Vascular Endothelial Growth Factor Induces CXCL1 Chemokine Release via JNK and PI-3K-Dependent Pathways in Human Lung Carcinoma Epithelial Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
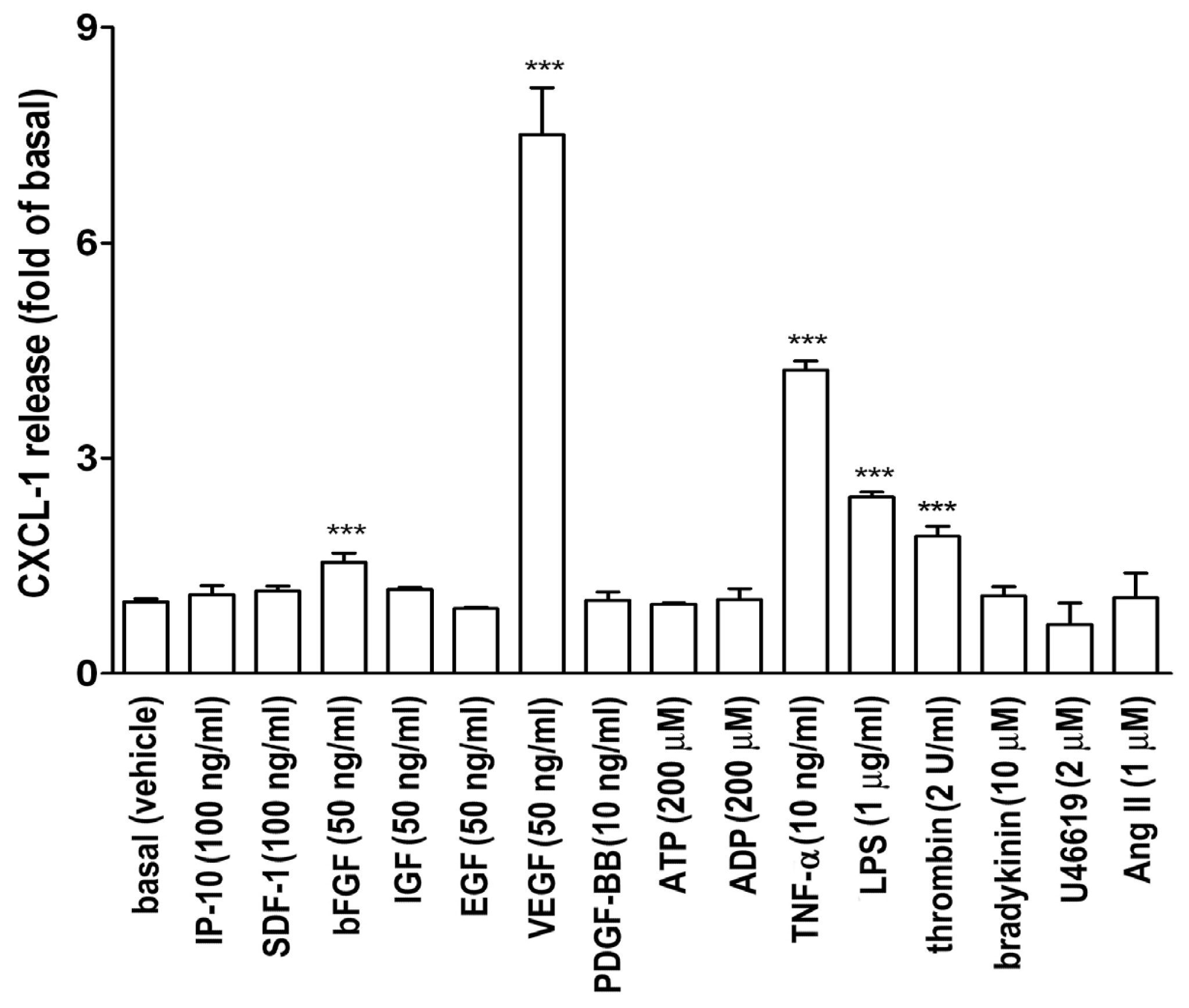
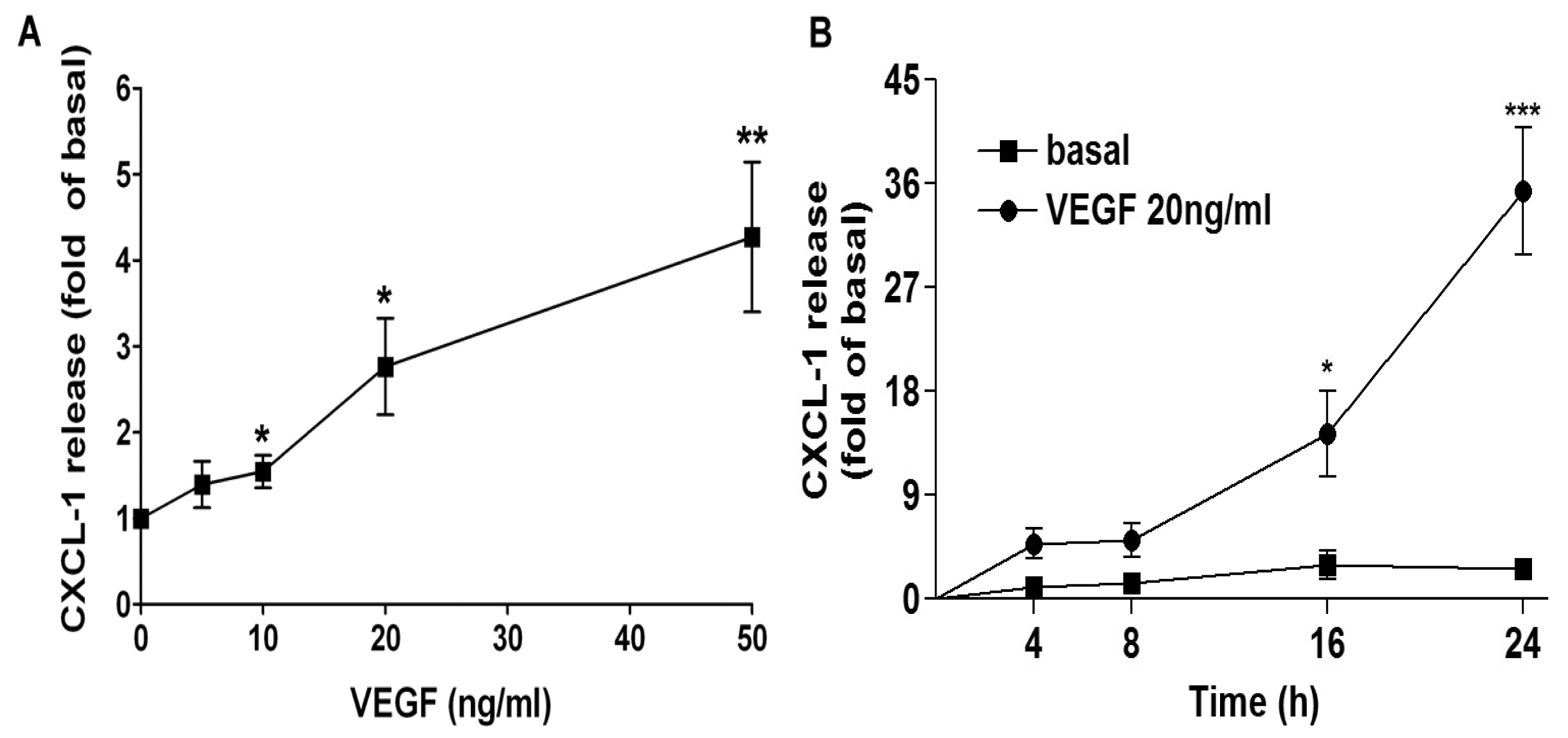
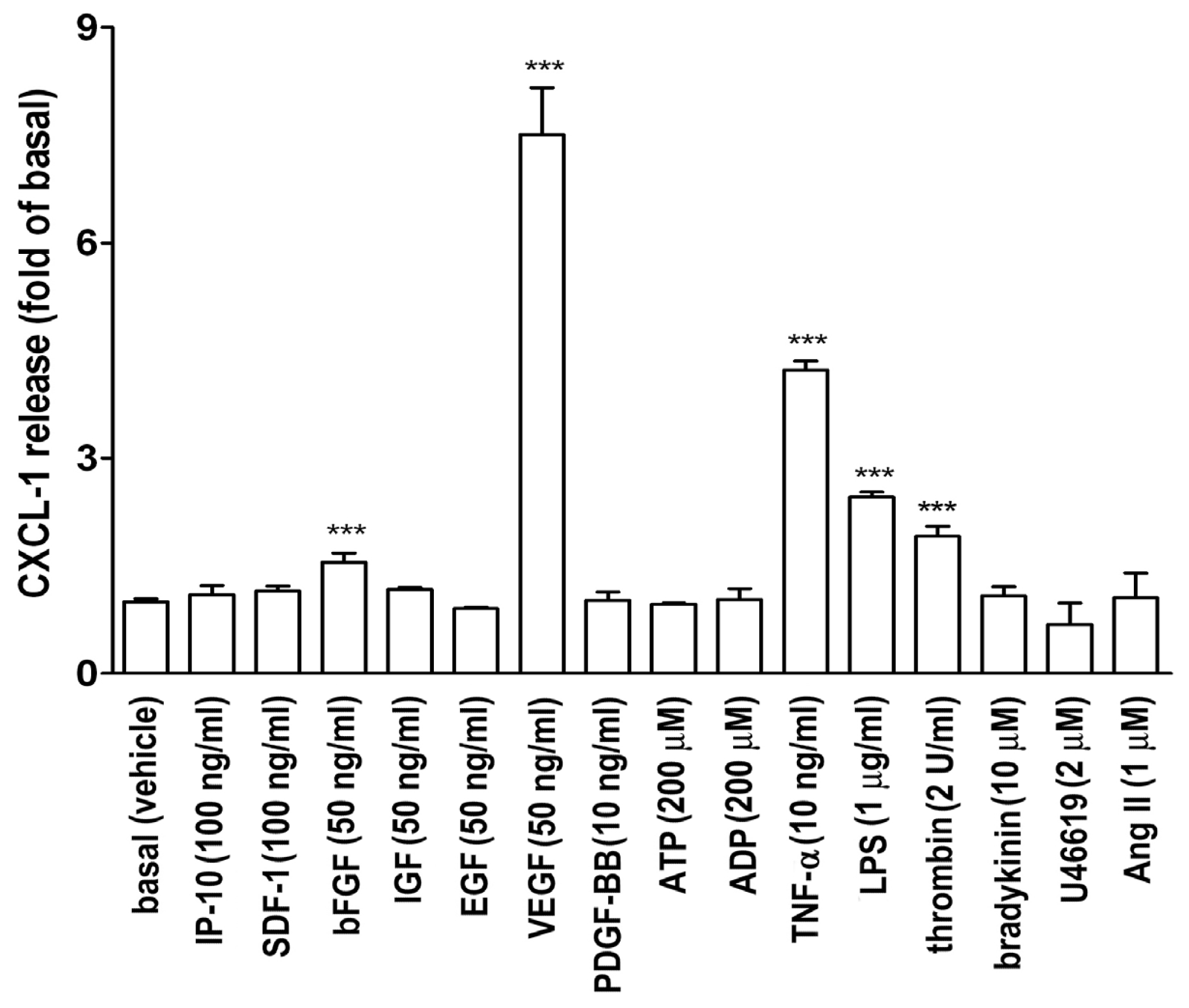
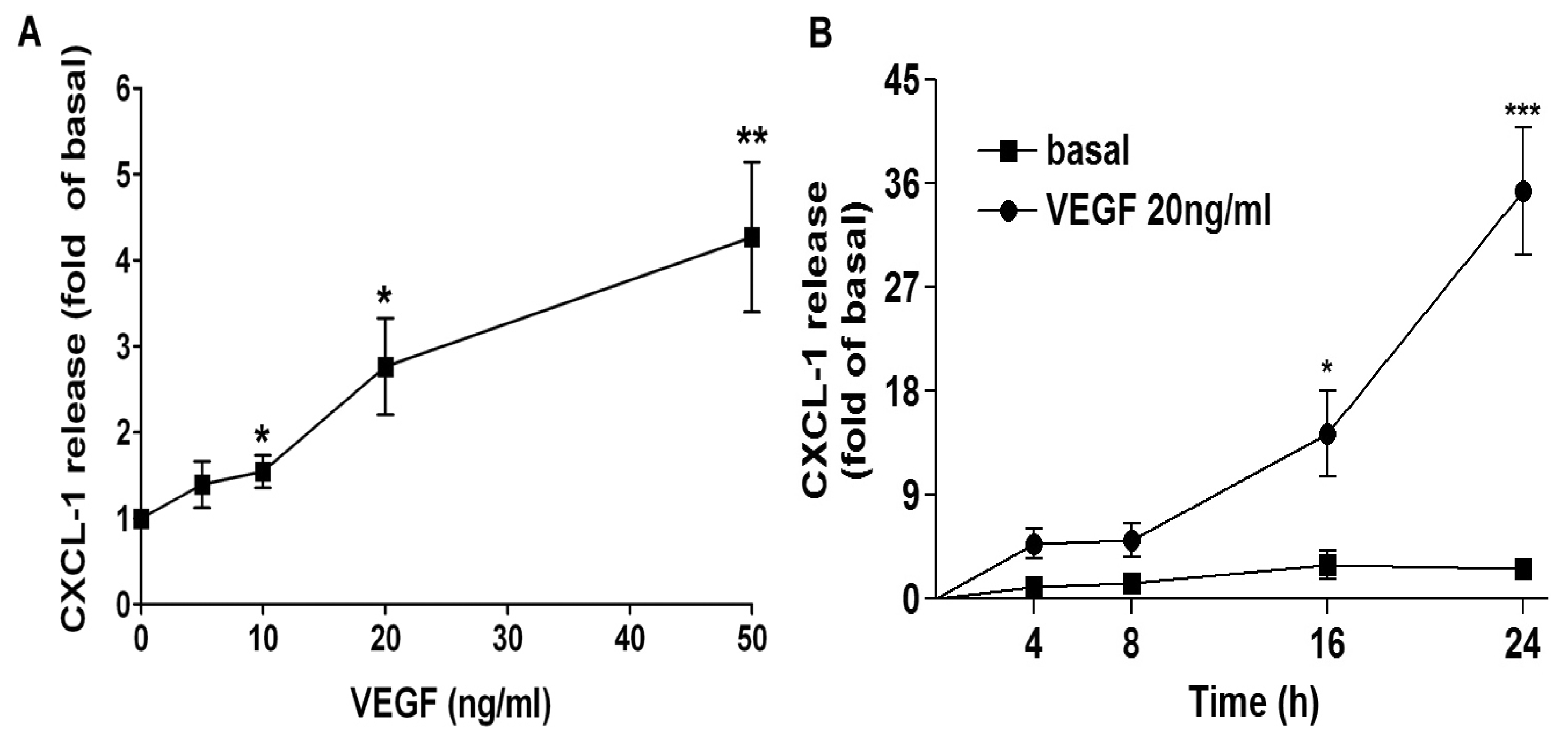
2.1. VEGF Markedly Induces CXCL1 Release in A549 Lung Epithelial Cells
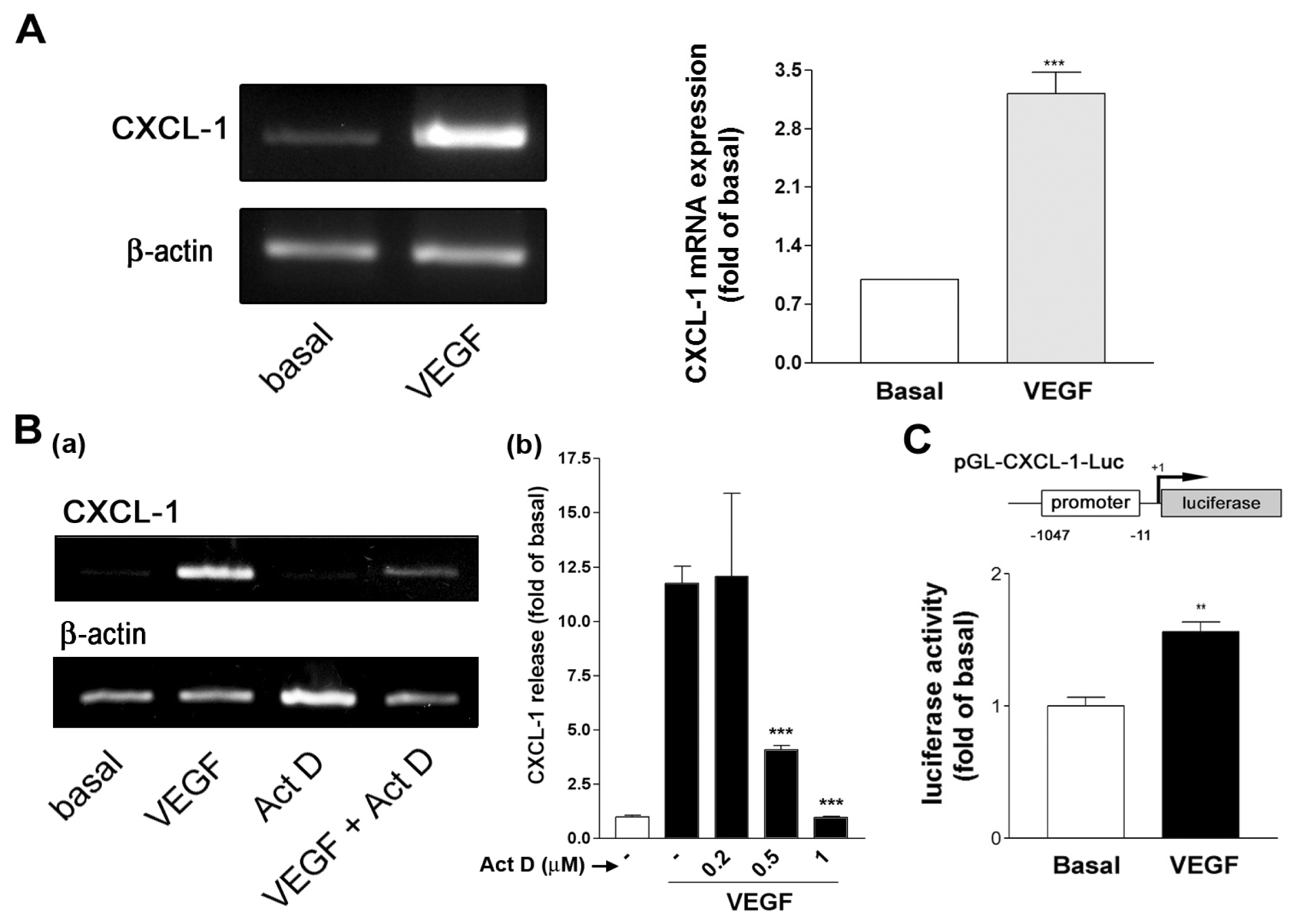
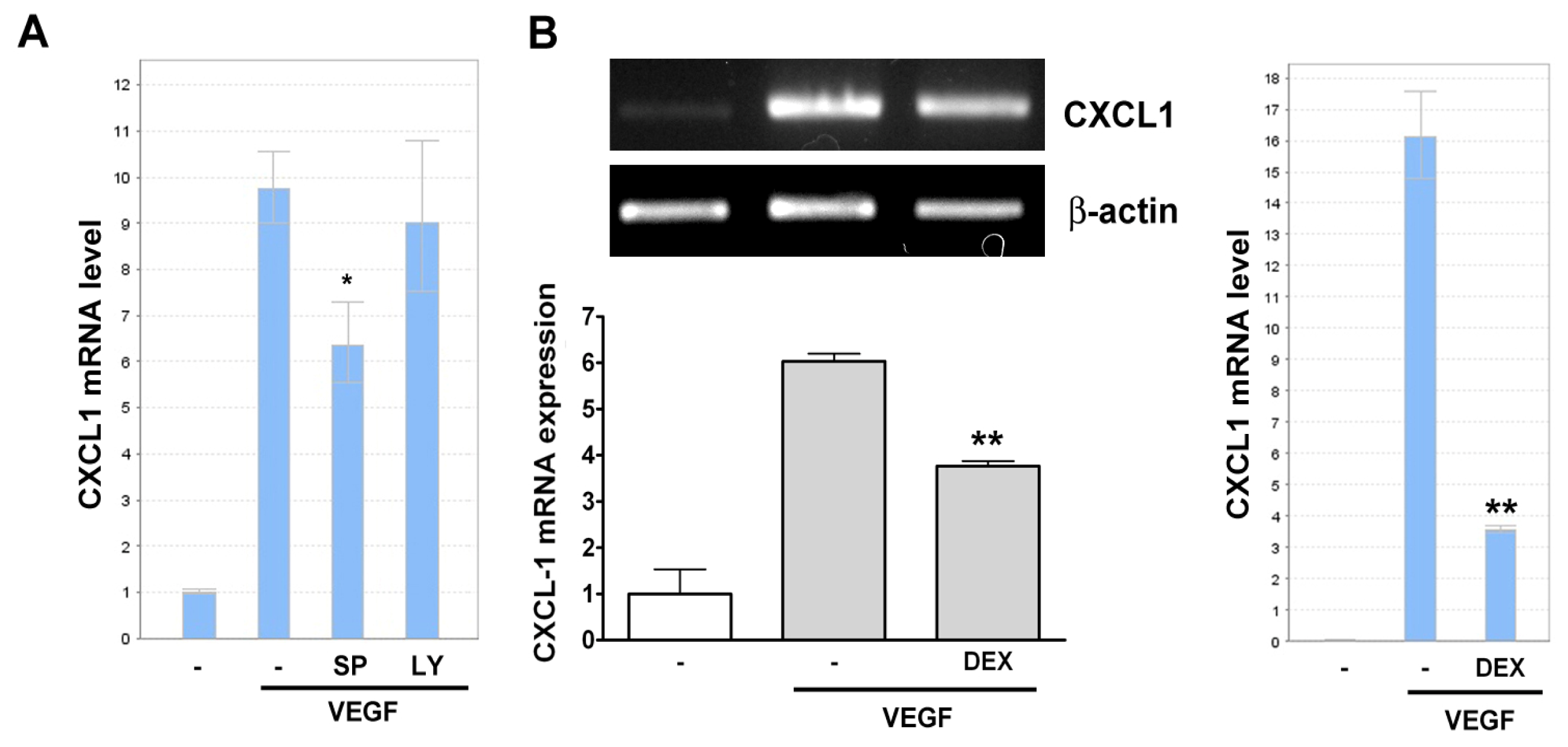
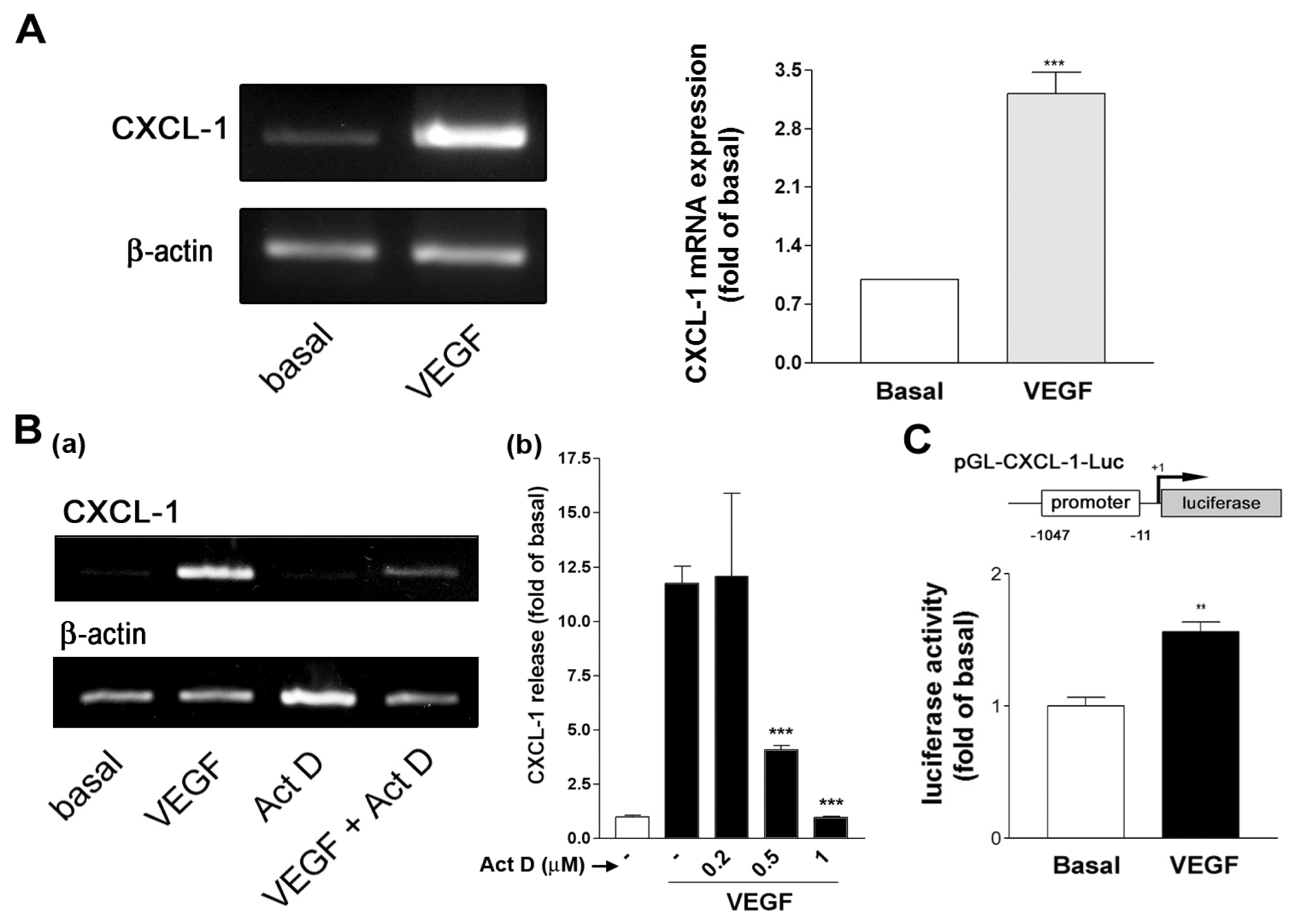
2.2. VEGF Transcriptionally Regulates CXCL1 Expression in A549 Lung Epithelial Cells
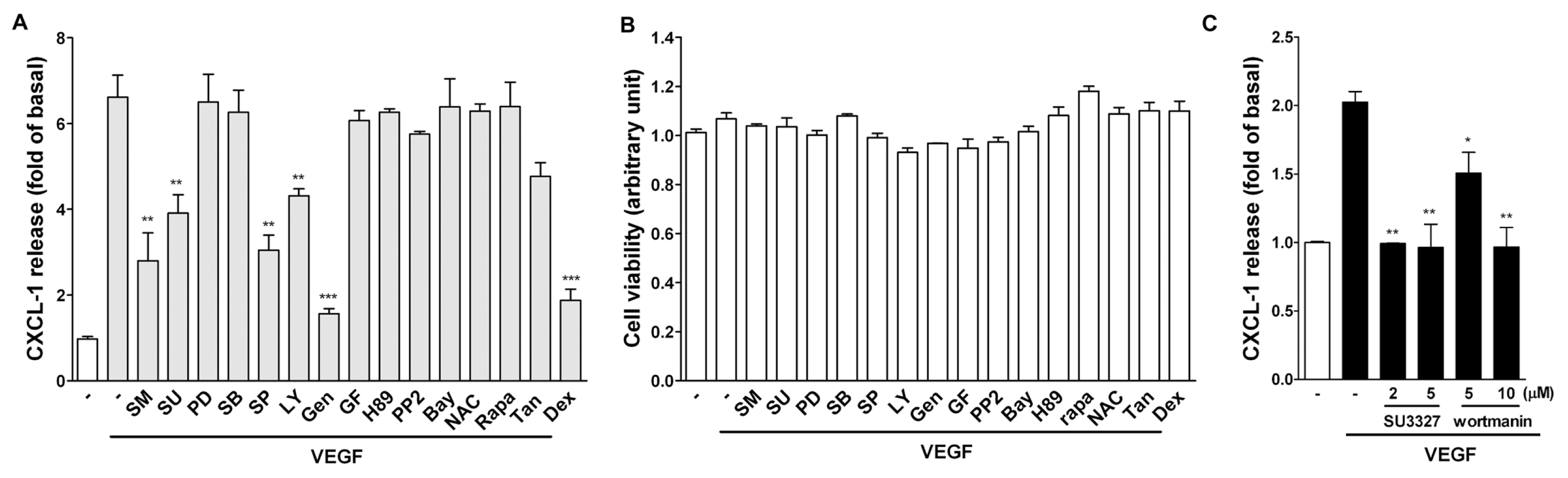
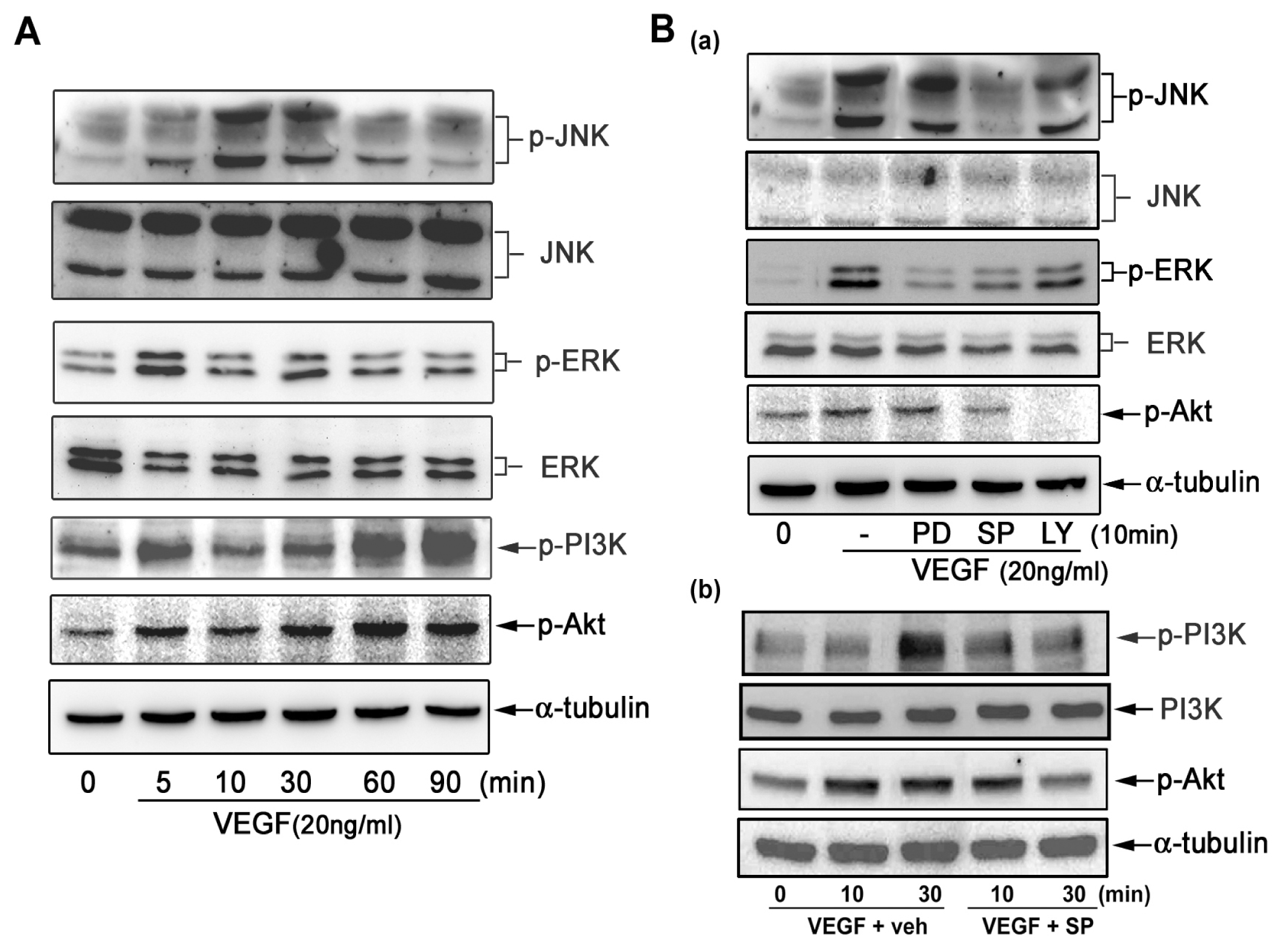
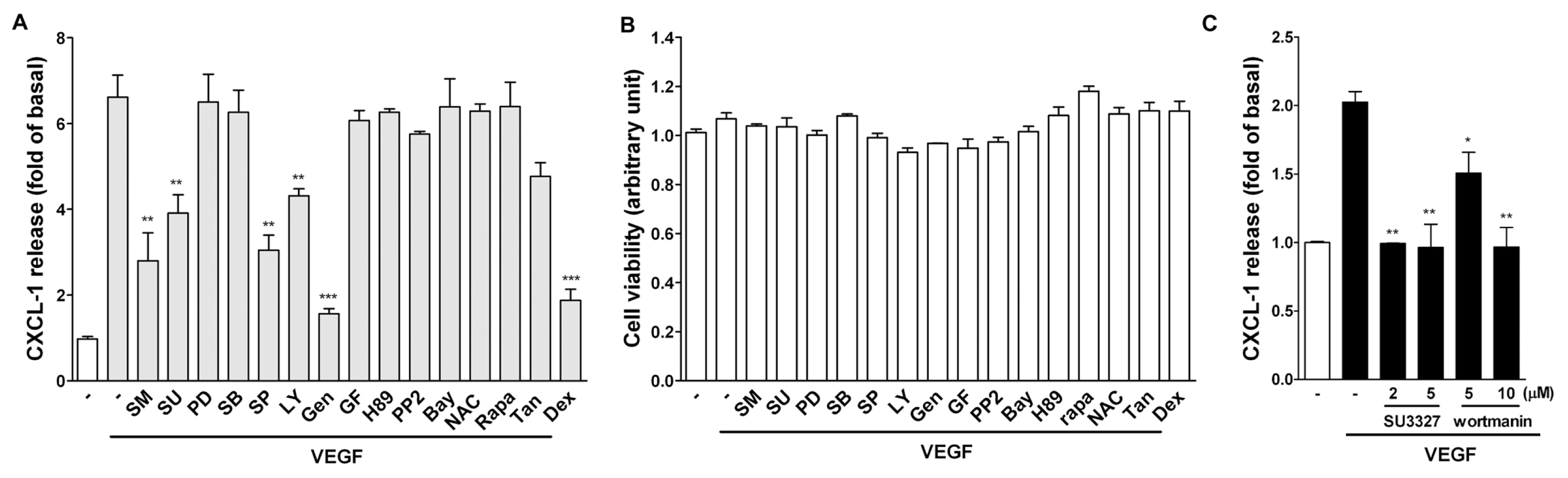
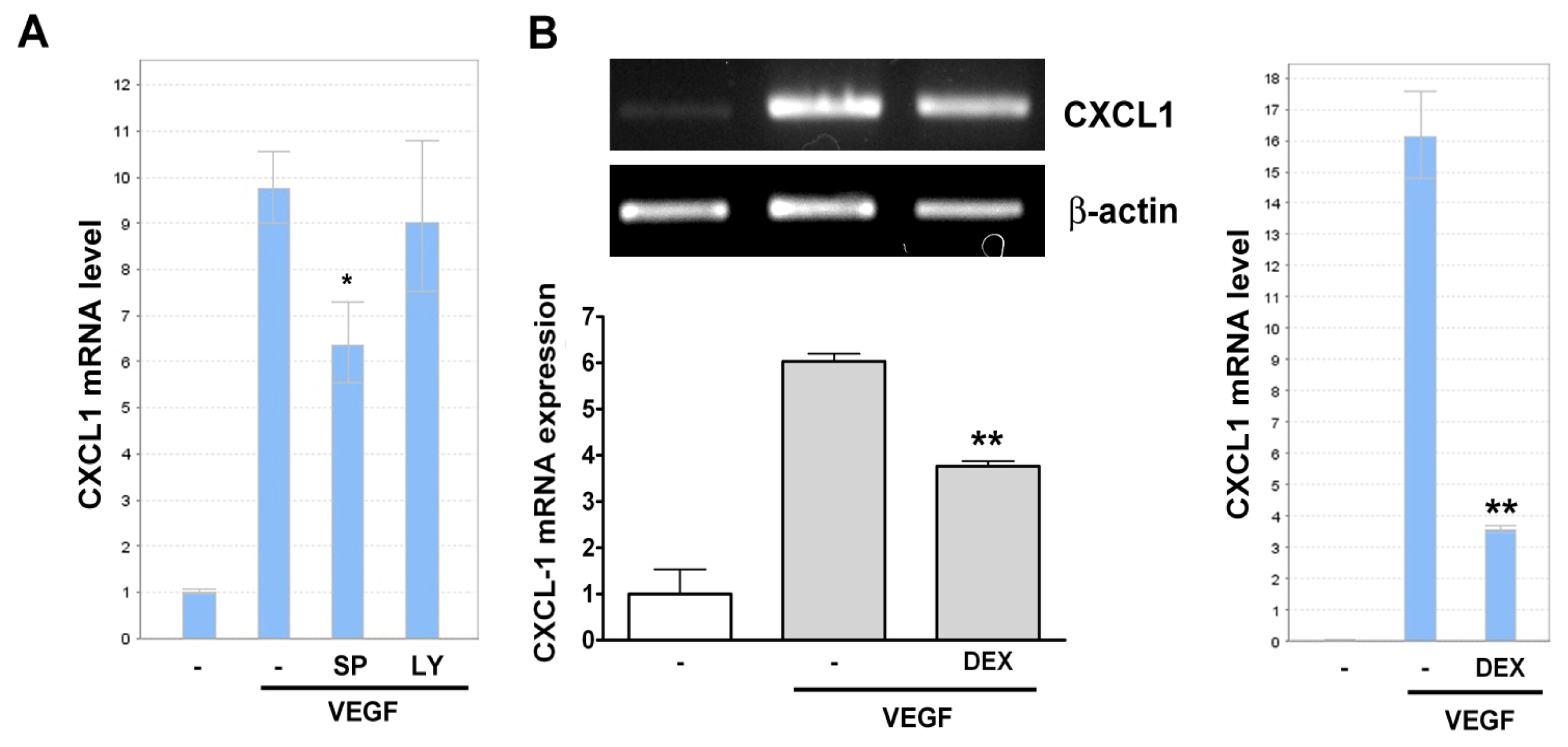
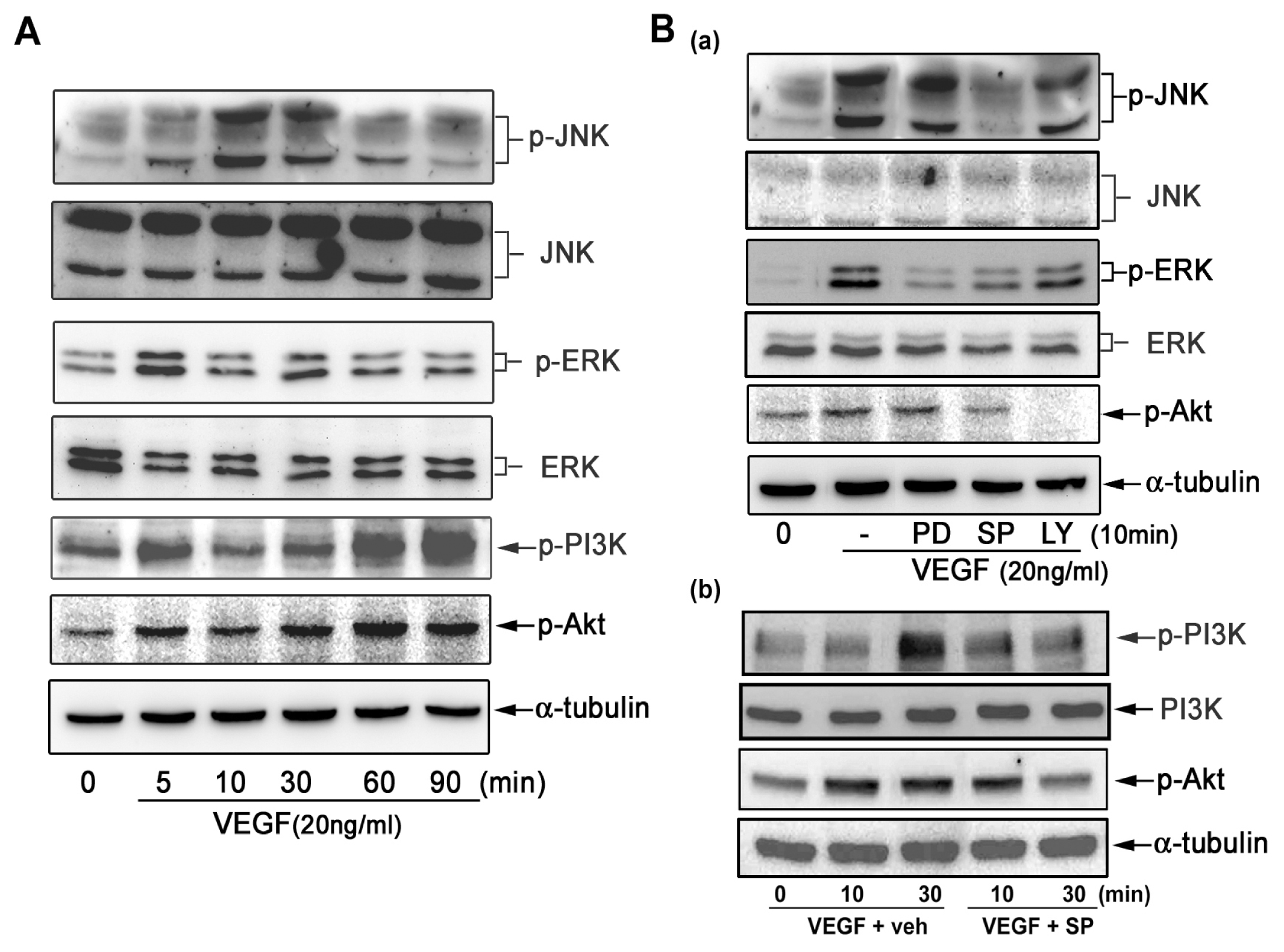
2.3. Effect of Signaling Inhibitors on VEGF-Induced CXCL1 Expression in A549 Cells
2.4. VEGF Directly Induces JNK and PI-3K Activation
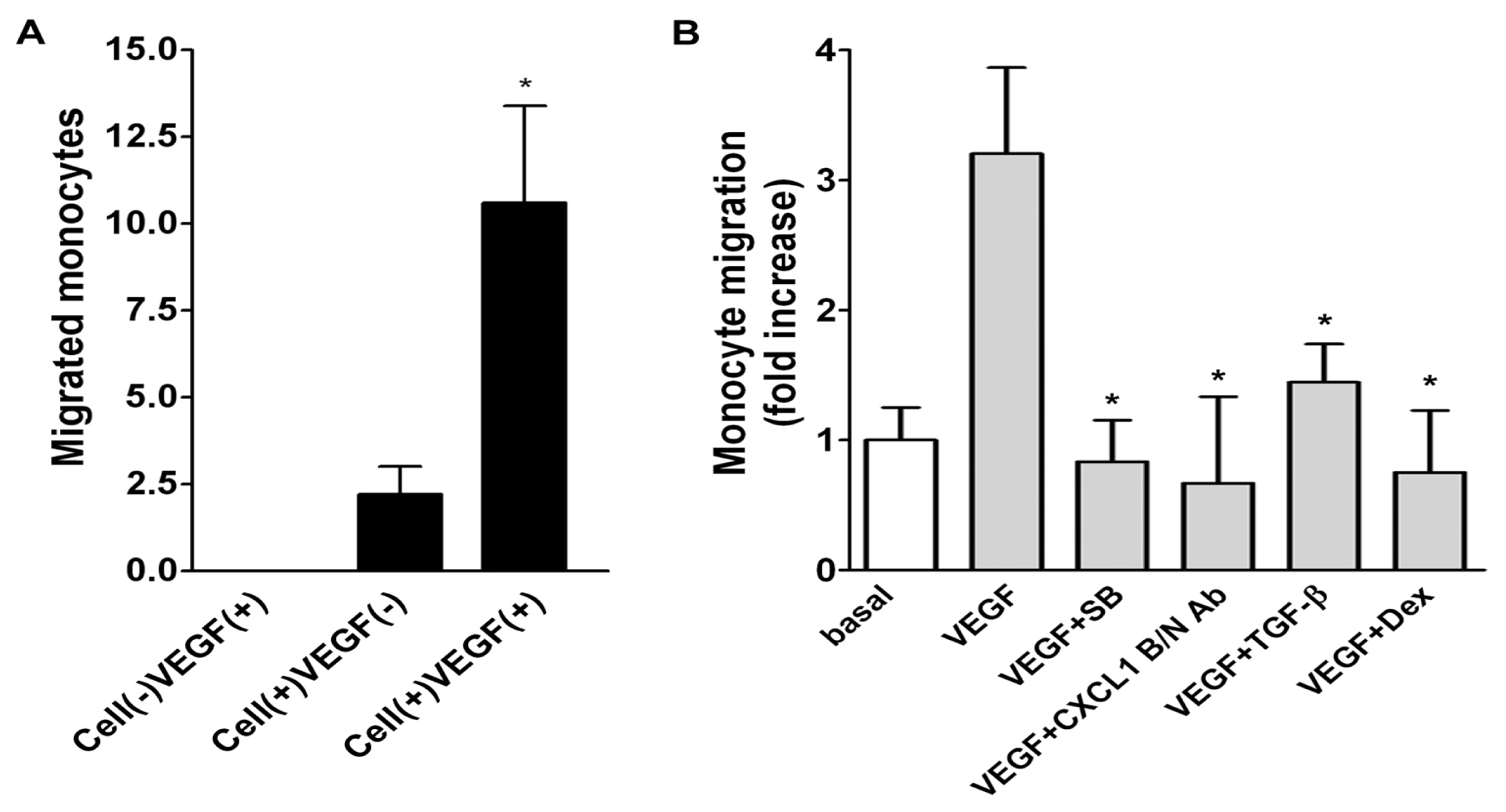
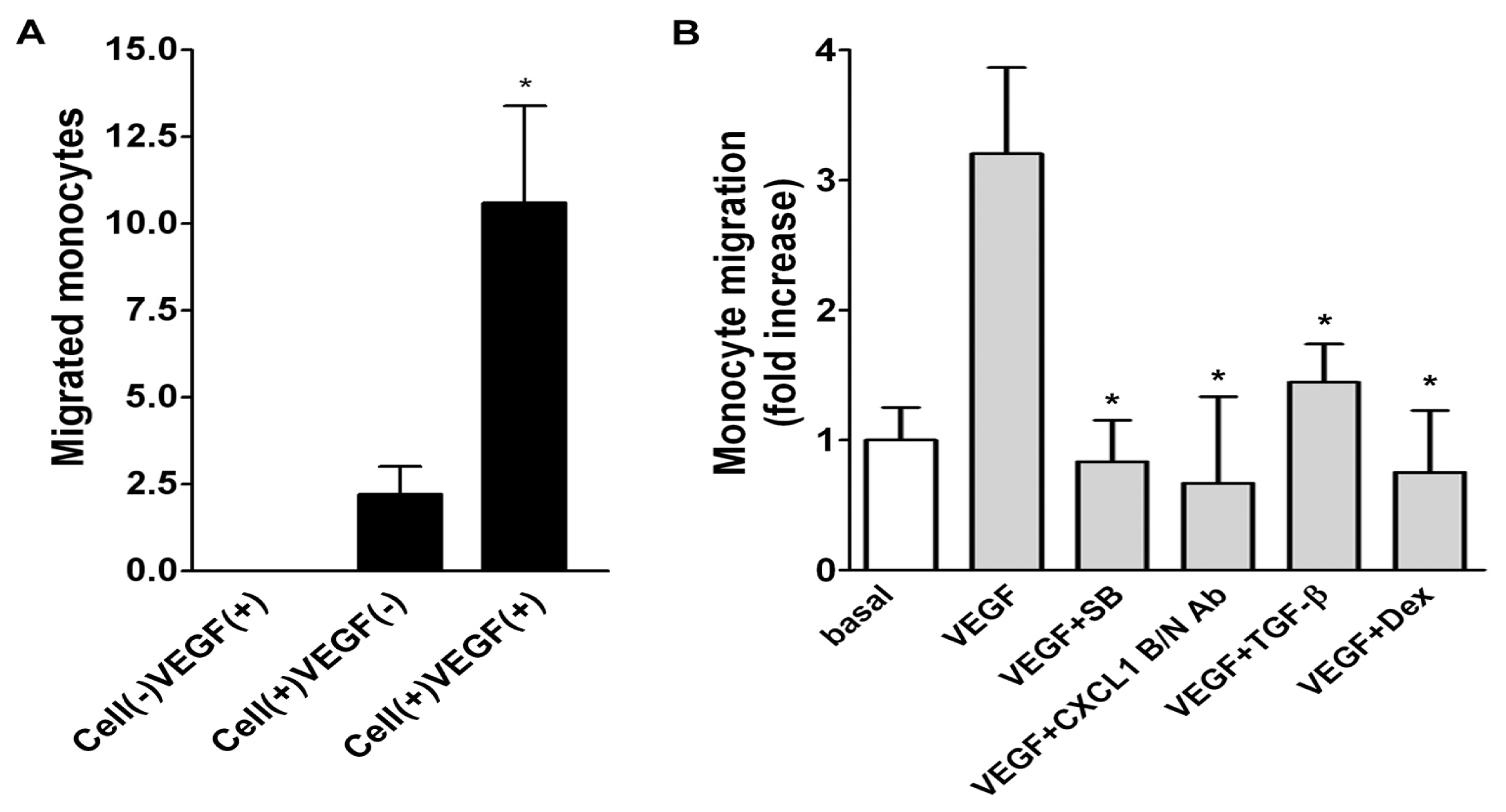
2.5. Role of Released CXCL1 by A549 Cells in Attracting Monocyte Migration
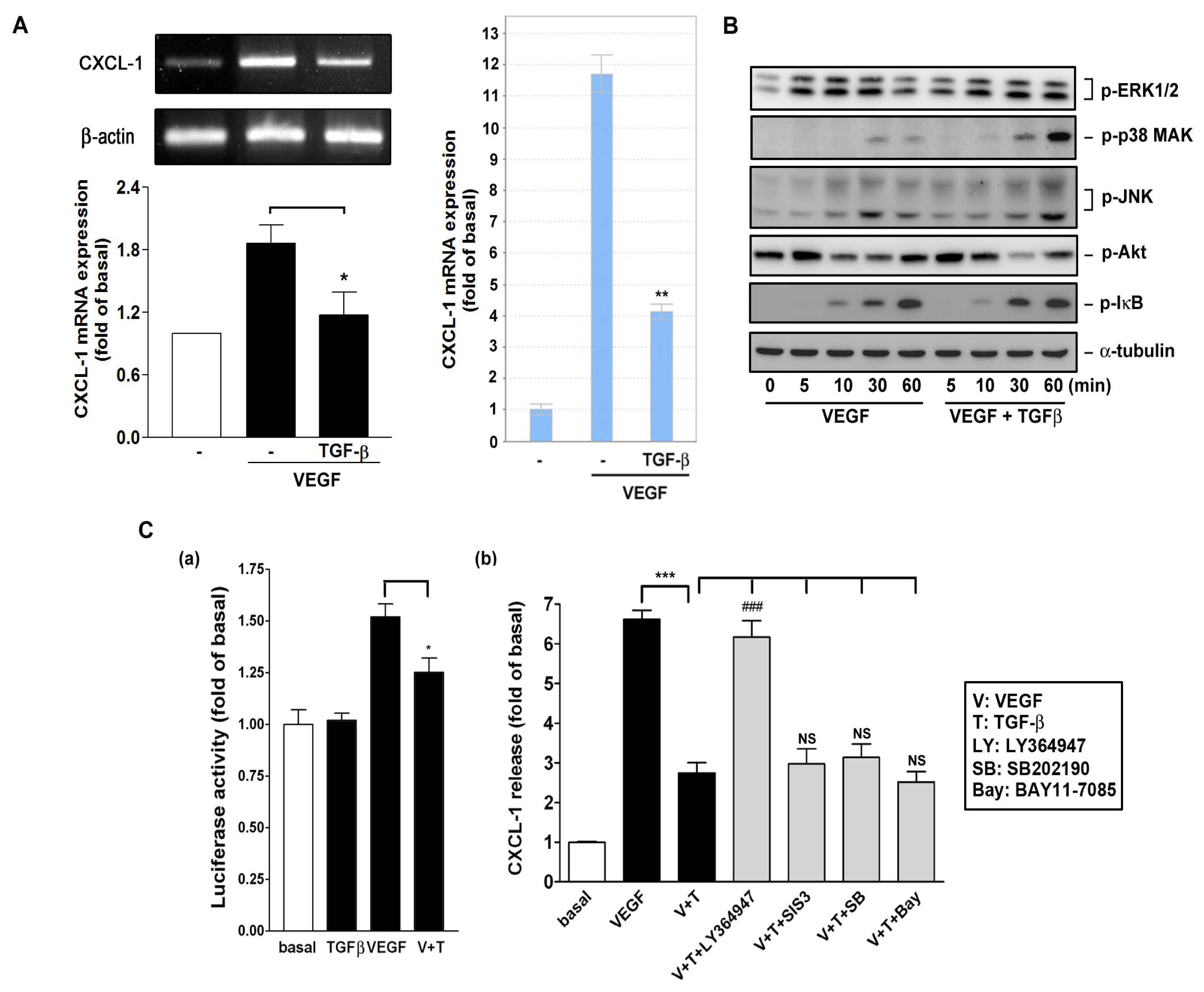
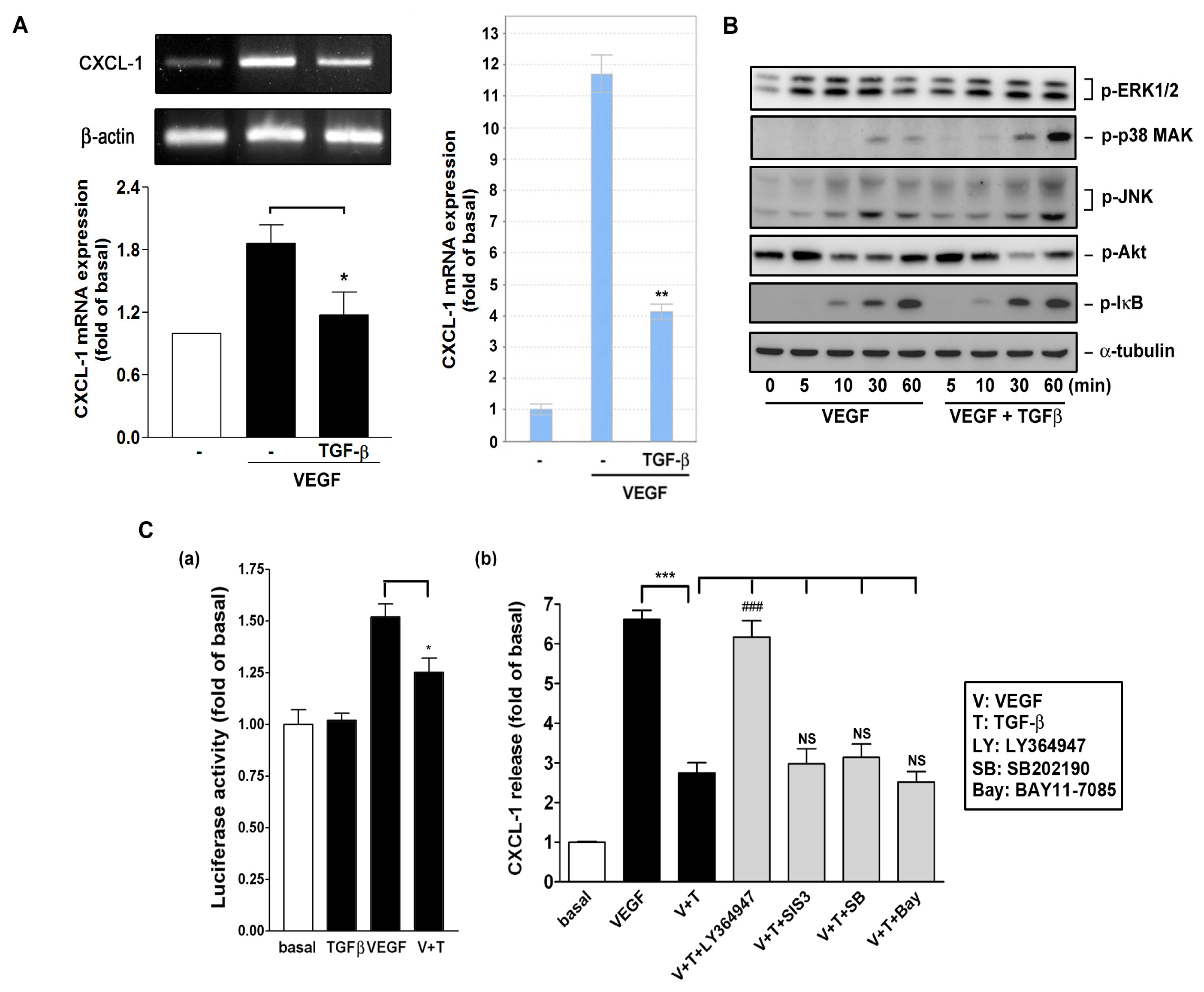
2.6. Effect of TGF-β on VEGF-Induced CXCL1 Expression
3. Discussion
4. Experimental Section
4.1. Materials
4.2. Cell Culture
4.3. Measurement of Secreted CXCL1 in Culture Medium by ELISA
4.4. Cell Viability Assay
4.5. Cell Lysate Preparation and Western Blot Analysis
4.6. Reverse Transcription-Polymerase Chain Reaction (RT-PCR) and Real-Time PCR Analysis of CXCL1 mRNA Expression
4.7. CXCL1 Reporter Construct, Transfection, and Luciferase Assay
4.8. Measurement of A549 Cells-Induced Monocyte Migration
4.9. Statistical Analysis
5. Conclusions
Acknowledgments
Conflict of Interest
References
- Charo, I.F.; Ransohoff, R.M. The many roles of chemokines and chemokine receptors in inflammation. N. Engl. J. Med 2006, 354, 610–621. [Google Scholar]
- Baggiolini, M. Chemokines in pathology and medicine. J. Intern. Med 2001, 250, 91–104. [Google Scholar]
- Nannuru, K.C.; Singh, S.; Singh, R.K. Chemokines and Metastasis. In The Tumor Microenvironment; Bagley, R.G., Ed.; Springer: New York, NY, USA, 2010; pp. 601–631. [Google Scholar]
- Scapini, P.; Morini, M.; Tecchio, C.; Minghelli, S.; di Carlo, E.; Tanghetti, E.; Albini, A.; Lowell, C.; Berton, G.; Noonan, D.M.; Cassatella, M.A. CXCL1/macrophage inflammatory protein-2-induced angiogenesis in vivo is mediated by neutrophil-derived vascular endothelial growth factor-A. J. Immunol 2004, 172, 5034–5040. [Google Scholar]
- Holmes, K.; Roberts, O.L.; Thomas, A.M.; Cross, M.J. Vascular endothelial growth factor receptor-2: Structure, function, intracellular signalling and therapeutic inhibition. Cell. Signal 2007, 19, 2003–2012. [Google Scholar]
- Geretti, E.; Shimizu, A.; Klagsbrun, M. Neuropilin structure governs VEGF and semaphorin binding and regulates angiogenesis. Angiogenesis 2008, 11, 31–39. [Google Scholar]
- Hutchings, H.; Ortega, N.; Plouët, J. Extracellular matrix-bound vascular endothelial growth factor promotes endothelial cell adhesion, migration, and survival through integrin ligation. FASEB J 2003, 17, 1520–1522. [Google Scholar]
- Liu, W.; Xu, J.; Wang, M.; Wang, Q.; Bi, Y.; Han, M. Tumor-derived vascular endothelial growth factor (VEGF)-a facilitates tumor metastasis through the VEGF-VEGFR1 signaling pathway. Int. J. Oncol 2011, 39, 1213–1220. [Google Scholar]
- Zhan, P.; Wang, J.; Lv, X.-J.; Wang, Q.; Qiu, L.-X.; Lin, X.-Q.; Yu, L.-K.; Song, Y. Prognostic value of vascular endothelial growth factor expression in patients with lung cancer: A systematic review with meta-analysis. J. Thorac. Oncol 2009, 4, 1094–1103. [Google Scholar]
- Gorski, D.H.; Beckett, M.A.; Jaskowiak, N.T.; Calvin, D.P.; Mauceri, H.J.; Salloum, R.M.; Seetharam, S.; Koons, A.; Hari, D.M.; Kufe, D.W.; et al. Blockage of the vascular endothelial growth factor stress response increases the antitumor effects of ionizing radiation. Cancer Res 1999, 59, 3374–3378. [Google Scholar]
- Becker, S.; Quay, J.; Koren, H.S.; Haskill, J.S. Constitutive and stimulated MCP-1, GRO alpha, beta, and gamma expression in human airway epithelium and bronchoalveolar macrophages. Am. J. Physiol 1994, 266, L278–L286. [Google Scholar]
- Huang, M.; Wang, J.; Lee, P.; Sharma, S.; Mao, J.T.; Meissner, H.; Uyemura, K.; Modlin, R.; Wollman, J.; Dubinett, S.M. Human non-small cell lung cancer cells express a type 2 cytokine pattern. Cancer Res 1995, 55, 3847–3853. [Google Scholar]
- Arenberg, D.A.; Keane, M.P.; DiGiovine, B.; Kunkel, S.L.; Morris, S.B.; Xue, Y.Y.; Burdick, M.D.; Glass, M.C.; Iannettoni, M.D.; Strieter, R.M. Epithelial-neutrophil activating peptide (ENA-78) is an important angiogenic factor in non-small cell lung cancer. J. Clin. Investig 1998, 102, 465–472. [Google Scholar]
- Numasaki, M.; Watanabe, M.; Suzuki, T.; Takahashi, H.; Nakamura, A.; McAllister, F.; Hishinuma, T.; Goto, J.; Lotze, M.T.; Kolls, J.K.; et al. IL-17 enhances the net angiogenic activity and in vivo growth of human non-small cell lung cancer in SCID mice through promoting CXCR-2-dependent angiogenesis. J. Immunol 2005, 175, 6177–6189. [Google Scholar]
- Caunt, M.; Hu, L.; Tang, T.; Brooks, P.C.; Ibrahim, S.; Karpatkin, S. Growth-regulated oncogene is pivotal in thrombin-induced angiogenesis. Cancer Res 2006, 66, 4125–4132. [Google Scholar]
- Alexander, S.P.; Mathie, A.; Peters, J.A. Guide to receptors and channels (GRAC). Br. J. Pharmacol 2011, 164, S1–S2. [Google Scholar]
- Blobe, G.C.; Schiemann, W.P.; Lodish, H.F. Role of transforming growth factor β in human disease. N. Engl. J. Med 2000, 342, 1350–1358. [Google Scholar]
- De Caestecker, M.P.; Piek, E.; Roberts, A.B. Role of transforming growth factor-β signaling in cancer. J. Natl. Cancer Inst 2000, 92, 1388–1402. [Google Scholar]
- Jeon, H.-S.; Jen, J. TGF-[beta] Signaling and the role of inhibitory smads in non-small cell lung cancer. J. Thorac. Oncol 2010, 5, 417–419. [Google Scholar]
- Shull, M.M.; Ormsby, I.; Kier, A.B.; Pawlowski, S.; Diebold, R.J.; Yin, M.; Allen, R.; Sidman, C.; Proetzel, G.; Calvin, D.; et al. Targeted disruption of the mouse transforming growth factor-beta 1 gene results in multifocal inflammatory disease. Nature 1992, 359, 693–699. [Google Scholar]
- Markowitz, S.D.; Roberts, A.B. Tumor suppressor activity of the TGF-beta pathway in human cancers. Cytokine Growth Factor Rev 1996, 7, 93–102. [Google Scholar]
- Roberts, A.B.; Wakefield, L.M. The two faces of transforming growth factor beta in carcinogenesis. Proc. Natl. Acad. Sci. USA 2003, 100, 8621–8623. [Google Scholar]
- Kelly, R.J.; Morris, J.C. Transforming growth factor-beta: A target for cancer therapy. J. Immunotoxicol 2010, 7, 15–26. [Google Scholar]
- Wislez, M.; Fujimoto, N.; Izzo, J.G.; Hanna, A.E.; Cody, D.D.; Langley, R.R.; Tang, H.; Burdick, M.D.; Sato, M.; Minna, J.D.; et al. High expression of ligands for chemokine receptor CXCR2 in alveolar epithelial neoplasia induced by oncogenic kras. Cancer Res 2006, 66, 4198–4207. [Google Scholar]
- Zhong, L.; Roybal, J.; Chaerkady, R.; Zhang, W.; Choi, K.; Alvarez, C.A.; Tran, H.; Creighton, C.J.; Yan, S.; Strieter, R.M.; et al. Identification of secreted proteins that mediate cell-cell interactions in an in vitro model of the lung cancer microenvironment. Cancer Res 2008, 68, 7237–7245. [Google Scholar]
- Anisowicz, A.; Messineo, M.; Lee, S.W.; Sager, R. An NF-kappa B-like transcription factor mediates IL-1/TNF-alpha induction of gro in human fibroblasts. J. Immunol 1991, 147, 520–527. [Google Scholar]
- Hao, Q.; Wang, L.; Tang, H. Vascular endothelial growth factor induces protein kinase D-dependent production of proinflammatory cytokines in endothelial cells. Am. J. Physiol 2009, 296, C821–C827. [Google Scholar]
- Koch, S.; Tugues, S.; Li, X.; Gualandi, L.; Claesson-Welsh, L. Signal transduction by vascular endothelial growth factor receptors. Biochem. J 2011, 437, 169–183. [Google Scholar]
- Mao, W.-F.; Shao, M.-H.; Gao, P.-T.; Ma, J.; Li, H.-J.; Li, G.-L.; Han, B.-H.; Yuan, C.-G. The important roles of RET, VEGFR2 and the RAF/MEK/ERK pathway in cancer treatment with sorafenib. Acta Pharmacol. Sin 2012, 33, 1311–1318. [Google Scholar]
- Leppa, S.; Bohmann, D. Diverse functions of JNK signaling and c-Jun in stress response and apoptosis. Oncogene 1999, 18, 6158–6162. [Google Scholar]
- Kyriakis, J.M.; Avruch, J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol. Rev 2001, 81, 807–869. [Google Scholar]
- Kuhns, D.B.; Gallin, J.I. Increased cell-associated IL-8 in human exudative and A23187-treated peripheral blood neutrophils. J. Immunol 1995, 154, 6556–6562. [Google Scholar]
- Pellmé, S.; Mörgelin, M.; Tapper, H.; Mellqvist, U.-H.; Dahlgren, C.; Karlsson, A. Localization of human neutrophil interleukin-8 (CXCL-8) to organelle(s) distinct from the classical granules and secretory vesicles. J. Leukoc. Biol 2006, 79, 564–573. [Google Scholar]
- Issa, R.; Xie, S.; Khorasani, N.; Sukkar, M.; Adcock, I.M.; Lee, K.-Y.; Chung, K.F. Corticosteroid inhibition of growth-related oncogene protein-α via mitogen-activated kinase phosphatase-1 in airway smooth muscle cells. J. Immunol 2007, 178, 7366–7375. [Google Scholar]
- Chen, C.C.; Manning, A.M. TGF-β1, IL-10 and IL-4 differentially modulate the cytokine-induced expression of IL-6 and IL-8 in human endothelial cells. Cytokine 1996, 8, 58–65. [Google Scholar]
- Bierie, B.; Stover, D.G.; Abel, T.W.; Chytil, A.; Gorska, A.E.; Aakre, M.; Forrester, E.; Yang, L.; Wagner, K.-U.; Moses, H.L. Transforming growth factor-β regulates mammary carcinoma cell survival and interaction with the adjacent microenvironment. Cancer Res 2008, 68, 1809–1819. [Google Scholar]
- Moustakas, A.; Heldin, C.-H. The regulation of TGFβ signal transduction. Development 2009, 136, 3699–3714. [Google Scholar]
- Biernacka, A.; Dobaczewski, M.; Frangogiannis, N.G. TGF-beta signaling in fibrosis. Growth Factors 2011, 29, 196–202. [Google Scholar]
- Derynck, R.; Zhang, Y.E. Smad-dependent and smad-independent pathways in TGF-[beta] family signalling. Nature 2003, 425, 577–584. [Google Scholar]
- Toonkel, R.L.; Borczuk, A.C.; Powell, C.A. TGF-[beta] Signaling pathway in lung adenocarcinoma invasion. J. Thorac. Oncol 2010, 5, 153–157. [Google Scholar]
- Inoue, T.; Ishida, T.; Takenoyama, M.; Sugio, K.; Sugimachi, K. The relationship between the immunodetection of transforming growth factor-beta in lung adenocarcinoma and longer survival rates. Surg. Oncol 1995, 4, 51–57. [Google Scholar]
- Shieh, J.-M.; Huang, T.-F.; Hung, C.-F.; Chou, K.-H.; Tsai, Y.-J.; Wu, W.-B. Activation of c-Jun N-terminal kinase is essential for mitochondrial membrane potential change and apoptosis induced by doxycycline in melanoma cells. Br. J. Pharmacol 2010, 160, 1171–1184. [Google Scholar]
- Chen, C.-P.; Hung, C.-F.; Lee, S.-C.; Lo, H.-M.; Wu, P.-H.; Wu, W.-B. Lycopene binding compromised PDGF-AA/-AB signaling and migration in smooth muscle cells and fibroblasts: prediction of the possible lycopene binding site within PDGF. Naunyn-Schmiedeberg’s Arch. Pharmacol 2010, 381, 401–414. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lo, H.-M.; Shieh, J.-M.; Chen, C.-L.; Tsou, C.-J.; Wu, W.-B. Vascular Endothelial Growth Factor Induces CXCL1 Chemokine Release via JNK and PI-3K-Dependent Pathways in Human Lung Carcinoma Epithelial Cells. Int. J. Mol. Sci. 2013, 14, 10090-10106. https://doi.org/10.3390/ijms140510090
Lo H-M, Shieh J-M, Chen C-L, Tsou C-J, Wu W-B. Vascular Endothelial Growth Factor Induces CXCL1 Chemokine Release via JNK and PI-3K-Dependent Pathways in Human Lung Carcinoma Epithelial Cells. International Journal of Molecular Sciences. 2013; 14(5):10090-10106. https://doi.org/10.3390/ijms140510090
Chicago/Turabian StyleLo, Huey-Ming, Jiunn-Min Shieh, Chih-Li Chen, Chih-Jen Tsou, and Wen-Bin Wu. 2013. "Vascular Endothelial Growth Factor Induces CXCL1 Chemokine Release via JNK and PI-3K-Dependent Pathways in Human Lung Carcinoma Epithelial Cells" International Journal of Molecular Sciences 14, no. 5: 10090-10106. https://doi.org/10.3390/ijms140510090
APA StyleLo, H.-M., Shieh, J.-M., Chen, C.-L., Tsou, C.-J., & Wu, W.-B. (2013). Vascular Endothelial Growth Factor Induces CXCL1 Chemokine Release via JNK and PI-3K-Dependent Pathways in Human Lung Carcinoma Epithelial Cells. International Journal of Molecular Sciences, 14(5), 10090-10106. https://doi.org/10.3390/ijms140510090