Calcium in Red Blood Cells—A Perilous Balance



{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Ca2+ Transport across the RBC Membrane
2.1. Ca2+ Extrusion Pathway: Plasma Membrane Ca2+ Pump
2.2. Ca2+ Influx Pathways
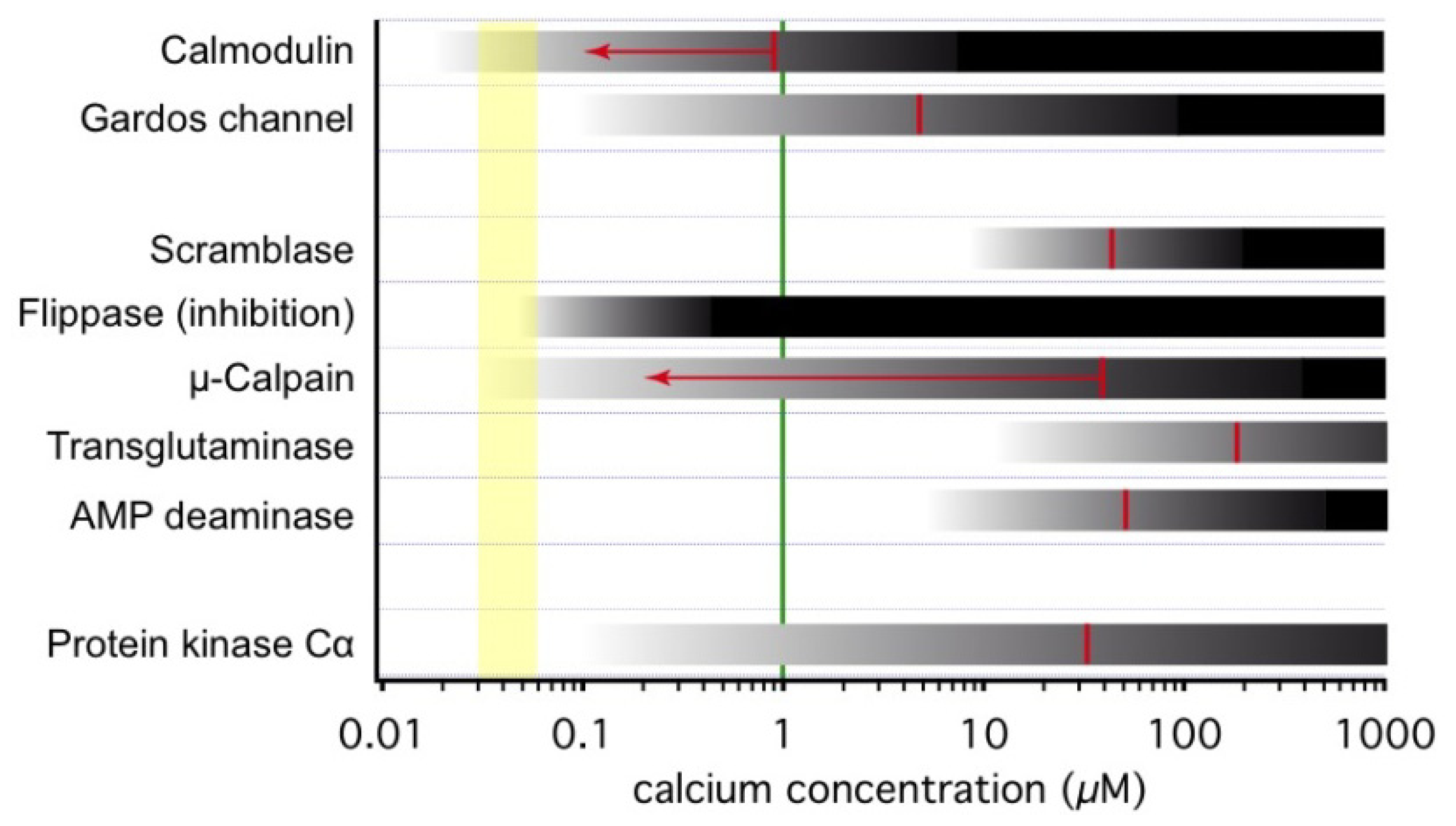
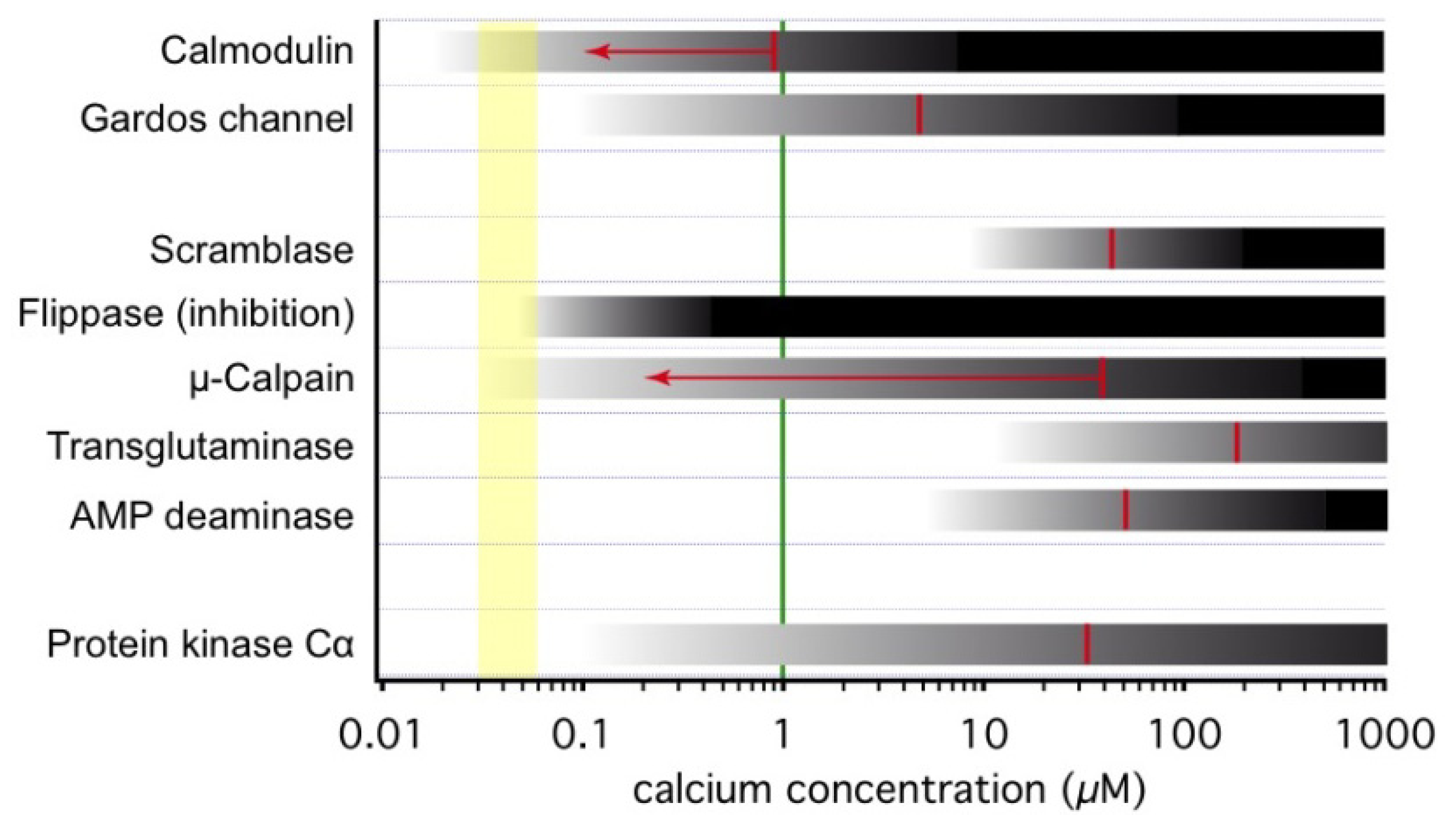
3. Ca2+-Sensitive Proteins in RBCs
3.1. Onset of Ca2+-Inducible Events and Ca2+ Sensors in RBCs
3.2. Ca2+-Dependent Phosphorylation
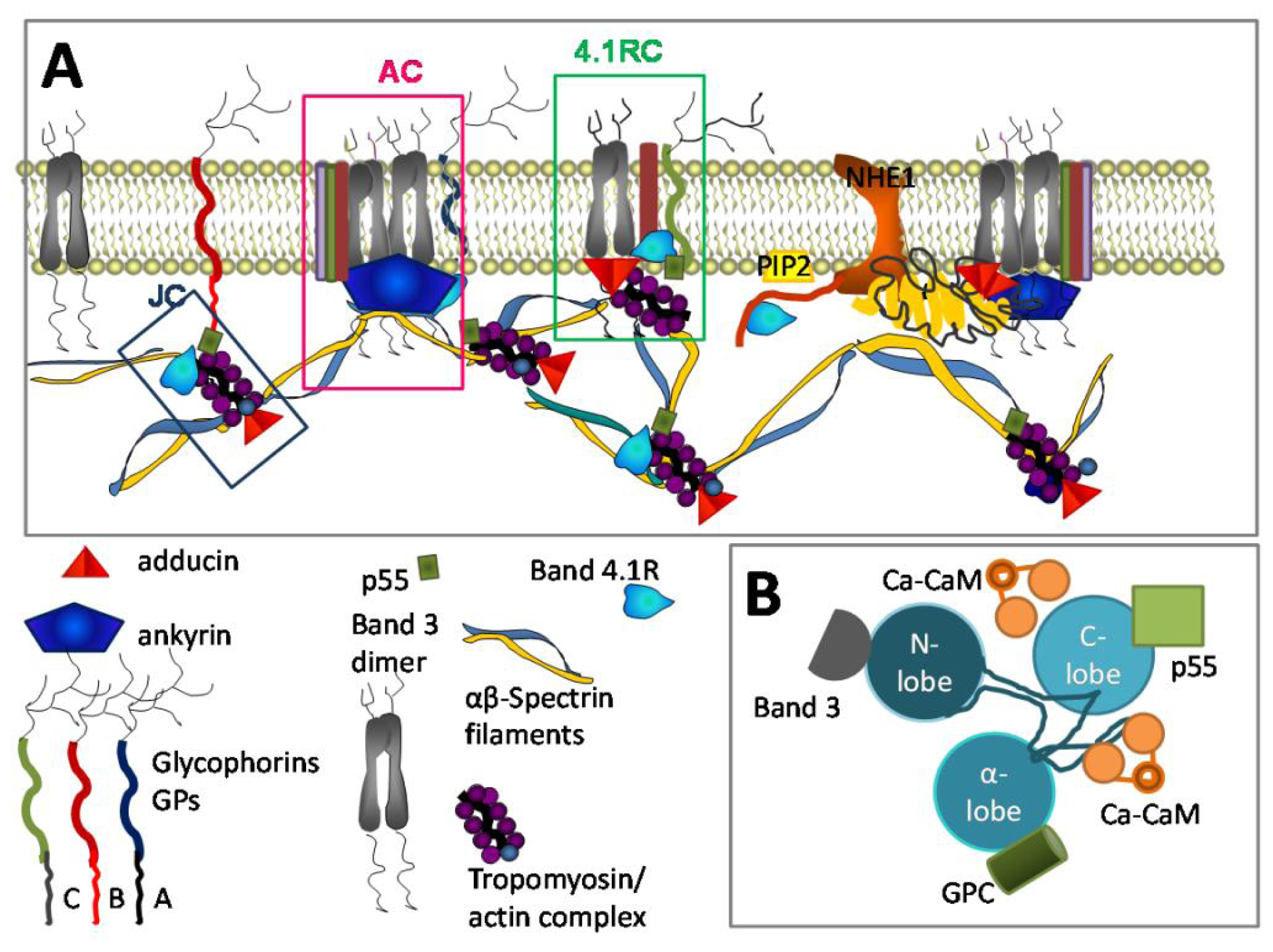
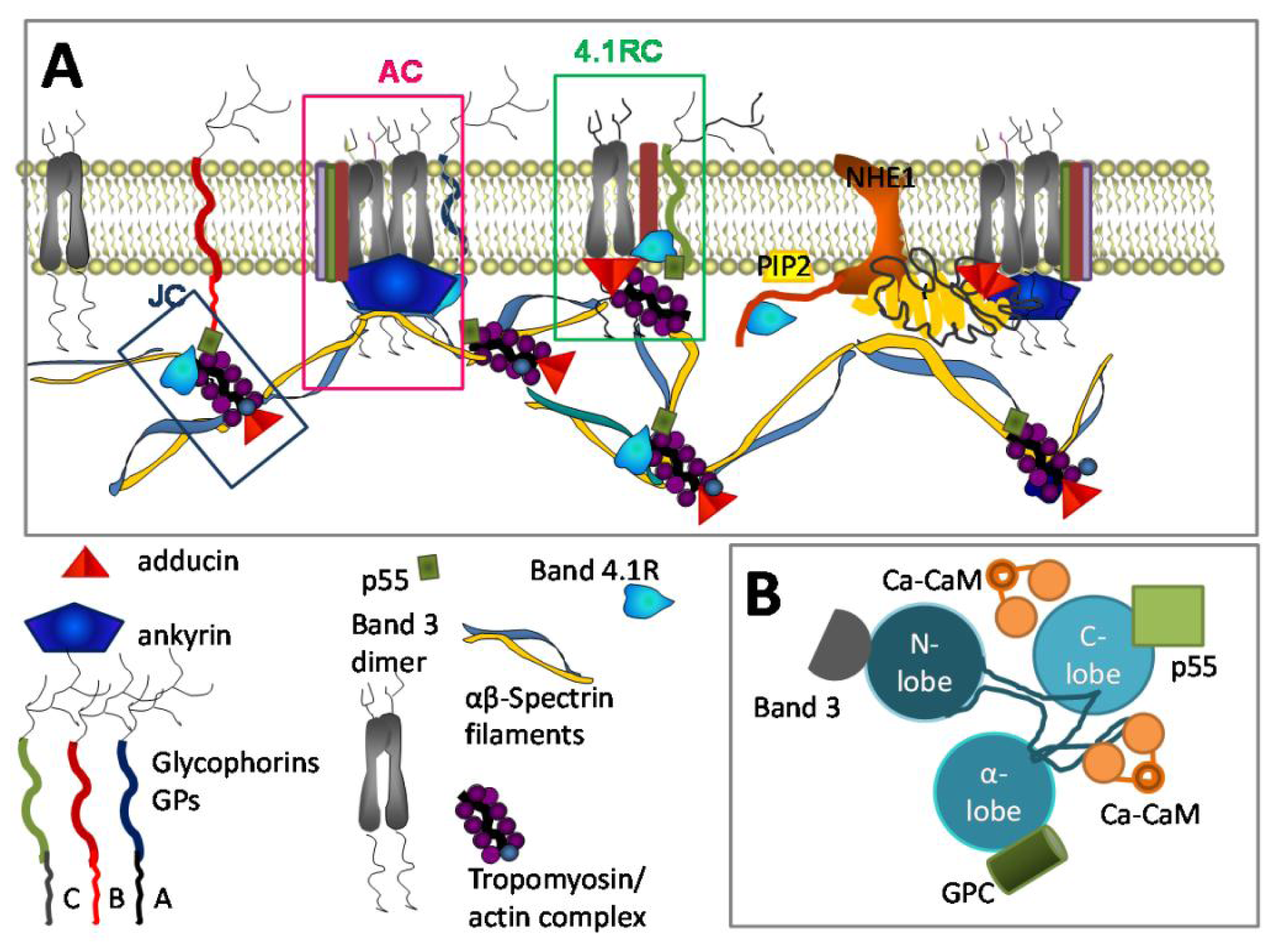
3.3. Ca2+ and RBC Cytoskeleton
3.4. Ca2+ and RBC Volume Regulation
3.5. Ca2+ and Lipid Bilayer
3.6. Ca2+ and Metabolism
3.7. Ca2+ and Redox State Preservation
3.8. Calpain and Its Targets in RBCs
3.9. Ca2+ and Inter-Cellular Interactions
4. The Physiological Role of Intracellular Ca2+: From RBC Birth to Clearance
4.1. Ca2+ in RBC Haematopoiesis
4.2. Ca2+ in Relation to the Physiological Function of RBCs
4.3. Ca2+ in RBC Clearance
5. Ca2+ Dysbalance and Haemolytic Anaemia
6. RBC Ca2+ Content and Medicinal Side Effects
6.1. Transfusion Medicine
6.2. Therapeutic Side Effects
7. Conclusion and Perspective
Conflict of Interest
References
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol 2000, 1, 11–21. [Google Scholar]
- Bootman, M.D.; Berridge, M.J.; Lipp, P. Cooking with calcium: The recipes for composing global signals from elementary events. Cell 1997, 91, 367–373. [Google Scholar]
- Bookchin, R.M.; Lew, V.L. Progressive inhibition of the Ca pump and Ca:Ca exchange in sickle red cells. Nature 1980, 284, 561–563. [Google Scholar]
- Tiffert, T.; Bookchin, R.M.; Lew, V.L. Calcium Homeostasis in Normal and Abnormal Human Red Cells. In Red Cell Membrane Transport in Health and Disease; Bernhardt, I., Ellory, C., Eds.; Springer Verlag: Heidelberg, Germany, 2003; pp. 373–405. [Google Scholar]
- Wilbrandt, W. A relation between the permeability of the red cell and its metabolism. Trans. Faraday Soc 1937, 33, 956–959. [Google Scholar]
- Pasini, E.M.; Kirkegaard, M.; Mortensen, P.; Lutz, H.U.; Thomas, A.W.; Mann, M. In-depth analysis of the membrane and cytosolic proteome of red blood cells. Blood 2006, 108, 791–801. [Google Scholar]
- Strehler, E.E.; Caride, A.J.; Filoteo, A.G.; Xiong, Y.; Penniston, J.T.; Enyedi, A. Plasma membrane Ca2+ ATPases as dynamic regulators of cellular calcium handling. Ann. N. Y. Acad. Sci 2007, 1099, 226–236. [Google Scholar]
- Tiffert, T.; Lew, V.L. Elevated intracellular Ca2+ reveals a functional membrane nucleotide pool in intact human red blood cells. J. Gen. Physiol 2011, 138, 381–391. [Google Scholar]
- Romero, P.J.; Romero, E.A. Differences in Ca2+ pumping activity between sub-populations of human red cells. Cell Calcium 1997, 21, 353–358. [Google Scholar]
- Romero, P.J.; Romero, E.A. The role of calcium metabolism in human red blood cell ageing: A proposal. Blood Cells Mol. Dis 1999, 25, 9–19. [Google Scholar]
- Schatzmann, H.J. Dependence on calcium concentration and stoichiometry of the calcium pump in human red cells. J. Physiol 1973, 235, 551–569. [Google Scholar]
- Chu, H.; Puchulu-Campanella, E.; Galan, J.A.; Taob, W.A.; Low, P.S.; Hoffman, J.F. Identification of cytoskeletal elements enclosing the ATP pools that fuel human red blood cell membrane cation pumps. Proc. Natl. Acad. Sci. USA 2012, 109, 12794–12799. [Google Scholar]
- Hoffman, J.F.; Dodson, A.; Proverbio, F. On the functional use of the membrane compartmentalized pool of ATP by the Na+ and Ca++ pumps in human red blood cell ghosts. J. Gen. Physiol 2009, 134, 351–361. [Google Scholar]
- Garrahan, P.J.; Rega, A.F. Activation of partial reactions of the Ca2+-ATPase from human red cells by Mg2+ and ATP. Biochim. Biophys. Acta 1978, 513, 59–65. [Google Scholar]
- Bredeston, L.M.; Rega, A.F. Phosphatidylcholine makes specific activity of the purified Ca2+-ATPase from plasma membranes independent of enzyme concentration. Biochim. Biophys. Acta 1999, 1420, 57–62. [Google Scholar]
- Colina, C.; Cervino, V.; Benaim, G. Ceramide and sphingosine have an antagonistic effect on the plasma-membrane Ca2+-ATPase from human erythrocytes. Biochem. J 2002, 362, 247–251. [Google Scholar]
- Oliveira, V.H.; Nascimento, K.S.O.; Freire, M.M.; Moreira, O.C.; Scofano, H.M.; Barrabin, H.; Mignaco, J.A. Mechanism of modulation of the plasma membrane Ca2+-ATPase by arachidonic acid. Prostaglandins Other Lipid Mediat 2008, 87, 47–53. [Google Scholar]
- Sarkadi, B.; Szasz, I.; Gerloczy, A.; Gardos, G. Transport parameters and stoichiometry of active calcium-ion extrusion in intact human red-cells. Biochim. Biophys. Acta 1977, 464, 93–107. [Google Scholar]
- Kaestner, L. Cation channels in erythrocytes—Historical and future perspective. Open Biol. J 2011, 4, 27–34. [Google Scholar]
- Christophersen, P.; Bennekou, P. Evidence for a voltage-gated, non-selective cation channel in the human red cell membrane. Biochim. Biophys. Acta 1991, 1065, 103–106. [Google Scholar]
- Bennekou, P. The voltage-gated non-selective cation channel from human red cells is sensitive to acetylcholine. Biochim. Biophys. Acta 1993, 1147, 165–167. [Google Scholar]
- Kaestner, L.; Bollensdorff, C.; Bernhardt, I. Non-selective voltage-activated cation channel in the human red blood cell membrane. Biochim. Biophys. Acta 1999, 1417, 9–15. [Google Scholar]
- Kaestner, L.; Christophersen, P.; Bernhardt, I.; Bennekou, P. The non-selective voltage-activated cation channel in the human red blood cell membrane: Reconciliation between two conflicting reports and further characterisation. Bioelectrochemistry 2000, 52, 117–125. [Google Scholar]
- Rodighiero, S.; de Simoni, A.; Formenti, A. The voltage-dependent nonselective cation current in human red blood cells studied by means of whole-cell and nystatin-perforated patch-clamp techniques. Biochim. Biophys. Acta 2004, 1660, 164–170. [Google Scholar]
- Bennekou, P.; Kristensen, B.I.; Christophersen, P. The human red cell voltage-regulated cation channel. The interplay with the chloride conductance, the Ca2+-activated K+ channel and the Ca2+ pump. J. Membr. Biol 2003, 195, 1–8. [Google Scholar]
- Bennekou, P.; Barksmann, T.L.; Kristensen, B.I.; Jensen, L.R.; Christophersen, P. Pharmacology of the human red cell voltage-dependent cation channel. Part II: Inactivation and blocking. Blood Cells Mol. Dis 2004, 33, 356–361. [Google Scholar]
- Bennekou, P.; Barksmann, T.L.; Christophersen, P.; Kristensen, B.I. The human red cell voltage-dependent cation channel. Part III: Distribution homogeneity and pH dependence. Blood Cells Mol. Dis 2006, 36, 10–14. [Google Scholar]
- Yang, L.; Andrews, D.A.; Low, P.S. Lysophosphatidic acid opens a Ca++ channel in human erythrocytes. Blood 2000, 95, 2420–2425. [Google Scholar]
- Andrews, D.A.; Yang, L.; Low, P.S. Phorbol ester stimulates a protein kinase C-mediated agatoxin-TK-sensitive calcium permeability pathway in human red blood cells. Blood 2002, 100, 3392–3399. [Google Scholar]
- Wagner-Britz, L.; Wang, J.; Kaestner, L.; Bernhardt, I. Protein Kinase Cα and P-Type Ca2+-Channel CaV2.1 in Red Blood Cells cCalcium Signaling. J. Cell. Physiol. Biochem 2013. in revision. [Google Scholar]
- Foller, M.; Kasinathan, R.S.; Koka, S.; Lang, C.; Shumilina, E.V.; Birnbaumer, L.; Lang, F.; Huber, S.M. TRPC6 contributes to the Ca2+ leak of human erythrocytes. Cell Physiol. Biochem 2008, 21, 183–192. [Google Scholar]
- Makhro, A.; Wang, J.; Vogel, J.; Boldyrev, A.A.; Gassmann, M.; Kaestner, L.; Bogdanova, A.Y. Functional NMDA receptors in rat erythrocytes. Am. J. Physiol. Cell Physiol 2010, 298, C1315–C1325. [Google Scholar]
- Makhro, A.; Hanggi, P.; Goede, J.; Wang, J.; Brüggemann, A.; Gassmann, M.; Kaestner, L.; Speer, O.; Bogdanova, A. N-Methyl d-Aspartate (NMDA) Receptors in Erythroid Precursor Cells and in Circulating Human Red Blood Cells Contributes to the Regulation of Intracellular Calcium Levels. Am. J. Physiol 2013. in revision. [Google Scholar]
- Madry, C.; Betz, H.; Geiger, J.R.P.; Laube, B. Supralinear potentiation of NR1/NR3A excitatory glycine receptors by Zn2+ and NR1 antagonist. Proc. Natl. Acad. Sci. USA 2008, 105, 12563–12568. [Google Scholar]
- Zarychanski, R.; Schulz, V.P.; Houston, B.L.; Maksimova, Y.; Houston, D.S.; Smith, B.; Rinehart, J.; Gallagher, P.G. Mutations in the mechanotransduction protein PIEZO1 are associated with hereditary xerocytosis. Blood 2012, 120, 1908–1915. [Google Scholar]
- Gottlieb, P.A.; Sachs, F. Piezo1: Properties of a cation selective mechanical channel. Channels (Austin) 2012, 6, 214–219. [Google Scholar]
- Gottlieb, P.A.; Bae, C.; Sachs, F. Gating the mechanical channel Piezo1: A comparison between whole-cell and patch recording. Channels (Austin) 2012, 6, 282–289. [Google Scholar]
- Foller, M.; Mahmud, H.; Gu, S.; Kucherenko, Y.; Gehring, E.-M.; Shumilina, E.; Floride, E.; Sprengel, R.; Lang, F. Modulation of suicidal erythrocyte cation channels by an AMPA antagonist. J. Cell. Mol. Med 2009, 13, 3680–3686. [Google Scholar]
- Browning, J.A.; Staines, H.M.; Robinson, H.C.; Powell, T.; Ellory, J.C.; Gibson, J.S. The effect of deoxygenation on whole-cell conductance of red blood cells from healthy individuals and patients with sickle cell disease. Blood 2007, 109, 2622–2629. [Google Scholar]
- Ma, Y.-L.; Rees, D.C.; Gibson, J.S.; Ellory, J.C. The conductance of red blood cells from sickle cell patients: Ion selectivity and inhibitors. J. Physiol. (Lond.) 2012, 590, 2095–2105. [Google Scholar]
- Lew, V.L.; Ortiz, O.E.; Bookchin, R.M. Stochastic nature and red cell population distribution of the sickling-induced Ca2+ permeability. J. Clin. Invest 1997, 99, 2727–2735. [Google Scholar]
- Bogdanova, A.; Makhro, A.; Wang, J.; Gassmann, M.; Kaestner, L. Responses of rat erythrocytes to homocysteine and homocysteic acid treatment. Clin. Biochem 2009, 42, 1858–1859. [Google Scholar]
- Clapham, D.E. Calcium signaling. Cell 2007, 131, 1047–1058. [Google Scholar]
- Meador, W.E.; Means, A.R.; Quiocho, F.A. Target enzyme recognition by calmodulin: 2.4 A structure of a calmodulin-peptide complex. Science 1992, 257, 1251–1255. [Google Scholar]
- Meador, W.E.; Means, A.R.; Quiocho, F.A. Modulation of calmodulin plasticity in molecular recognition on the basis of X-ray structures. Science 1993, 262, 1718–1721. [Google Scholar]
- Cho, W.; Stahelin, R.V. Membrane-protein interactions in cell signaling and membrane trafficking. Annu. Rev. Biophys. Biomol. Struct 2005, 34, 119–151. [Google Scholar]
- Thomas, D.; Tovey, S.C.; Collins, T.J.; Bootman, M.D.; Berridge, M.J.; Lipp, P. A comparison of fluorescent Ca2+ indicator properties and their use in measuring elementary and global Ca2+ signals. Cell Calcium 2000, 28, 213–223. [Google Scholar]
- Kaestner, L.; Tabellion, W.; Weiss, E.; Bernhardt, I.; Lipp, P. Calcium imaging of individual erythrocytes: Problems and approaches. Cell Calcium 2006, 39, 13–19. [Google Scholar]
- Jarrett, H.W.; Kyte, J. Human erythrocyte calmodulin. Further chemical characterization and the site of its interaction with the membrane. J. Biol. Chem 1979, 254, 8237–8244. [Google Scholar]
- Leinders, T.; van Kleef, R.G.; Vijverberg, H.P. Single Ca2+-activated K+ channels in human erythrocytes: Ca2+ dependence of opening frequency but not of open lifetimes. Biochim. Biophys. Acta 1992, 1112, 67–74. [Google Scholar]
- Stout, J.G.; Zhou, Q.; Wiedmer, T.; Sims, P.J. Change in conformation of plasma membrane phospholipid scramblase induced by occupancy of its Ca2+ binding site. Biochemistry 1998, 37, 14860–14866. [Google Scholar]
- Woon, L.A.; Holland, J.W.; Kable, E.P.; Roufogalis, B.D. Ca2+ sensitivity of phospholipid scrambling in human red cell ghosts. Cell Calcium 1999, 25, 313–320. [Google Scholar]
- Bitbol, M.; Fellmann, P.; Zachowski, A.; Devaux, P.F. Ion regulation of phosphatidylserine and phosphatidylethanolamine outside-inside translocation in human erythrocytes. Biochim. Biophys. Acta 1987, 904, 268–282. [Google Scholar]
- Murakami, T.; Hatanaka, M.; Murachi, T. The cytosol of human erythrocytes contains a highly Ca2+-sensitive thiol protease (calpain I) and its specific inhibitor protein (calpastatin). J. Biochem 1981, 90, 1809–1816. [Google Scholar]
- Salamino, F.; de Tullio, R.; Mengotti, P.; Viotti, P.L.; Melloni, E.; Pontremoli, S. Site-directed activation of calpain is promoted by a membrane-associated natural activator protein. Biochem. J 1993, 290, 191–197. [Google Scholar]
- Bergamini, C.M.; Signorini, M. Studies on tissue transglutaminases: Interaction of erythrocyte type-2 transglutaminase with GTP. Biochem. J 1993, 291, 37–39. [Google Scholar]
- Almaraz, L.; García-Sancho, J.; Lew, V.L. Calcium-induced conversion of adenine nucleotides to inosine monophosphate in human red cells. J. Physiol. (Lond.) 1988, 407, 557–567. [Google Scholar]
- Kohout, S.C.; Corbalán-García, S.; Torrecillas, A.; Goméz-Fernandéz, J.C.; Falke, J.J. C2 domains of protein kinase C isoforms alpha, beta, and gamma: Activation parameters and calcium stoichiometries of the membrane-bound state. Biochemistry 2002, 41, 11411–11424. [Google Scholar]
- Kuhlman, P.A.; Hughes, C.A.; Bennett, V.; Fowler, V.M. A new function for adducin. Calcium/calmodulin-regulated capping of the barbed ends of actin filaments. J. Biol. Chem 1996, 271, 7986–7991. [Google Scholar]
- Kifor, G.; Toon, M.R.; Janoshazi, A.; Solomon, A.K. Interaction between red cell membrane band 3 and cytosolic carbonic anhydrase. J. Membr. Biol 1993, 134, 169–179. [Google Scholar]
- Li, X.; Alvarez, B.; Casey, J.R.; Reithmeier, R.A.F.; Fliegel, L. Carbonic anhydrase II binds to and enhances activity of the Na+/H+ exchanger. J. Biol. Chem 2002, 277, 36085–36091. [Google Scholar]
- Nunomura, W.; Denker, S.P.; Barber, D.L.; Takakuwa, Y.; Gascard, P. Characterization of cytoskeletal protein 4.1R interaction with NHE1 (Na+/H+ exchanger isoform 1). Biochem. J 2012, 446, 427–435. [Google Scholar]
- An, X.; Zhang, X.; Debnath, G.; Baines, A.J.; Mohandas, N. Phosphatidylinositol-4,5- biphosphate (PIP2) differentially regulates the interaction of human erythrocyte protein 4.1 (4.1R) with membrane proteins. Biochemistry 2006, 45, 5725–5732. [Google Scholar]
- Nunomura, W.; Sasakura, D.; Shiba, K.; Nakamura, S.; Kidokoro, S.-I.; Takakuwa, Y. Structural stabilization of protein 4.1R FERM domain upon binding to apo-calmodulin: Novel insights into the biological significance of the calcium-independent binding of calmodulin to protein 4.1R. Biochem. J 2011, 440, 367–374. [Google Scholar]
- Lipp, P.; Reither, G. Protein kinase C: The “masters” of calcium and lipid. Cold Spring Harb. Perspect. Biol. 2011, 3. [Google Scholar] [CrossRef]
- Govekar, R.B.; Zingde, S.M. Protein kinase C isoforms in human erythrocytes. Ann. Hematol 2001, 80, 531–534. [Google Scholar]
- Newton, A.C. Protein kinase C: Structural and spatial regulation by phosphorylation, cofactors, and macromolecular interactions. Chem. Rev 2001, 101, 2353–2364. [Google Scholar]
- Steinberg, S.F. Structural basis of protein kinase C isoform function. Physiol. Rev 2008, 88, 1341–1378. [Google Scholar]
- Wright, L.C.; Chen, S.; Roufogalis, B.D. Regulation of the activity and phosphorylation of the plasma membrane Ca2+-ATPase by protein kinase C in intact human erythrocytes. Arch. Biochem. Biophys 1993, 306, 277–284. [Google Scholar]
- Cohen, C.M.; Gascard, P. Regulation and post-translational modification of erythrocyte membrane and membrane-skeletal proteins. Semin. Hematol 1992, 29, 244–292. [Google Scholar]
- De Oliveira, S.; Silva-Herdade, A.S.; Saldanha, C. Modulation of erythrocyte deformability by PKC activity. Clin. Hemorheol. Microcirc 2008, 39, 363–373. [Google Scholar]
- George, A.; Pushkaran, S.; Konstantinidis, D.G.; Koochaki, S.; Malik, P.; Mohandas, N.; Zheng, Y.; Joiner, C.H.; Kalfa, T.A. Erythrocyte NADPH oxidase activity modulated by Rac GTPases, PKC, and plasma cytokines contributes to oxidative stress in sickle cell disease. Blood 2013, 121, 2099–2107. [Google Scholar]
- Ceolotto, G.; Conlin, P.; Clari, G.; Semplicini, A.; Canessa, M. Protein kinase C and insulin regulation of red blood cell Na+/H+ exchange. Am. J. Physiol 1997, 272, C818–C826. [Google Scholar]
- Mohandas, N.; Gallagher, P.G. Red cell membrane: Past, present, and future. Blood 2008, 112, 3939–3948. [Google Scholar]
- Bruce, L.J.; Beckmann, R.; Ribeiro, M.L.; Peters, L.L.; Chasis, J.A.; Delaunay, J.; Mohandas, N.; Anstee, D.J.; Tanner, M.J.A. A band 3-based macrocomplex of integral and peripheral proteins in the RBC membrane. Blood 2003, 101, 4180–4188. [Google Scholar]
- Salomao, M.; Zhang, X.; Yang, Y.; Lee, S.; Hartwig, J.H.; Chasis, J.A.; Mohandas, N.; An, X. Protein 4.1R-dependent multiprotein complex: New insights into the structural organization of the red blood cell membrane. Proc. Natl. Acad. Sci. USA 2008, 105, 8026–8031. [Google Scholar]
- Rivera, A.; de Franceschi, L.; Peters, L.L.; Gascard, P.; Mohandas, N.; Brugnara, C. Effect of complete protein 4.1R deficiency on ion transport properties of murine erythrocytes. Am. J. Physiol. Cell Physiol 2006, 291, C880–C886. [Google Scholar]
- Hoeflich, K.P.; Ikura, M. Calmodulin in action: Diversity in target recognition and activation mechanisms. Cell 2002, 108, 739–742. [Google Scholar]
- Chin, D.; Means, A.R. Calmodulin: A prototypical calcium sensor. Trends Cell Biol 2000, 10, 322–328. [Google Scholar]
- Vetter, S.W.; Leclerc, E. Phosphorylation of serine residues affects the conformation of the calmodulin binding domain of human protein 4.1. Eur. J. Biochem. FEBS 2001, 268, 4292–4299. [Google Scholar]
- Gauthier, E.; Guo, X.; Mohandas, N.; An, X. Phosphorylation-dependent perturbations of the 4.1R-associated multiprotein complex of the erythrocyte membrane. Biochemistry 2011, 50, 4561–4567. [Google Scholar]
- Dyrda, A.; Cytlak, U.; Ciuraszkiewicz, A.; Lipinska, A.; Cueff, A.; Bouyer, G.; Egée, S.; Bennekou, P.; Lew, V.L.; Thomas, S.L.Y. Local membrane deformations activate Ca2+-dependent K+ and anionic currents in intact human red blood cells. PLoS One 2010, 5, e9447. [Google Scholar]
- Manno, S.; Takakuwa, Y.; Mohandas, N. Modulation of erythrocyte membrane mechanical function by protein 4.1 phosphorylation. J. Biol. Chem 2005, 280, 7581–7587. [Google Scholar]
- George, A.; Pushkaran, S.; Li, L.; An, X.; Zheng, Y.; Mohandas, N.; Joiner, C.H.; Kalfa, T.A. Altered phosphorylation of cytoskeleton proteins in sickle red blood cells: The role of protein kinase C, Rac GTPases, and reactive oxygen species. Blood Cells Mol. Dis 2010, 45, 41–45. [Google Scholar]
- Lorand, L.; Murthy, S.N.P.; Khan, A.A.; Xue, W.; Lockridge, O.; Chishti, A.H. Transglutaminase-mediated remodeling of the human erythrocyte membrane skeleton: Relevance for erythrocyte diseases with shortened cell lifespan. Adv. Enzymol. Relat. Areas Mol. Biol 2011, 78, 385–414. [Google Scholar]
- Guizouarn, H.; Gabillat, N.; Motais, R.; Borgese, F. Multiple transport functions of a red blood cell anion exchanger, tAE1: Its role in cell volume regulation. J. Physiol. (Lond.) 2001, 535, 497–506. [Google Scholar]
- Gardos, G. The function of calcium in the potassium permeability of human erythrocytes. Biochim. Biophys. Acta 1958, 30, 653–654. [Google Scholar]
- Hamill, O.P. Potassium channel currents in human red blood cells. J. Physiol. (Lond.) 1981, 319, 97P–98P. [Google Scholar]
- Hamill, O.P. Potassium and Chloride Channels in Red Blood Cells. In Single Channel Recording; Sakmann, B., Neher, E., Eds.; Plenum Press: New York, NY, USA and London, UK, 1983; pp. 451–471. [Google Scholar]
- Grygorczyk, R.; Schwarz, W. Properties of the Ca2+-activated K+ conductance of human red cells as revealed by the patch-clamp technique. Cell Calcium 1983, 4, 499–510. [Google Scholar]
- Hoffman, J.F.; Joiner, W.; Nehrke, K.; Potapova, O.; Foye, K.; Wickrema, A. The hSK4 (KCNN4) isoform is the Ca2+-activated K+ channel (Gardos channel) in human red blood cells. Proc. Natl. Acad. Sci. USA 2003, 100, 7366–7371. [Google Scholar]
- Kaestner, L.; Bernhardt, I. Ion channels in the human red blood cell membrane: Their further investigation and physiological relevance. Bioelectrochemistry 2002, 55, 71–74. [Google Scholar]
- Kaestner, L.; Tabellion, W.; Lipp, P.; Bernhardt, I. Prostaglandin E2 activates channel-mediated calcium entry in human erythrocytes: An indication for a blood clot formation supporting process. Thromb. Haemostasis 2004, 92, 1269–1272. [Google Scholar]
- Bassé, F.; Stout, J.G.; Sims, P.J.; Wiedmer, T. Isolation of an erythrocyte membrane protein that mediates Ca2+-dependent transbilayer movement of phospholipid. J. Biol. Chem 1996, 271, 17205–17210. [Google Scholar]
- Verkleij, A.J.; Zwaal, R.F.; Roelofsen, B.; Comfurius, P.; Kastelijn, D.; van Deenen, L.L. The asymmetric distribution of phospholipids in the human red cell membrane. A combined study using phospholipases and freeze-etch electron microscopy. Biochim. Biophys. Acta 1973, 323, 178–193. [Google Scholar]
- Zhou, Q.; Zhao, J.; Stout, J.G.; Luhm, R.A.; Wiedmer, T.; Sims, P.J. Molecular cloning of human plasma membrane phospholipid scramblase. A protein mediating transbilayer movement of plasma membrane phospholipids. J. Biol. Chem 1997, 272, 18240–18244. [Google Scholar]
- Morrot, G.; Hervé, P.; Zachowski, A.; Fellmann, P.; Devaux, P.F. Aminophospholipid translocase of human erythrocytes: Phospholipid substrate specificity and effect of cholesterol. Biochemistry 1989, 28, 3456–3462. [Google Scholar]
- Beitner, R. Control of glycolytic enzymes through binding to cell structures and by glucose-1,6-bisphosphate under different conditions. The role of Ca2+ and calmodulin. Int. J. Biochem 1993, 25, 297–305. [Google Scholar]
- Nakashima, K.; Fujii, S.; Kaku, K.; Kaneko, T. Calcium-calmodulin dependent phosphorylation of erythrocyte pyruvate kinase. Biochem. Biophys. Res. Commun 1982, 104, 285–289. [Google Scholar]
- Campanella, M.E.; Chu, H.; Low, P.S. Assembly and regulation of a glycolytic enzyme complex on the human erythrocyte membrane. Proc. Natl. Acad. Sci. USA 2005, 102, 2402–2407. [Google Scholar]
- Zipser, Y.; Piade, A.; Barbul, A.; Korenstein, R.; Kosower, N.S. Ca2+ promotes erythrocyte band 3 tyrosine phosphorylation via dissociation of phosphotyrosine phosphatase from band 3. Biochem. J 2002, 368, 137–144. [Google Scholar]
- Chu, H.; Low, P.S. Mapping of glycolytic enzyme-binding sites on human erythrocyte band 3. Biochem. J 2006, 400, 143–151. [Google Scholar]
- Chu, H.; Breite, A.; Ciraolo, P.; Franco, R.S.; Low, P.S. Characterization of the deoxyhemoglobin binding site on human erythrocyte band 3: Implications for O2 regulation of erythrocyte properties. Blood 2008, 111, 932–938. [Google Scholar]
- Tiffert, T.; Etzion, Z.; Bookchin, R.M.; Lew, V.L. Effects of deoxygenation on active and passive Ca2+ transport and cytoplasmic Ca2+ buffering in normal human red cells. J. Physiol. (Lond.) 1993, 464, 529–544. [Google Scholar]
- Tiffert, T.; Lew, V.L. Cytoplasmic calcium buffers in intact humanred cells. J. Physiol. (Lond.) 1997, 500, 139–154. [Google Scholar]
- Alderton, W.K.; Cooper, C.E.; Knowles, R.G. Nitric oxide synthases: Structure, function and inhibition. Biochem. J 2001, 357, 593–615. [Google Scholar]
- Spratt, D.E.; Newman, E.; Mosher, J.; Ghosh, D.K.; Salerno, J.C.; Guillemette, J.G. Binding and activation of nitric oxide synthase isozymes by calmodulin EF hand pairs. FEBS J 2006, 273, 1759–1771. [Google Scholar]
- Kleinbongard, P.; Schulz, R.; Rassaf, T.; Lauer, T.; Dejam, A.; Jax, T.; Kumara, I.; Gharini, P.; Kabanova, S.; Ozüyaman, B.; et al. Red blood cells express a functional endothelial nitric oxide synthase. Blood 2006, 107, 2943–2951. [Google Scholar]
- Ozüyaman, B.; Grau, M.; Kelm, M.; Merx, M.W.; Kleinbongard, P. RBC NOS: Regulatory mechanisms and therapeutic aspects. Trends Mol. Med 2008, 14, 314–322. [Google Scholar]
- Ulker, P.; Yaras, N.; Yalcin, O.; Celik-Ozenci, C.; Johnson, P.C.; Meiselman, H.J.; Baskurt, O.K. Shear stress activation of nitric oxide synthase and increased nitric oxide levels in human red blood cells. Nitric Oxide 2011, 24, 184–191. [Google Scholar]
- Cave, A.C.; Brewer, A.C.; Narayanapanicker, A.; Ray, R.; Grieve, D.J.; Walker, S.; Shah, A.M. NADPH oxidases in cardiovascular health and disease. Antioxid. Redox Signal 2006, 8, 691–728. [Google Scholar]
- Mihov, D.; Vogel, J.; Gassmann, M.; Bogdanova, A.Y. Erythropoietin activates nitric oxide synthase in murine erythrocytes. Am. J. Physiol. Cell Physiol 2009, 297, C378–C388. [Google Scholar]
- Inomata, M.; Nakamura, M.; Imajoh-Ohmi, S.; Kawashima, S. A variety of calpain/calpastatin systems in mammalian erythrocytes. Biochim. Biophys. Acta 1993, 1178, 207–214. [Google Scholar]
- Goll, D.E.; Thompson, V.F.; Li, H.; Wei, W.; Cong, J. The calpain system. Physiol. Rev 2003, 83, 731–801. [Google Scholar]
- Campbell, R.L.; Davies, P.L. Structure–function relationships in calpains. Biochem. J 2012, 447, 335–351. [Google Scholar]
- Hatanaka, M.; Yoshimura, N.; Murakami, T.; Kannagi, R.; Murachi, T. Evidence for membrane-associated calpain I in human erythrocytes. Detection by an immunoelectrophoretic blotting method using monospecific antibody. Biochemistry 1984, 23, 3272–3276. [Google Scholar]
- Samis, J.A.; Elce, J.S. Immunogold electron-microscopic localization of calpain I in human erythrocytes. Thromb. Haemost 1989, 61, 250–253. [Google Scholar]
- Molinari, M.; Anagli, J.; Carafoli, E. Ca2+-activated neutral protease is active in the erythrocyte membrane in its nonautolyzed 80-kDa form. J. Biol. Chem 1994, 269, 27992–27995. [Google Scholar]
- Mortensen, A.M.; Novak, R.F. Dynamic changes in the distribution of the calcium-activated neutral protease in human red blood cells following cellular insult and altered Ca2+ homeostasis. Toxicol. Appl. Pharmacol 1992, 117, 180–188. [Google Scholar]
- Melloni, E.; Salamino, F.; Sparatore, B.; Michetti, M.; Pontremoli, S. Ca2+-dependent neutral proteinase from human erythrocytes: Activation by Ca2+ ions and substrate and regulation by the endogenous inhibitor. Biochem. Int 1984, 8, 477–489. [Google Scholar]
- Wieschhaus, A.; Khan, A.; Zaidi, A.; Rogalin, H.; Hanada, T.; Liu, F.; de Franceschi, L.; Brugnara, C.; Rivera, A.; Chishti, A.H. Calpain-1 knockout reveals broad effects on erythrocyte deformability and physiology. Biochem. J 2012, 448, 141–152. [Google Scholar]
- Molinari, M.; Maki, M.; Carafoli, E. Purification of mu-calpain by a novel affinity chromatography approach. New insights into the mechanism of the interaction of the protease with targets. J. Biol. Chem 1995, 270, 14576–14581. [Google Scholar]
- Schwarz-Benmeir, N.; Glaser, T.; Barnoy, S.; Kosower, N.S. Calpastatin in erythrocytes of young and old individuals. Biochem. J 1994, 304, 365–370. [Google Scholar]
- Glaser, T.; Schwarz-Benmeir, N.; Barnoy, S.; Barak, S.; Eshhar, Z.; Kosower, N.S. Calpain (Ca2+-dependent thiol protease) in erythrocytes of young and old individuals. Proc. Natl. Acad. Sci. USA 1994, 91, 7879–7883. [Google Scholar]
- Rademaker, M.; Thomas, R.H.; Kirby, J.D.; Kovacs, I.B. Calcium influx into red blood cells: The effect of sera from patients with systemic sclerosis. Clin. Exp. Rheumatol 1991, 9, 247–251. [Google Scholar]
- Hung, T.C.; Pham, S.; Steed, D.L.; Webster, M.W.; Butter, D.B. Alterations in erythrocyte rheology in patients with severe peripheral vascular disease: 1. Cell volume dependence of erythrocyte rigidity. Angiology 1991, 42, 210–217. [Google Scholar]
- Jendryczko, A.; Pardela, M. Abnormal effect of sera from patients with atherosclerosis on calcium influx into normal erythrocytes. Cor. Vasa 1992, 34, 428–433. [Google Scholar]
- Piagnerelli, M.; Boudjeltia, K.Z.; Vanhaeverbeek, M.; Vincent, J.-L. Red blood cell rheology in sepsis. Intensive Care Med 2003, 29, 1052–1061. [Google Scholar]
- Baskurt, O.; Neu, B.; Meiselman, H.J. Red Blood Cell Aggregation; CRC Press: Boca Raton, FL, USA, 2012. [Google Scholar]
- Duke, W.W. The relation of blood platelets to hemorrhagic disease. Description of a method for determining the bleeding time and coagulation time and report of three cases of hemorrhagic disease relieved by transfusion. J. Am. Med. Assoc 1910, 55, 1185–1192. [Google Scholar]
- Hellem, A.J.; Borchgrevink, C.F.; Ames, S.B. The role of red cells in haemostasis: The relation between haematocrit, bleeding time and platelet adhesiveness. Br. J. Haematol 1961, 7, 42–50. [Google Scholar]
- Andrews, D.A.; Low, P.S. Role of red blood cells in thrombosis. Curr. Opin. Hematol 1999, 6, 76–82. [Google Scholar]
- Chung, S.M.; Bae, O.N.; Lim, K.M.; Noh, J.Y.; Lee, M.Y.; Jung, Y.S.; Chung, J.H. Lysophosphatidic acid induces thrombogenic activity through phosphatidylserine exposure and procoagulant microvesicle generation in human erythrocytes. Arterioscl. Thromb. Vasc. Biol 2007, 27, 414–421. [Google Scholar]
- Noh, J.-Y.; Lim, K.-M.; Bae, O.-N.; Chung, S.-M.; Lee, S.-W.; Joo, K.-M.; Lee, S.-D.; Chung, J.-H. Procoagulant and prothrombotic activation of human erythrocytes by phosphatidic acid. AJP Heart Circ. Physiol 2010, 299, H347–H355. [Google Scholar]
- Steffen, P.; Jung, A.; Nguyen, D.B.; Müller, T.; Bernhardt, I.; Kaestner, L.; Wagner, C. Stimulation of human red blood cells leads to Ca2+-mediated intercellular adhesion. Cell Calcium 2011, 50, 54–61. [Google Scholar]
- Kaestner, L.; Steffen, P.; Nguyen, D.B.; Wang, J.; Wagner-Britz, L.; Jung, A.; Wagner, C.; Bernhardt, I. Lysophosphatidic acid induced red blood cell aggregation in vitro. Bioelectrochemistry 2012, 87, 89–95. [Google Scholar]
- Wautier, M.-P.; Nemer, El W.; Gane, P.; Rain, J.-D.; Cartron, J.-P.; Colin, Y.; le van Kim, C.; Wautier, J.-L. Increased adhesion to endothelial cells of erythrocytes from patients with polycythemia vera is mediated by laminin alpha5 chain and Lu/BCAM. Blood 2007, 110, 894–901. [Google Scholar]
- Yedgar, S.; Kaul, D.K.; Barshtein, G. RBC adhesion to vascular endothelial cells: More potent than RBC aggregation in inducing circulatory disorders. Microcirculation 2008, 15, 581–583. [Google Scholar]
- Wautier, M.-P.; Héron, E.; Picot, J.; Colin, Y.; Hermine, O.; Wautier, J.-L. Red blood cell phosphatidylserine exposure is responsible for increased erythrocyte adhesion to endothelium in central retinal vein occlusion. J. Thromb. Haemost 2011, 9, 1049–1055. [Google Scholar]
- Miller, B.A.; Cheung, J.Y. Mechanisms of erythropoietin signal transduction: Involvement of calcium channels. Proc. Soc. Exp. Biol. Med 1994, 206, 263–267. [Google Scholar]
- Schaefer, A.; Magócsi, M.; Marquardt, H. Signalling mechanisms in erythropoiesis: The enigmatic role of calcium. Cell. Signal 1997, 9, 483–495. [Google Scholar]
- Cheung, J.Y.; Zhang, X.Q.; Bokvist, K.; Tillotson, D.L.; Miller, B.A. Modulation of calcium channels in human erythroblasts by erythropoietin. Blood 1997, 89, 92–100. [Google Scholar]
- Miller, B.A.; Cheung, J.Y.; Tillotson, D.L.; Hope, S.M.; Scaduto, R.C. Erythropoietin stimulates a rise in intracellular-free calcium concentration in single BFU-E derived erythroblasts at specific stages of differentiation. Blood 1989, 73, 1188–1194. [Google Scholar]
- De Haro, C.; de Herreros, A.G.; Ochoa, S. Protein phosphorylation and translational control in reticulocytes: Activation of the heme-controlled translational inhibitor by calcium ions and phospholipid. Curr. Top. Cell. Regul 1985, 27, 63–81. [Google Scholar]
- Liu, J.; Guo, X.; Mohandas, N.; Chasis, J.A.; An, X. Membrane remodeling during reticulocyte maturation. Blood 2010, 115, 2021–2027. [Google Scholar]
- Bookchin, R.M.; Lew, V.L.; Roth, E.F. Elevated Red Cell Calcium: Innocent Bystander or Kiss of Death? In Cellular and Molecular Aspects of Aging: The Red Cell as a Model; Eaton, J.W., Ed.; John Wiley & Sons: New York, NY, USA, 1985; pp. 369–375. [Google Scholar]
- Clark, M.R. Senescence of red blood cells: Progress and problems. Physiol. Rev 1988, 68, 503–554. [Google Scholar]
- Friederichs, E.; Meiselman, H.J. Effects of calcium permeabilization on RBC rheologic behavior. Biorheology 1994, 31, 207–215. [Google Scholar]
- Bosman, G.J.; Willekens, F.L.; Werre, J.M. Erythrocyte aging: A more than superficial resemblance to apoptosis? Cell Physiol. Biochem 2005, 16, 1–8. [Google Scholar]
- Mohandas, N.; Groner, W. Cell membrane and volume changes during red cell development and aging. Ann. N. Y. Acad. Sci 1989, 554, 217–224. [Google Scholar]
- Lutz, H.U. Innate immune and non-immune mediators of erythrocyte clearance. Cell. Mol. Biol. (Noisy-le-grand) 2004, 50, 107–116. [Google Scholar]
- Lew, V.L.; Daw, N.; Etzion, Z.; Tiffert, T.; Muoma, A.; Vanagas, L.; Bookchin, R.M. Effects of age-dependent membrane transport changes on the homeostasis of senescent human red blood cells. Blood 2007, 110, 1334–1342. [Google Scholar]
- Rice, L.; Alfrey, C.P. The negative regulation of red cell mass by neocytolysis: Physiologic and pathophysiologic manifestations. Cell Physiol. Biochem 2005, 15, 245–250. [Google Scholar]
- Risso, A.; Turello, M.; Biffoni, F.; Antonutto, G. Red blood cell senescence and neocytolysis in humans after high altitude acclimatization. Blood Cells Mol. Dis 2007, 38, 83–92. [Google Scholar]
- Chang, C.-C.; Chen, Y.; Modi, K.; Awar, O.; Alfrey, C.; Rice, L. Changes of red blood cell surface markers in a blood doping model of neocytolysis. J. Investig. Med 2009, 57, 650–654. [Google Scholar]
- Nguyen, D.B.; Wagner-Britz, L.; Maia, S.; Steffen, P.; Wagner, C.; Kaestner, L.; Bernhardt, I. Regulation of phosphatidylserine exposure in red blood cells. Cell Physiol. Biochem 2011, 28, 847–856. [Google Scholar]
- Eaton, J.W.; Skelton, T.D.; Swofford, H.S.; Kolpin, C.E.; Jacob, H.S. Elevated erythrocyte calcium in sickle cell disease. Nature 1973, 246, 105–106. [Google Scholar]
- Etzion, Z.; Tiffert, T.; Bookchin, R.M.; Lew, V.L. Effects of deoxygenation on active and passive Ca2+ transport and on the cytoplasmic Ca2+ levels of sickle cell anemia red cells. J. Clin. Invest 1993, 92, 2489–2498. [Google Scholar]
- Joiner, C.H.; Jiang, M.; Franco, R.S. Deoxygenation-induced cation fluxes in sickle cells. IV. Modulation by external calcium. Am. J. Physiol 1995, 269, C403–C409. [Google Scholar]
- Wiley, J.S. Increased erythrocyte cation permeability in thalassemia and conditions of marrow stress. J. Clin. Invest 1981, 67, 917–922. [Google Scholar]
- Shalev, O.; Mogilner, S.; Shinar, E.; Rachmilewitz, E.A.; Schrier, S.L. Impaired erythrocyte calcium homeostasis in beta-thalassemia. Blood 1984, 64, 564–566. [Google Scholar]
- Bookchin, R.M.; Ortiz, O.E.; Shalev, O.; Tsurel, S.; Rachmilewitz, E.A.; Hockaday, A.; Lew, V.L. Calcium transport and ultrastructure of red cells in beta-thalassemia intermedia. Blood 1988, 72, 1602–1607. [Google Scholar]
- Sabina, R.L.; Waldenström, A.; Ronquist, G. The contribution of Ca+ calmodulin activation of human erythrocyte AMP deaminase (isoform E) to the erythrocyte metabolic dysregulation of familial phosphofructokinase deficiency. Haematologica 2006, 91, 652–655. [Google Scholar]
- Bookchin, R.M.; Ortiz, O.E.; Somlyo, A.V.; Somlyo, A.P.; Sepulveda, M.I.; Hockaday, A.; Lew, V.L. Calcium-accumulating inside-out vesicles in sickle cell anemia red cells. Trans. Assoc. Am. Phys 1985, 98, 10–20. [Google Scholar]
- Lew, V.L.; Hockaday, A.; Sepulveda, M.I.; Somlyo, A.P.; Somlyo, A.V.; Ortiz, O.E.; Bookchin, R.M. Compartmentalization of sickle-cell calcium in endocytic inside-out vesicles. Nature 1985, 315, 586–589. [Google Scholar]
- Eaton, W.A.; Hofrichter, J. Sickle cell hemoglobin polymerization. Adv. Protein Chem 1990, 40, 263–279. [Google Scholar]
- Vandorpe, D.H.; Xu, C.; Shmukler, B.E.; Otterbein, L.E.; Trudel, M.; Sachs, F.; Gottlieb, P.A.; Brugnara, C.; Alper, S.L. Hypoxia activates a Ca2+-permeable cation conductance sensitive to carbon monoxide and to GsMTx-4 in human and mouse sickle erythrocytes. PLoS One 2010, 5, e8732. [Google Scholar]
- Ortiz, O.E.; Lew, V.L.; Bookchin, R.M. Calcium accumulated by sickle cell anemia red cells does not affect their potassium (86Rb+) flux components. Blood 1986, 67, 710–715. [Google Scholar]
- Rhoda, M.D.; Apovo, M.; Beuzard, Y.; Giraud, F. Ca2+ permeability in deoxygenated sickle cells. Blood 1990, 75, 2453–2458. [Google Scholar]
- Clark, M.R.; Rossi, M.E. Permeability characteristics of deoxygenated sickle cells. Blood 1990, 76, 2139–2145. [Google Scholar]
- De Franceschi, L.; Franco, R.S.; Bertoldi, M.; Brugnara, C.; Matte, A.; Siciliano, A.; Wieschhaus, A.J.; Chishti, A.H.; Joiner, C.H. Pharmacological inhibition of calpain-1 prevents red cell dehydration and reduces Gardos channel activity in a mouse model of sickle cell disease. FASEB J 2013, 27, 750–759. [Google Scholar]
- Siciliano, A.; Turrini, F.; Bertoldi, M.; Matte, A.; Pantaleo, A.; Olivieri, O.; de Franceschi, L. Deoxygenation affects tyrosine phosphoproteome of red cell membrane from patients with sickle cell disease. Blood Cells Mol. Dis 2010, 44, 233–242. [Google Scholar]
- Rank, B.H.; Hebbel, R.P.; Carlsson, J. Oxidation of membrane thiols in sickle erythrocytes. Prog. Clin. Biol. Res 1984, 165, 473–477. [Google Scholar]
- Wood, K.C.; Hebbel, R.P.; Lefer, D.J.; Granger, D.N. Critical role of endothelial cell-derived nitric oxide synthase in sickle cell disease-induced microvascular dysfunction. Free Radic. Biol. Med 2006, 40, 1443–1453. [Google Scholar]
- Hebbel, R.P. Perspectives series: Cell adhesion in vascular biology. Adhesive interactions of sickle erythrocytes with endothelium. J. Clin. Invest 1997, 99, 2561–2564. [Google Scholar]
- Antonelou, M.H.; Tzounakas, V.L.; Velentzas, A.D.; Stamoulis, K.E.; Kriebardis, A.G.; Papassideri, I.S. Effects of pre-storage leukoreduction on stored red blood cells signaling: A time-course evaluation from shape to proteome. J. Proteomics 2012, 76, 220–238. [Google Scholar]
- Schrier, S.L.; Sohmer, P.R.; Moore, G.L.; Ma, L.; Junga, I. Red blood cell membrane abnormalities during storage: Correlation with in vivo survival. Transfusion 1982, 22, 261–265. [Google Scholar]
- Wolfe, L. The red cell membrane and the storage lesion. Clin. Haematol 1985, 14, 259–276. [Google Scholar]
- Chin-Yee, I.H.; Gray-Statchuk, L.; Milkovich, S.; Ellis, C.G. Transfusion of stored red blood cells adhere in the rat microvasculature. Transfusion 2009, 49, 2304–2310. [Google Scholar]
- Chaudhary, R.; Katharia, R. Oxidative injury as contributory factor for red cells storage lesion during twenty eight days of storage. Blood Transfus 2012, 10, 59–62. [Google Scholar]
- Kaestner, L.; Juzeniene, A.; Moan, J. Erythrocytes-the “house elves” of photodynamic therapy. Photochem. Photobiol. Sci 2004, 3, 981–989. [Google Scholar]
- Kaestner, L. Evaluation of human erythrocytes as model cells in photodynamic therapy. Gen. Physiol. Biophys 2003, 22, 455–465. [Google Scholar]
- Muller, O.; Tian, Q.; Zantl, R.; Kahl, V.; Lipp, P.; Kaestner, L. A system for optical high resolution screening of electrical excitable cells. Cell Calcium 2010, 47, 224–233. [Google Scholar]
- Kaestner, L. Calcium Signalling. Approaches and Findings in the Heart and Blood; Springer: Heidelberg, Germany, 2013. [Google Scholar]
- Minetti, G.; Egée, S.; Mörsdorf, D.; Steffen, P.; Makhro, A.; Achilli, C.; Ciana, A.; Wang, J.; Bouyer, G.; Bernhardt, I.; et al. Red cell investigations: Art and artefacts. Blood Rev 2013, 27, 91–101. [Google Scholar] [Green Version]
- Wang, J.; Wagner-Britz, L.; Bogdanova, A.; Ruppenthal, S.; Wiesen, K.; Kaiser, E.; Tian, Q.; Krause, E.; Bernhardt, I.; Lipp, P.; et al. Morphologically homogeneous red blood cells present a heterogeneous response to hormonal stimulation. PLoS One 2013. submitted for publication. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Bogdanova, A.; Makhro, A.; Wang, J.; Lipp, P.; Kaestner, L. Calcium in Red Blood Cells—A Perilous Balance. Int. J. Mol. Sci. 2013, 14, 9848-9872. https://doi.org/10.3390/ijms14059848
Bogdanova A, Makhro A, Wang J, Lipp P, Kaestner L. Calcium in Red Blood Cells—A Perilous Balance. International Journal of Molecular Sciences. 2013; 14(5):9848-9872. https://doi.org/10.3390/ijms14059848
Chicago/Turabian StyleBogdanova, Anna, Asya Makhro, Jue Wang, Peter Lipp, and Lars Kaestner. 2013. "Calcium in Red Blood Cells—A Perilous Balance" International Journal of Molecular Sciences 14, no. 5: 9848-9872. https://doi.org/10.3390/ijms14059848
APA StyleBogdanova, A., Makhro, A., Wang, J., Lipp, P., & Kaestner, L. (2013). Calcium in Red Blood Cells—A Perilous Balance. International Journal of Molecular Sciences, 14(5), 9848-9872. https://doi.org/10.3390/ijms14059848