stable | |
| Radon level Bq/m3 | Lifetime risk of lung cancer death from radon exposure in homes (%) | ||
|---|---|---|---|
| Never smokers | Current smokers | General population | |
| 740 | 3.6 | 26.3 | 10.5 |
| 370 | 1.8 | 15 | 5.6 |
| 296 | 1.5 | 12 | 4.5 |
| 148 | 0.7 | 6.2 | 2.3 |
| 74 | 0.4 | 3.2 | 1.2 |
| 46.25 | 0.2 | 2 | 0.7 |
| 14.8 | 0.1 | 0.6 | 0.2 |
| Exposure a | Cancer/cell type | G to T hotspot transversion | Number of observed mutations/number of cancers studied | Exon(s) sequenced | Reference |
|---|---|---|---|---|---|
| - | NS | Not evident | 7/19 | 5–9 | [60] |
| 1382 WLM | 11 LCC; 41 SCC | (16/29) | 29/52 | 5–9 | [62] |
| - | 23 AC | Not evident | 0/23 | 5–9 | [63] |
| 4 Gy (~1460 WLM) | NHBE cells | Not evident | - | 7 | [69] |
| ~1160 WLM (estimated mean) | 19 AC; 19 SmCC; 9 SCC; 2 Mixed; 1 ASC | Not evident | 5/50 | 7 | [65] |
| <50 or >140 Bq·m−3 | 73 SmCC; 59 SCC; 86 AC; 25 Other | Not evident | 58/243 | 5–8 | [70] |
| ~1100 WLM | 19 AC; 19 SmCC; 9 SCC; 3 Mixed | Not evident | 5/50 | 7 | [66] |
| - | 16 SCC; 11 AC; 1 SmCC; 1 LCC | Rare (1/29) | 12/29 | 5–7 | [68] |
| - | 29 SCC | Rare (2/29) | Not reported/29 | 7 | [67] |
| 11.1–51.8 Bq·m−3 | NS | Not evident | 0/4 (Only 4/16 could be analysed) | 4–8 | [71] |
| Exposure | Cell type | Dose range | Dose rate | Energy (MeV) | Investigated abnormality | Reference |
|---|---|---|---|---|---|---|
| 212Bi | CHO-K1; xrs-5 | 1–5 Gy | 0.125–0.5 Gy/h | CellSurvival; CAs; HPRT mutations | [149] | |
| 222Rn | Blood lymphocyte | 2–18 cGy | 15 cGy/h | Chromatid deletions | [150] | |
| 238Pu | Multipotential murine marrow | 0.25–1 Gy | - | 3.3 | CAs | [151] |
| 238Pu | Multipotential human marrow | 0.25–1 Gy | - | CAs | [42] | |
| 238Pu | 10T1/2; 3T3 | 2.5–5 cGy | - | 5.3 | CAs; SCEs | [152] |
| 238Pu | CHO | 0.16–4.9 mGy | 0.147 Gy/min | 3.7 | SCEs | [153] |
| 238Pu | HFL1 | 0.4–12.9 cGy | 3.65 cGy/s | 3.5 | SCEs | [31] |
| 238Pu | HFL1 | 1.8–12.9 cGy | 3.65 cGy/s | 3.5 | SCE | [32] |
| 238Pu | HFL1 | 0.4–19 cGy | 3.65 cGy/s | 3.5 | ROS generation | [48] |
| 238Pu | HFL1 | 1–19 cGy | 3.65 cGy/s | 3.5 | ROS, TP53, CDC2, CDKN1A and TGF-β1 generation; cell proliferation | [154] |
| 238Pu | AG1521; AG1522; GM5758; GM6419; GM8333 | 0.3–75 cGy | 9.9 cGy/min | 3.65 | TP53, CDC2, CDKN1A, RAD51 and CCNB1 generation | [155] |
| 214Po | Blood lymphocyte | 0.03–41.4 mGy | - | 7.68 | CA | [156] |
| 4He | HBPE | 0.6 Gy | - | 4.0 | Colony formation; malignant transformation; TP53 mutations; CA; invasion ability | [76] |
| 222Rn | Blood lymphocyte | 0–127 mGy | - | 5.5 | CA; CBMN | [157] |
| 222Rn | Blood lymphocyte | 0.9–5.2 mGy | - | 5.5 | CA | [158] |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Robertson, A.; Allen, J.; Laney, R.; Curnow, A. The Cellular and Molecular Carcinogenic Effects of Radon Exposure: A Review. Int. J. Mol. Sci. 2013, 14, 14024-14063. https://doi.org/10.3390/ijms140714024
Robertson A, Allen J, Laney R, Curnow A. The Cellular and Molecular Carcinogenic Effects of Radon Exposure: A Review. International Journal of Molecular Sciences. 2013; 14(7):14024-14063. https://doi.org/10.3390/ijms140714024
Chicago/Turabian StyleRobertson, Aaron, James Allen, Robin Laney, and Alison Curnow. 2013. "The Cellular and Molecular Carcinogenic Effects of Radon Exposure: A Review" International Journal of Molecular Sciences 14, no. 7: 14024-14063. https://doi.org/10.3390/ijms140714024
APA StyleRobertson, A., Allen, J., Laney, R., & Curnow, A. (2013). The Cellular and Molecular Carcinogenic Effects of Radon Exposure: A Review. International Journal of Molecular Sciences, 14(7), 14024-14063. https://doi.org/10.3390/ijms140714024