Skeletal Muscle Function during Exercise—Fine-Tuning of Diverse Subsystems by Nitric Oxide




Abstract
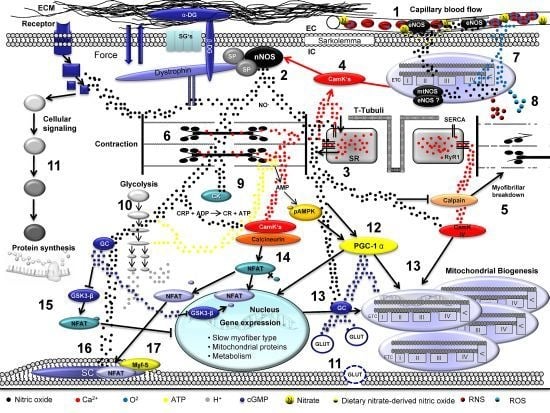
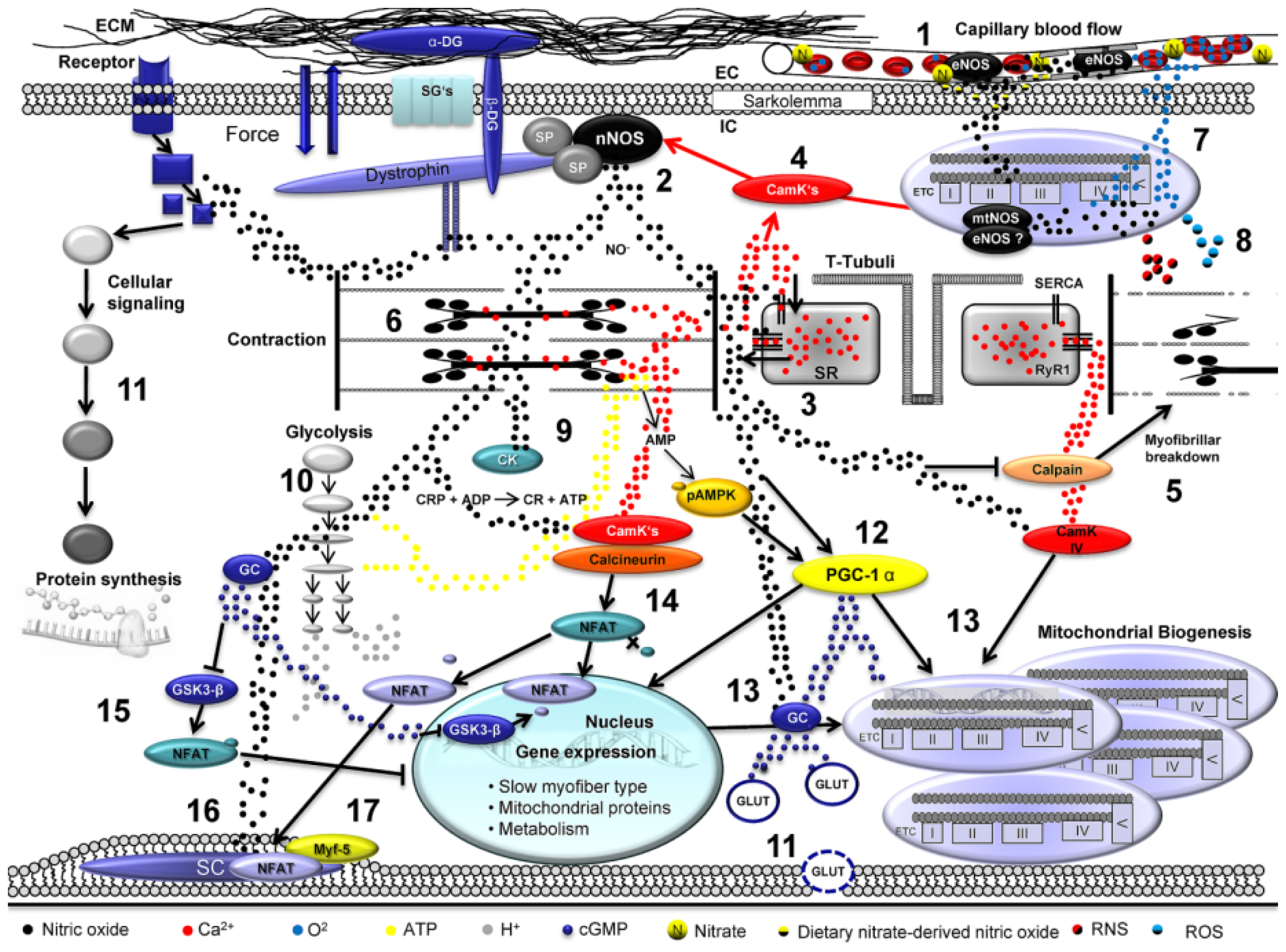
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Basics of Nitric Oxide
3. Reaction Routes of NO
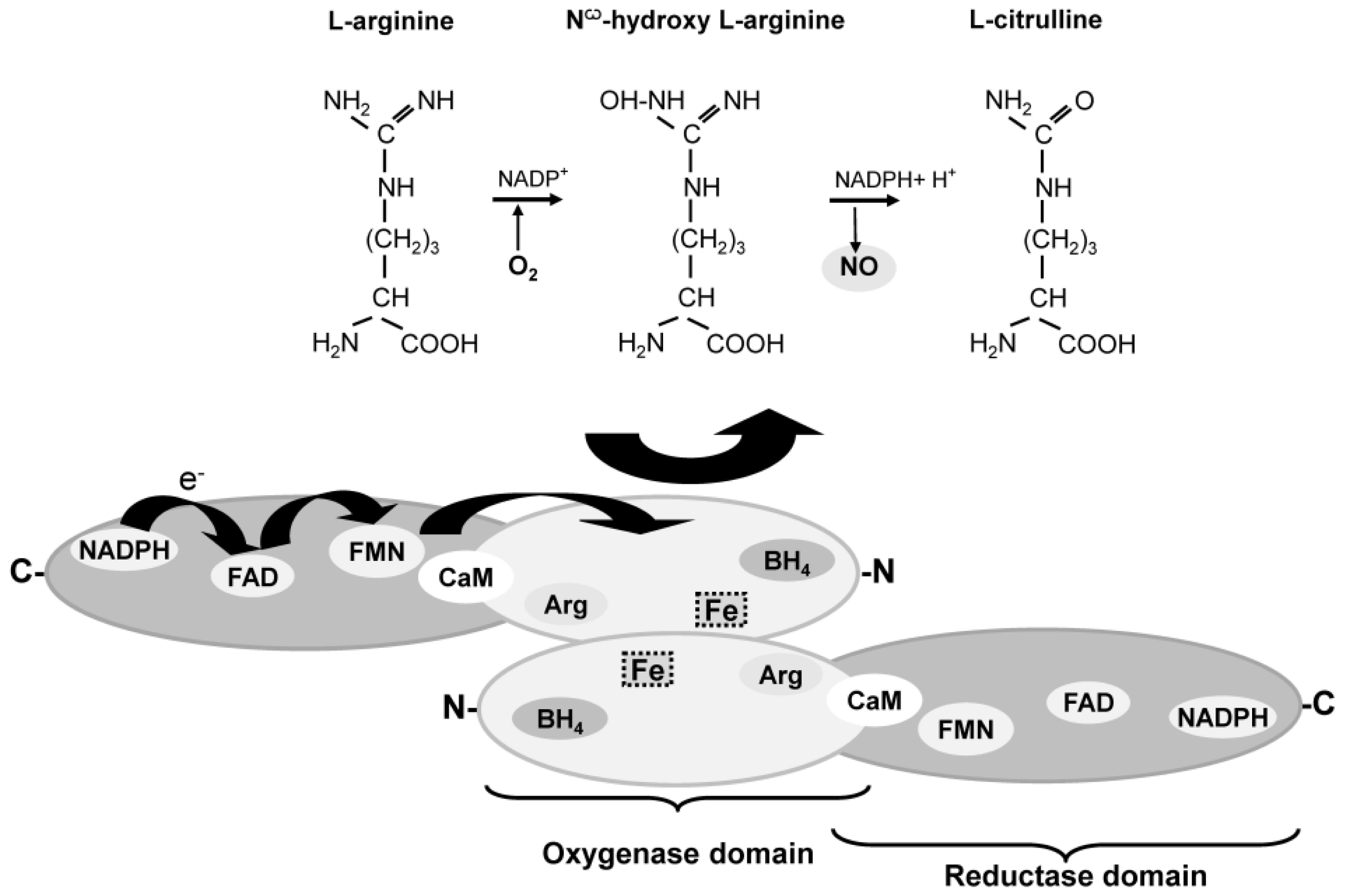
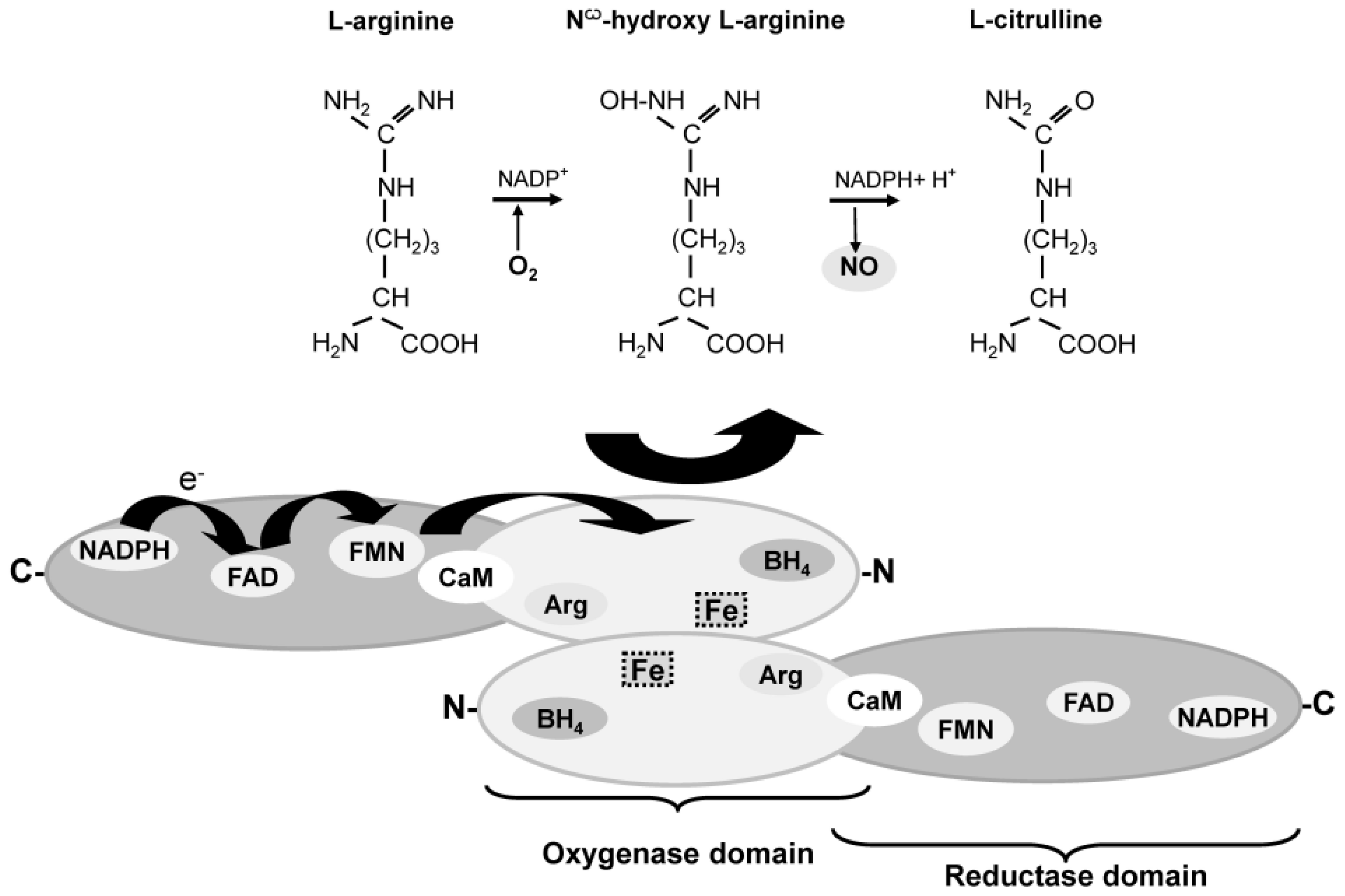
4. Enzymatic and Non-Enzymatic Synthesis of NO
5. Nitric Oxide Synthase (NOS) Isoforms
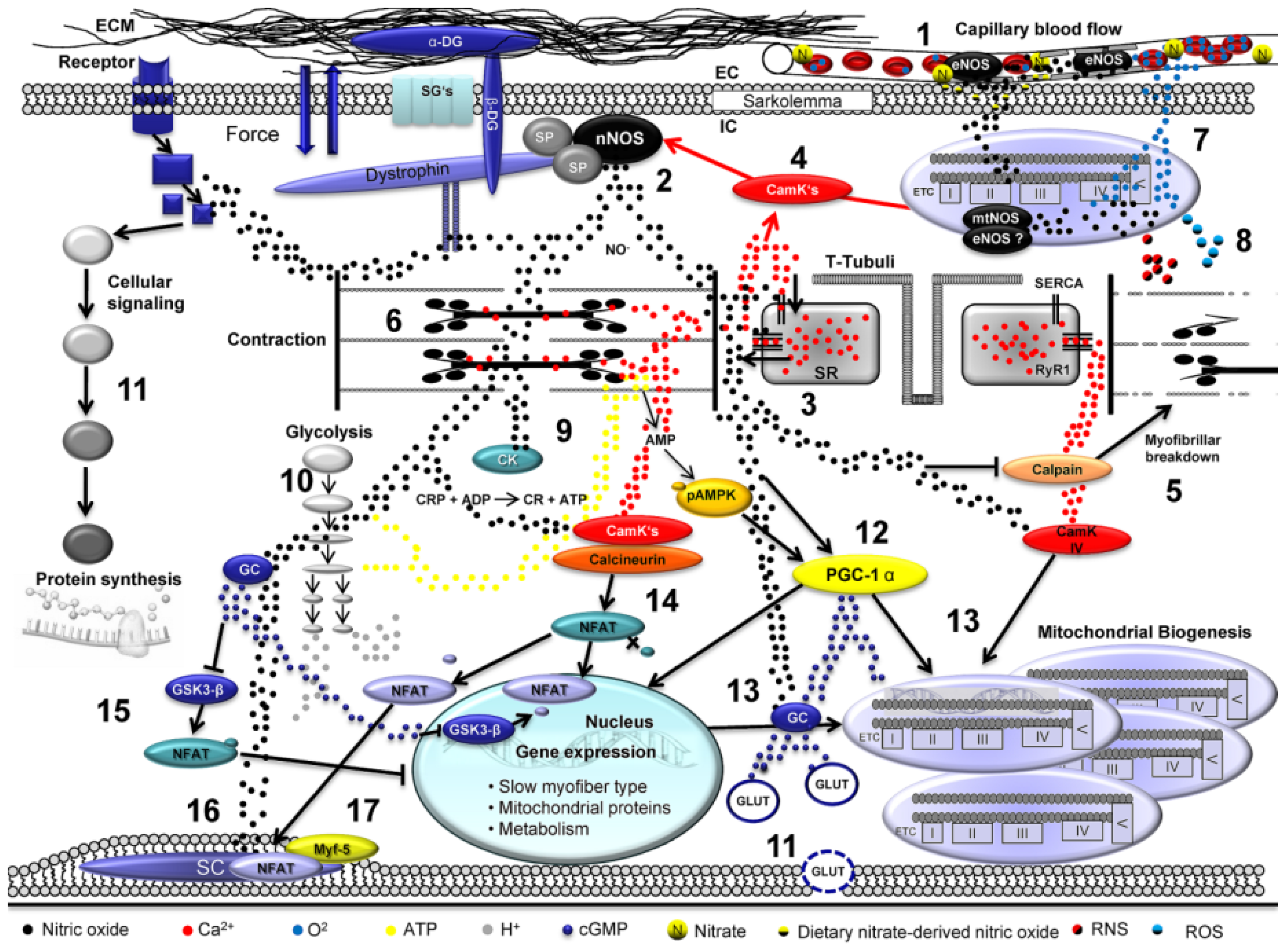
6. Localizations of NOS Isoforms and Their Occurrence in Skeletal Muscle Tissue
7. Cytoskeletal Components as Scaffold of NO Signaling
8. Signaling Involved in NO-Induced Modulation of Skeletal Muscle Contractility
9. Reactive Oxygen Species/Reactive Nitrogen Species and Antioxidative Enzymes in Skeletal Muscle
10. NO-Mediated Modulation of Metabolism
11. NO Signaling in Skeletal Muscle Hypertrophy
12. NO and NO-Dependent Signaling in Satellite Cells
13. NO-Induced Modulation of Exercise-Induced Skeletal Muscle Metabolic Demands and Myofiber Type Conversions
14. Conclusions
Conflict of Interest
References
- Stamler, J.S.; Meissner, G. Physiology of nitric oxide in skeletal muscle. Physiol. Rev 2001, 81, 209–237. [Google Scholar]
- Reid, M.B.; Haack, K.E.; Franchek, K.M.; Valberg, P.A.; Kobzik, L.; West, M.S. Reactive oxygen in skeletal muscle. I. Intracellular oxidant kinetics and fatigue in vitro. J. Appl. Physiol 1992, 73, 1797–1804. [Google Scholar]
- Balon, T.W.; Nadler, J.L. Nitric oxide release is present from incubated skeletal muscle preparations. J. Appl. Physiol 1994, 77, 2519–2521. [Google Scholar]
- Palomero, J.; Pye, D.; Kabayo, T.; Spiller, D.G.; Jackson, M.J. In situ detection and measurement of intracellular reactive oxygen species in single isolated mature skeletal muscle fibers by real time fluorescence microscopy. Antioxid. Redox. Signal 2008, 10, 1463–1474. [Google Scholar]
- Powers, S.K.; Jackson, M.J. Exercise-induced oxidative stress: Cellular mechanisms and impact on muscle force production. Physiol. Rev 2008, 88, 1243–1276. [Google Scholar]
- Pye, D.; Palomero, J.; Kabayo, T.; Jackson, M.J. Real-time measurement of nitric oxide in single mature mouse skeletal muscle fibres during contractions. J. Physiol 2007, 581, 309–318. [Google Scholar]
- Molavi, B.; Mehta, J.L. Oxidative stress in cardiovascular disease: Molecular basis of its deleterious effects, its detection, and therapeutic considerations. Curr. Opin. Cardiol 2004, 19, 488–493. [Google Scholar]
- Rao, G.N.; Berk, B.C. Active oxygen species stimulate vascular smooth muscle cell growth and proto-oncogene expression. Circ. Res 1992, 70, 593–599. [Google Scholar]
- Droge, W. Free radicals in the physiological control of cell function. Physiol. Rev 2002, 82, 47–95. [Google Scholar]
- Jackson, M.J.; Papa, S.; Bolanos, J.; Bruckdorfer, R.; Carlsen, H.; Elliott, R.M.; Flier, J.; Griffiths, H.R.; Heales, S.; Holst, B.; et al. Antioxidants, reactive oxygen and nitrogen species, gene induction and mitochondrial function. Mol. Aspects Med 2002, 23, 209–285. [Google Scholar]
- Furchgott, R.F.; Zawadzki, J.V. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature 1980, 288, 373–376. [Google Scholar]
- Palmer, R.M.; Ferrige, A.G.; Moncada, S. Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature 1987, 327, 524–526. [Google Scholar]
- Anderson, T.J.; Uehata, A.; Gerhard, M.D.; Meredith, I.T.; Knab, S.; Delagrange, D.; Lieberman, E.H.; Ganz, P.; Creager, M.A.; Yeung, A.C. Close relation of endothelial function in the human coronary and peripheral circulations. J. Am. Coll. Cardiol 1995, 26, 1235–1241. [Google Scholar]
- Oelze, M.; Mollnau, H.; Hoffmann, N.; Warnholtz, A.; Bodenschatz, M.; Smolenski, A.; Walter, U.; Skatchkov, M.; Meinertz, T.; Munzel, T. Vasodilator-stimulated phosphoprotein serine 239 phosphorylation as a sensitive monitor of defective nitric oxide/cGMP signaling and endothelial dysfunction. Circ. Res 2000, 87, 999–1005. [Google Scholar]
- Nathan, C.; Shiloh, M.U. Reactive oxygen and nitrogen intermediates in the relationship between mammalian hosts and microbial pathogens. Proc. Natl. Acad. Sci. USA 2000, 97, 8841–8848. [Google Scholar]
- Pervin, S.; Singh, R.; Chaudhuri, G. Nitric oxide-induced cytostasis and cell cycle arrest of a human breast cancer cell line (MDA-MB-231): Potential role of cyclin D1. Proc. Natl. Acad. Sci. USA 2001, 98, 3583–3588. [Google Scholar]
- Kolb, H.; Kolb-Bachofen, V. Nitric oxide in autoimmune disease: Cytotoxic or regulatory mediator? Immunol. Today 1998, 19, 556–561. [Google Scholar]
- Stamler, J.; Mendelsohn, M.E.; Amarante, P.; Smick, D.; Andon, N.; Davies, P.F.; Cooke, J.P.; Loscalzo, J. N-acetylcysteine potentiates platelet inhibition by endothelium-derived relaxing factor. Circ. Res 1989, 65, 789–795. [Google Scholar]
- Radomski, M.W.; Palmer, R.M.; Moncada, S. The anti-aggregating properties of vascular endothelium: Interactions between prostacyclin and nitric oxide. Br. J. Pharmacol 1987, 92, 639–646. [Google Scholar]
- Anderson, J.E. A role for nitric oxide in muscle repair: Nitric oxide-mediated activation of muscle satellite cells. Mol. Biol. Cell 2000, 11, 1859–1874. [Google Scholar]
- Hendgen-Cotta, U.; Grau, M.; Rassaf, T.; Gharini, P.; Kelm, M.; Kleinbongard, P. Reductive gas-phase chemiluminescence and flow injection analysis for measurement of the nitric oxide pool in biological matrices. Methods Enzymol 2008, 441, 295–315. [Google Scholar]
- Stamler, J.S.; Singel, D.J.; Loscalzo, J. Biochemistry of nitric oxide and its redox-activated forms. Science 1992, 258, 1898–1902. [Google Scholar]
- Freeman, B. Free radical chemistry of nitric oxide. Looking at the dark side. Chest 1994, 105, 79S–84S. [Google Scholar]
- Mellion, B.T.; Ignarro, L.J.; Ohlstein, E.H.; Pontecorvo, E.G.; Hyman, A.L.; Kadowitz, P.J. Evidence for the inhibitory role of guanosine 3′,5′-monophosphate in ADP-induced human platelet aggregation in the presence of nitric oxide and related vasodilators. Blood 1981, 57, 946–955. [Google Scholar]
- Lincoln, T.M. Cyclic GMP and mechanisms of vasodilation. Pharmacol. Ther 1989, 41, 479–502. [Google Scholar]
- Lincoln, T.M.; Dey, N.; Sellak, H. Invited review: cGMP-dependent protein kinase signaling mechanisms in smooth muscle: From the regulation of tone to gene expression. J. Appl. Physiol 2001, 91, 1421–1430. [Google Scholar]
- Hofmann, F.; Ammendola, A.; Schlossmann, J. Rising behind NO: cGMP-dependent protein kinases. J. Cell Sci 2000, 113, 1671–1676. [Google Scholar]
- Garthwaite, J.; Charles, S.L.; Chess-Williams, R. Endothelium-derived relaxing factor release on activation of NMDA receptors suggests role as intercellular messenger in the brain. Nature 1988, 336, 385–388. [Google Scholar]
- Zhuo, M.; Hawkins, R.D. Long-term depression: A learning-related type of synaptic plasticity in the mammalian central nervous system. Rev. Neurosci 1995, 6, 259–277. [Google Scholar]
- Munzel, T.; Feil, R.; Mulsch, A.; Lohmann, S.M.; Hofmann, F.; Walter, U. Physiology and pathophysiology of vascular signaling controlled by guanosine 3′,5′-cyclic monophosphate-dependent protein kinase [corrected]. Circulation 2003, 108, 2172–2183. [Google Scholar]
- Bruckdorfer, R. The basics about nitric oxide. Mol. Aspects Med 2005, 26, 3–31. [Google Scholar]
- Thoonen, R.; Sips, P.Y.; Bloch, K.D.; Buys, E.S. Pathophysiology of hypertension in the absence of nitric oxide/cyclic GMP signaling. Curr. Hypertens. Rep 2013, 15, 47–58. [Google Scholar]
- Buono, R.; Vantaggiato, C.; Pisa, V.; Azzoni, E.; Bassi, M.T.; Brunelli, S.; Sciorati, C.; Clementi, E. Nitric oxide sustains long-term skeletal muscle regeneration by regulating fate of satellite cells via signaling pathways requiring Vangl2 and cyclic GMP. Stem Cells 2012, 30, 197–209. [Google Scholar]
- Foster, M.W.; McMahon, T.J.; Stamler, J.S. S-nitrosylation in health and disease. Trends Mol. Med 2003, 9, 160–168. [Google Scholar]
- Martinez-Ruiz, A.; Lamas, S. S-nitrosylation: A potential new paradigm in signal transduction. Cardiovasc. Res 2004, 62, 43–52. [Google Scholar]
- Broillet, M.C. S-nitrosylation of proteins. Cell Mol. Life Sci 1999, 55, 1036–1042. [Google Scholar]
- Stamler, J.S.; Simon, D.I.; Osborne, J.A.; Mullins, M.E.; Jaraki, O.; Michel, T.; Singel, D.J.; Loscalzo, J. S-nitrosylation of proteins with nitric oxide: Synthesis and characterization of biologically active compounds. Proc. Natl. Acad. Sci. USA 1992, 89, 444–448. [Google Scholar]
- Hess, D.T.; Matsumoto, A.; Kim, S.O.; Marshall, H.E.; Stamler, J.S. Protein S-nitrosylation: purview and parameters. Nat. Rev. Mol. Cell Biol 2005, 6, 150–166. [Google Scholar]
- Lima, B.; Forrester, M.T.; Hess, D.T.; Stamler, J.S. S-nitrosylation in cardiovascular signaling. Circ. Res 2010, 106, 633–646. [Google Scholar]
- Palmer, R.M.; Rees, D.D.; Ashton, D.S.; Moncada, S. L-arginine is the physiological precursor for the formation of nitric oxide in endothelium-dependent relaxation. Biochem. Biophys. Res. Commun 1988, 153, 1251–1256. [Google Scholar]
- Filho, J.C.; Bergstrom, J.; Stehle, P.; Furst, P. Simultaneous measurements of free amino acid patterns of plasma, muscle and erythrocytes in healthy human subjects. Clin Nutr 1997, 16, 299–305. [Google Scholar]
- Stuehr, D.J.; Kwon, N.S.; Nathan, C.F.; Griffith, O.W.; Feldman, P.L.; Wiseman, J. N omega-hydroxy-L-arginine is an intermediate in the biosynthesis of nitric oxide from L-arginine. J. Biol. Chem 1991, 266, 6259–6263. [Google Scholar]
- Spiegelhalder, B.; Eisenbrand, G.; Preussmann, R. Influence of dietary nitrate on nitrite content of human saliva: Possible relevance to in vivo formation of N-nitroso compounds. Food Cosmet. Toxicol 1976, 14, 545–548. [Google Scholar]
- Tannenbaum, S.R.; Correa, P. Nitrate and gastric cancer risks. Nature 1985, 317, 675–676. [Google Scholar]
- Zweier, J.L.; Samouilov, A.; Kuppusamy, P. Non-enzymatic nitric oxide synthesis in biological systems. Biochim. Biophys. Acta 1999, 1411, 250–262. [Google Scholar]
- Cosby, K.; Partovi, K.S.; Crawford, J.H.; Patel, R.P.; Reiter, C.D.; Martyr, S.; Yang, B.K.; Waclawiw, M.A.; Zalos, G.; Xu, X.; et al. Nitrite reduction to nitric oxide by deoxyhemoglobin vasodilates the human circulation. Nat. Med 2003, 9, 1498–1505. [Google Scholar]
- Gladwin, M.T.; Shelhamer, J.H.; Schechter, A.N.; Pease-Fye, M.E.; Waclawiw, M.A.; Panza, J.A.; Ognibene, F.P.; Cannon, R.O., III. Role of circulating nitrite and S-nitrosohemoglobin in the regulation of regional blood flow in humans. Proc. Natl. Acad. Sci. USA 2000, 97, 11482–11487. [Google Scholar]
- Isbell, T.S.; Gladwin, M.T.; Patel, R.P. Hemoglobin oxygen fractional saturation regulates nitrite-dependent vasodilation of aortic ring bioassays. Am. J. Physiol. Heart Circ. Physiol 2007, 293, H2565–H2572. [Google Scholar]
- Totzeck, M.; Hendgen-Cotta, U.B.; Luedike, P.; Berenbrink, M.; Klare, J.P.; Steinhoff, H.J.; Semmler, D.; Shiva, S.; Williams, D.; Kipar, A.; et al. Nitrite regulates hypoxic vasodilation via myoglobin-dependent nitric oxide generation. Circulation 2012, 126, 325–334. [Google Scholar]
- Shiva, S.; Huang, Z.; Grubina, R.; Sun, J.; Ringwood, L.A.; MacArthur, P.H.; Xu, X.; Murphy, E.; Darley-Usmar, V.M.; Gladwin, M.T. Deoxymyoglobin is a nitrite reductase that generates nitric oxide and regulates mitochondrial respiration. Circ. Res 2007, 100, 654–661. [Google Scholar]
- Rassaf, T.; Flogel, U.; Drexhage, C.; Hendgen-Cotta, U.; Kelm, M.; Schrader, J. Nitrite reductase function of deoxymyoglobin: oxygen sensor and regulator of cardiac energetics and function. Circ. Res 2007, 100, 1749–1754. [Google Scholar]
- Haynes, V.; Elfering, S.; Traaseth, N.; Giulivi, C. Mitochondrial nitric-oxide synthase: Enzyme expression, characterization, and regulation. J. Bioenerg. Biomembr 2004, 36, 341–346. [Google Scholar]
- Giulivi, C.; Poderoso, J.J.; Boveris, A. Production of nitric oxide by mitochondria. J. Biol. Chem 1998, 273, 11038–11043. [Google Scholar]
- Brenman, J.E.; Xia, H.; Chao, D.S.; Black, S.M.; Bredt, D.S. Regulation of neuronal nitric oxide synthase through alternative transcripts. Dev. Neurosci 1997, 19, 224–231. [Google Scholar]
- Silvagno, F.; Xia, H.; Bredt, D.S. Neuronal nitric-oxide synthase-mu, an alternatively spliced isoform expressed in differentiated skeletal muscle. J. Biol. Chem 1996, 271, 11204–11208. [Google Scholar]
- Amancharla, M.R.; Rodarte, J.R.; Boriek, A.M. Modeling the kinematics of the canine midcostal diaphragm. Am. J. Physiol. Regul. Integr. Comp. Physiol 2001, 280, R588–R597. [Google Scholar]
- Ignarro, L.J. Heme-dependent activation of soluble guanylate cyclase by nitric oxide: Regulation of enzyme activity by porphyrins and metalloporphyrins. Semin. Hematol 1989, 26, 63–76. [Google Scholar]
- Kleinbongard, P.; Schulz, R.; Rassaf, T.; Lauer, T.; Dejam, A.; Jax, T.; Kumara, I.; Gharini, P.; Kabanova, S.; Ozuyaman, B.; et al. Red blood cells express a functional endothelial nitric oxide synthase. Blood 2006, 107, 2943–2951. [Google Scholar]
- Cortese-Krott, M.M.; Rodriguez-Mateos, A.; Sansone, R.; Kuhnle, G.G.; Thasian-Sivarajah, S.; Krenz, T.; Horn, P.; Krisp, C.; Wolters, D.; Heiss, C.; et al. Human red blood cells at work: Identification and visualization of erythrocytic eNOS activity in health and disease. Blood 2012, 120, 4229–4237. [Google Scholar]
- Suhr, F.; Brenig, J.; Muller, R.; Behrens, H.; Bloch, W.; Grau, M. Moderate exercise promotes human RBC-NOS activity, NO production and deformability through Akt kinase pathway. PLoS One 2012, 7, e45982. [Google Scholar]
- Grau, M.; Pauly, S.; Ali, J.; Walpurgis, K.; Thevis, M.; Bloch, W.; Suhr, F. RBC-NOS-dependent S-nitrosylation of cytoskeletal proteins improves RBC dformability. PLoS One 2013, 8, e56759. [Google Scholar]
- Cho, H.J.; Xie, Q.W.; Calaycay, J.; Mumford, R.A.; Swiderek, K.M.; Lee, T.D.; Nathan, C. Calmodulin is a subunit of nitric oxide synthase from macrophages. J. Exp. Med 1992, 176, 599–604. [Google Scholar]
- Nathan, C. Inducible nitric oxide synthase: What difference does it make? J. Clin. Invest 1997, 100, 2417–2423. [Google Scholar]
- Forstermann, U.; Closs, E.I.; Pollock, J.S.; Nakane, M.; Schwarz, P.; Gath, I.; Kleinert, H. Nitric oxide synthase isozymes. Characterization, purification, molecular cloning, and functions. Hypertension 1994, 23, 1121–1131. [Google Scholar]
- Adams, V.; Yu, J.; Mobius-Winkler, S.; Linke, A.; Weigl, C.; Hilbrich, L.; Schuler, G.; Hambrecht, R. Increased inducible nitric oxide synthase in skeletal muscle biopsies from patients with chronic heart failure. Biochem. Mol. Med 1997, 61, 152–160. [Google Scholar]
- Ungureanu-Longrois, D.; Balligand, J.L.; Kelly, R.A.; Smith, T.W. Myocardial contractile dysfunction in the systemic inflammatory response syndrome: Role of a cytokine-inducible nitric oxide synthase in cardiac myocytes. J. Mol. Cell Cardiol 1995, 27, 155–167. [Google Scholar]
- Gielen, S.; Adams, V.; Linke, A.; Erbs, S.; Mobius-Winkler, S.; Schubert, A.; Schuler, G.; Hambrecht, R. Exercise training in chronic heart failure: Correlation between reduced local inflammation and improved oxidative capacity in the skeletal muscle. Eur. J. Cardiovasc. Prev. Rehabil 2005, 12, 393–400. [Google Scholar]
- Schulze, P.C.; Gielen, S.; Schuler, G.; Hambrecht, R. Chronic heart failure and skeletal muscle catabolism: Effects of exercise training. Int. J. Cardiol 2002, 85, 141–149. [Google Scholar]
- Akita, Y.; Otani, H.; Matsuhisa, S.; Kyoi, S.; Enoki, C.; Hattori, R.; Imamura, H.; Kamihata, H.; Kimura, Y.; Iwasaka, T. Exercise-induced activation of cardiac sympathetic nerve triggers cardioprotection via redox-sensitive activation of eNOS and upregulation of iNOS. Am. J. Physiol. Heart Circ. Physiol 2007, 292, H2051–H2059. [Google Scholar]
- Xu, W.; Charles, I.G.; Moncada, S.; Gorman, P.; Sheer, D.; Liu, L.; Emson, P. Mapping of the genes encoding human inducible and endothelial nitric oxide synthase (NOS2 and NOS3) to the pericentric region of chromosome 17 and to chromosome 7, respectively. Genomics 1994, 21, 419–422. [Google Scholar]
- Alderton, W.K.; Cooper, C.E.; Knowles, R.G. Nitric oxide synthases: structure, function and inhibition. Biochem. J 2001, 357, 593–615. [Google Scholar]
- Bredt, D.S.; Snyder, S.H. Isolation of nitric oxide synthetase, a calmodulin-requiring enzyme. Proc. Natl. Acad. Sci. USA 1990, 87, 682–685. [Google Scholar]
- Yui, Y.; Hattori, R.; Kosuga, K.; Eizawa, H.; Hiki, K.; Kawai, C. Purification of nitric oxide synthase from rat macrophages. J. Biol. Chem 1991, 266, 12544–12547. [Google Scholar]
- Pollock, J.S.; Forstermann, U.; Mitchell, J.A.; Warner, T.D.; Schmidt, H.H.; Nakane, M.; Murad, F. Purification and characterization of particulate endothelium-derived relaxing factor synthase from cultured and native bovine aortic endothelial cells. Proc. Natl. Acad. Sci. USA 1991, 88, 10480–10484. [Google Scholar]
- Asano, K.; Chee, C.B.; Gaston, B.; Lilly, C.M.; Gerard, C.; Drazen, J.M.; Stamler, J.S. Constitutive and inducible nitric oxide synthase gene expression, regulation, and activity in human lung epithelial cells. Proc. Natl. Acad. Sci. USA 1994, 91, 10089–10093. [Google Scholar]
- Nathan, C. Nitric oxide as a secretory product of mammalian cells. FASEB J 1992, 6, 3051–3064. [Google Scholar]
- Snyder, L.M.; Fortier, N.L.; Trainor, J.; Jacobs, J.; Leb, L.; Lubin, B.; Chiu, D.; Shohet, S.; Mohandas, N. Effect of hydrogen peroxide exposure on normal human erythrocyte deformability, morphology, surface characteristics, and spectrin-hemoglobin cross-linking. J. Clin. Invest 1985, 76, 1971–1977. [Google Scholar]
- Shaul, P.W. Regulation of endothelial nitric oxide synthase: Location, location, location. Annu. Rev. Physiol 2002, 64, 749–774. [Google Scholar]
- Chang, W.J.; Iannaccone, S.T.; Lau, K.S.; Masters, B.S.; McCabe, T.J.; McMillan, K.; Padre, R.C.; Spencer, M.J.; Tidball, J.G.; Stull, J.T. Neuronal nitric oxide synthase and dystrophin-deficient muscular dystrophy. Proc. Natl. Acad. Sci. USA 1996, 93, 9142–9147. [Google Scholar]
- Frandsen, U.; Lopez-Figueroa, M.; Hellsten, Y. Localization of nitric oxide synthase in human skeletal muscle. Biochem. Biophys. Res. Commun 1996, 227, 88–93. [Google Scholar]
- Ho, K.M.; McMurray, G.; Brading, A.F.; Noble, J.G.; Ny, L.; Andersson, K.E. Nitric oxide synthase in the heterogeneous population of intramural striated muscle fibres of the human membranous urethral sphincter. J. Urol 1998, 159, 1091–1096. [Google Scholar]
- Nakane, M.; Schmidt, H.H.; Pollock, J.S.; Forstermann, U.; Murad, F. Cloned human brain nitric oxide synthase is highly expressed in skeletal muscle. FEBS Lett 1993, 316, 175–180. [Google Scholar]
- Blottner, D.; Luck, G. Nitric oxide synthase (NOS) in mouse skeletal muscle development and differentiated myoblasts. Cell Tissue Res 1998, 292, 293–302. [Google Scholar]
- Christova, T.; Grozdanovic, Z.; Gossrau, R. Nitric oxide synthase (NOS) I during postnatal development in rat and mouse skeletal muscle. Acta Histochem 1997, 99, 311–324. [Google Scholar]
- Guo, Y.; Greenwood, M.T.; Petrof, B.J.; Hussain, S.N. Expression and regulation of protein inhibitor of neuronal nitric oxide synthase in ventilatory muscles. Am. J. Respir. Cell Mol. Biol 1999, 20, 319–326. [Google Scholar]
- Kobzik, L.; Reid, M.B.; Bredt, D.S.; Stamler, J.S. Nitric oxide in skeletal muscle. Nature 1994, 372, 546–548. [Google Scholar]
- Gath, I.; Closs, E.I.; Godtel-Armbrust, U.; Schmitt, S.; Nakane, M.; Wessler, I.; Forstermann, U. Inducible NO synthase II and neuronal NO synthase I are constitutively expressed in different structures of guinea pig skeletal muscle: Implications for contractile function. FASEB J 1996, 10, 1614–1620. [Google Scholar]
- El, D.Q.; Guo, Y.; Comtois, A.; Zhu, E.; Greenwood, M.T.; Bredt, D.S.; Hussain, S.N. Ontogenesis of nitric oxide synthases in the ventilatory muscles. Am. J. Respir. Cell Mol. Biol 1998, 18, 844–852. [Google Scholar]
- Kapur, S.; Bedard, S.; Marcotte, B.; Cote, C.H.; Marette, A. Expression of nitric oxide synthase in skeletal muscle: A novel role for nitric oxide as a modulator of insulin action. Diabetes 1997, 46, 1691–1700. [Google Scholar]
- Gossrau, R. Caveolin-3 and nitric oxide synthase I in healthy and diseased skeletal muscle. Acta Histochem 1998, 100, 99–112. [Google Scholar]
- Kusner, L.L.; Kaminski, H.J. Nitric oxide synthase is concentrated at the skeletal muscle endplate. Brain Res 1996, 730, 238–242. [Google Scholar]
- Grozdanovic, Z.; Gosztonyi, G.; Gossrau, R. Nitric oxide synthase I (NOS-I) is deficient in the sarcolemma of striated muscle fibers in patients with Duchenne muscular dystrophy, suggesting an association with dystrophin. Acta Histochem 1996, 98, 61–69. [Google Scholar]
- Wakayama, Y.; Inoue, M.; Murahashi, M.; Shibuya, S.; Jimi, T.; Kojima, H.; Oniki, H. Ultrastructural localization of alpha 1-syntrophin and neuronal nitric oxide synthase in normal skeletal myofiber, and their relation to each other and to dystrophin. Acta Neuropathol 1997, 94, 455–464. [Google Scholar]
- Williams, G.; Brown, T.; Becker, L.; Prager, M.; Giroir, B.P. Cytokine-induced expression of nitric oxide synthase in C2C12 skeletal muscle myocytes. Am. J. Physiol 1994, 267, R1020–R1025. [Google Scholar]
- Kobzik, L.; Stringer, B.; Balligand, J.L.; Reid, M.B.; Stamler, J.S. Endothelial type nitric oxide synthase in skeletal muscle fibers: mitochondrial relationships. Biochem. Biophys. Res. Commun 1995, 211, 375–381. [Google Scholar]
- Bates, T.E.; Loesch, A.; Burnstock, G.; Clark, J.B. Mitochondrial nitric oxide synthase: A ubiquitous regulator of oxidative phosphorylation? Biochem. Biophys. Res. Commun 1996, 218, 40–44. [Google Scholar]
- Grozdanovic, Z.; Nakos, G.; Dahrmann, G.; Mayer, B.; Gossrau, R. Species-independent expression of nitric oxide synthase in the sarcolemma region of visceral and somatic striated muscle fibers. Cell Tissue Res 1995, 281, 493–499. [Google Scholar]
- Heinzel, B.; John, M.; Klatt, P.; Bohme, E.; Mayer, B. Ca2+/calmodulin-dependent formation of hydrogen peroxide by brain nitric oxide synthase. Biochem. J 1992, 281, 627–630. [Google Scholar]
- Laursen, J.B.; Somers, M.; Kurz, S.; McCann, L.; Warnholtz, A.; Freeman, B.A.; Tarpey, M.; Fukai, T.; Harrison, D.G. Endothelial regulation of vasomotion in apoE-deficient mice: Implications for interactions between peroxynitrite and tetrahydrobiopterin. Circulation 2001, 103, 1282–1288. [Google Scholar]
- Cai, H.; Harrison, D.G. Endothelial dysfunction in cardiovascular diseases: The role of oxidant stress. Circ. Res 2000, 87, 840–844. [Google Scholar]
- Xia, Y.; Tsai, A.L.; Berka, V.; Zweier, J.L. Superoxide generation from endothelial nitric-oxide synthase. A Ca2+/calmodulin-dependent and tetrahydrobiopterin regulatory process. J. Biol. Chem 1998, 273, 25804–25808. [Google Scholar]
- Vasquez-Vivar, J.; Kalyanaraman, B.; Martasek, P.; Hogg, N.; Masters, B.S.; Karoui, H.; Tordo, P.; Pritchard, K.A., Jr. Superoxide generation by endothelial nitric oxide synthase: The influence of cofactors. Proc. Natl. Acad. Sci. USA 1998, 95, 9220–9225. [Google Scholar]
- Wink, D.A.; Cook, J.A.; Pacelli, R.; Liebmann, J.; Krishna, M.C.; Mitchell, J.B. Nitric oxide (NO) protects against cellular damage by reactive oxygen species. Toxicol. Lett. 1995, 82–83, 221–226. [Google Scholar]
- Brenman, J.E.; Chao, D.S.; Gee, S.H.; McGee, A.W.; Craven, S.E.; Santillano, D.R.; Wu, Z.; Huang, F.; Xia, H.; Peters, M.F.; et al. Interaction of nitric oxide synthase with the postsynaptic density protein PSD-95 and alpha1-syntrophin mediated by PDZ domains. Cell 1996, 84, 757–767. [Google Scholar]
- Rando, T.A. Role of nitric oxide in the pathogenesis of muscular dystrophies: A “two hit” hypothesis of the cause of muscle necrosis. Microsc. Res. Tech 2001, 55, 223–235. [Google Scholar]
- Carmignac, V.; Durbeej, M. Cell-matrix interactions in muscle disease. J. Pathol 2012, 226, 200–218. [Google Scholar]
- Van Deutekom, J.C.; van Ommen, G.J. Advances in Duchenne muscular dystrophy gene therapy. Nat. Rev. Genet 2003, 4, 774–783. [Google Scholar]
- Brenman, J.E.; Chao, D.S.; Xia, H.; Aldape, K.; Bredt, D.S. Nitric oxide synthase complexed with dystrophin and absent from skeletal muscle sarcolemma in Duchenne muscular dystrophy. Cell 1995, 82, 743–752. [Google Scholar]
- Lai, Y.; Thomas, G.D.; Yue, Y.; Yang, H.T.; Li, D.; Long, C.; Judge, L.; Bostick, B.; Chamberlain, J.S.; Terjung, R.L.; et al. Dystrophins carrying spectrin-like repeats 16 and 17 anchor nNOS to the sarcolemma and enhance exercise performance in a mouse model of muscular dystrophy. J. Clin. Invest 2009, 119, 624–635. [Google Scholar]
- Lai, Y.; Zhao, J.; Yue, Y.; Duan, D. α2 and α3 helices of dystrophin R16 and R17 frame a microdomain in the α1 helix of dystrophin R17 for neuronal NOS binding. Proc. Natl. Acad. Sci. USA 2013, 110, 525–530. [Google Scholar]
- Wehling-Henricks, M.; Tidball, J.G. Neuronal nitric oxide synthase-rescue of dystrophin/utrophin double knockout mice does not require nNOS localization to the cell membrane. PLoS One 2011, 6, e25071. [Google Scholar]
- Keesey, J.C. Clinical evaluation and management of myasthenia gravis. Muscle Nerve 2004, 29, 484–505. [Google Scholar]
- Meinen, S.; Lin, S.; Ruegg, M.A.; Punga, A.R. Fatigue and muscle atrophy in a mouse model of myasthenia gravis is paralleled by loss of sarcolemmal nNOS. PLoS One 2012, 7, e44148. [Google Scholar]
- Berchtold, M.W.; Brinkmeier, H.; Muntener, M. Calcium ion in skeletal muscle: Its crucial role for muscle function, plasticity, and disease. Physiol. Rev 2000, 80, 1215–1265. [Google Scholar]
- Grozdanovic, Z.; Baumgarten, H.G. Nitric oxide synthase in skeletal muscle fibers: A signaling component of the dystrophin-glycoprotein complex. Histol. Histopathol 1999, 14, 243–256. [Google Scholar]
- Beltman, J.G.; de Haan, A.; Haan, H.; Gerrits, H.L.; van Mechelen, W.; Sargeant, A.J. Metabolically assessed muscle fibre recruitment in brief isometric contractions at different intensities. Eur. J. Appl. Physiol 2004, 92, 485–492. [Google Scholar]
- Reid, M.B. Role of nitric oxide in skeletal muscle: Synthesis, distribution and functional importance. Acta Physiol. Scand 1998, 162, 401–409. [Google Scholar]
- Betzenhauser, M.J.; Marks, A.R. Ryanodine receptor channelopathies. Pflugers Arch 2010, 460, 467–480. [Google Scholar]
- Andersson, D.C.; Betzenhauser, M.J.; Reiken, S.; Meli, A.C.; Umanskaya, A.; Xie, W.; Shiomi, T.; Zalk, R.; LaCampagne, A.; Marks, A.R. Ryanodine receptor oxidation causes intracellular calcium leak and muscle weakness in aging. Cell Metab 2011, 14, 196–207. [Google Scholar]
- Bellinger, A.M.; Reiken, S.; Dura, M.; Murphy, P.W.; Deng, S.X.; Landry, D.W.; Nieman, D.; Lehnart, S.E.; Samaru, M.; LaCampagne, A.; et al. Remodeling of ryanodine receptor complex causes “leaky” channels: A molecular mechanism for decreased exercise capacity. Proc. Natl. Acad. Sci. USA 2008, 105, 2198–2202. [Google Scholar]
- Gehlert, S.; Bungartz, G.; Willkomm, L.; Korkmaz, Y.; Pfannkuche, K.; Schiffer, T.; Bloch, W.; Suhr, F. Intense resistance exercise induces early and transient increases in ryanodine receptor 1 phosphorylation in human skeletal muscle. PLoS One 2012, 7, e49326. [Google Scholar]
- Bellinger, A.M.; Mongillo, M.; Marks, A.R. Stressed out: The skeletal muscle ryanodine receptor as a target of stress. J. Clin. Invest 2008, 118, 445–453. [Google Scholar]
- Bellinger, A.M.; Reiken, S.; Carlson, C.; Mongillo, M.; Liu, X.; Rothman, L.; Matecki, S.; LaCampagne, A.; Marks, A.R. Hypernitrosylated ryanodine receptor calcium release channels are leaky in dystrophic muscle. Nat. Med 2009, 15, 325–330. [Google Scholar]
- Sun, J.; Xin, C.; Eu, J.P.; Stamler, J.S.; Meissner, G. Cysteine-3635 is responsible for skeletal muscle ryanodine receptor modulation by NO. Proc. Natl. Acad. Sci. USA 2001, 98, 11158–11162. [Google Scholar]
- Suhr, F.; Gehlert, S.; Braun, K.; Bungartz, G.; Kern, P.; Willkomm, L.; Pfannkuche, K.; Krüger, M.; Bloch, W. Institute of Cardiovascular Research and Sport Medicine, German Sport University Cologne: Cologne, Germany, Unpublished work; 2013.
- Shen, W.; Hintze, T.H.; Wolin, M.S. Nitric oxide. An important signaling mechanism between vascular endothelium and parenchymal cells in the regulation of oxygen consumption. Circulation 1995, 92, 3505–3512. [Google Scholar]
- Morrison, R.J.; Miller, C.C., III; Reid, M.B. Nitric oxide effects on shortening velocity and power production in the rat diaphragm. J. Appl. Physiol. 1996, 80, 1065–1069. [Google Scholar]
- Perkins, W.J.; Han, Y.S.; Sieck, G.C. Skeletal muscle force and actomyosin ATPase activity reduced by nitric oxide donor. J. Appl. Physiol 1997, 83, 1326–1332. [Google Scholar]
- Marechal, G.; Beckers-Bleukx, G. Effect of nitric oxide on the maximal velocity of shortening of a mouse skeletal muscle. Pflugers Arch 1998, 436, 906–913. [Google Scholar]
- Morrison, R.J.; Miller, C.C., III; Reid, M.B. Nitric oxide effects on force-velocity characteristics of the rat diaphragm. Comp. Biochem. Physiol. A 1998, 119, 203–209. [Google Scholar]
- Evangelista, A.M.; Rao, V.S.; Filo, A.R.; Marozkina, N.V.; Doctor, A.; Jones, D.R.; Gaston, B.; Guilford, W.H. Direct regulation of striated muscle myosins by nitric oxide and endogenous nitrosothiols. PLoS One 2010, 5, e11209. [Google Scholar]
- Halliwell, B.; Cross, C.E. Oxygen-derived species: Their relation to human disease and environmental stress. Environ. Health Perspect 1994, 102, 5–12. [Google Scholar]
- Vollaard, N.B.; Shearman, J.P.; Cooper, C.E. Exercise-induced oxidative stress: Myths, realities and physiological relevance. Sports Med 2005, 35, 1045–1062. [Google Scholar]
- Boveris, A.; Chance, B. The mitochondrial generation of hydrogen peroxide. General properties and effect of hyperbaric oxygen. Biochem. J 1973, 134, 707–716. [Google Scholar]
- Xia, R.; Webb, J.A.; Gnall, L.L.; Cutler, K.; Abramson, J.J. Skeletal muscle sarcoplasmic reticulum contains a NADH-dependent oxidase that generates superoxide. Am. J. Physiol Cell Physiol 2003, 285, C215–C221. [Google Scholar]
- Espinosa, A.; Leiva, A.; Pena, M.; Muller, M.; Debandi, A.; Hidalgo, C.; Carrasco, M.A.; Jaimovich, E. Myotube depolarization generates reactive oxygen species through NAD(P)H oxidase; ROS-elicited Ca2+ stimulates ERK, CREB, early genes. J. Cell Physiol 2006, 209, 379–388. [Google Scholar]
- Hidalgo, C.; Sanchez, G.; Barrientos, G.; Aracena-Parks, P. A transverse tubule NADPH oxidase activity stimulates calcium release from isolated triads via ryanodine receptor type 1 S-glutathionylation. J. Biol. Chem 2006, 281, 26473–26482. [Google Scholar]
- Sen, C.K.; Packer, L. Antioxidant and redox regulation of gene transcription. FASEB J 1996, 10, 709–720. [Google Scholar]
- Reid, M.B. Invited review: Redox modulation of skeletal muscle contraction: What we know and what we don’t. J. Appl. Physiol 2001, 90, 724–731. [Google Scholar]
- Supinski, G.S.; Callahan, L.A. Free radical-mediated skeletal muscle dysfunction in inflammatory conditions. J. Appl. Physiol 2007, 102, 2056–2063. [Google Scholar]
- Reid, M.B.; Khawli, F.A.; Moody, M.R. Reactive oxygen in skeletal muscle. III. Contractility of unfatigued muscle. J. Appl. Physiol 1993, 75, 1081–1087. [Google Scholar]
- Ji, L.L.; Stratman, F.W.; Lardy, H.A. Antioxidant enzyme systems in rat liver and skeletal muscle. Influences of selenium deficiency, chronic training, and acute exercise. Arch. Biochem. Biophys 1988, 263, 150–160. [Google Scholar]
- Powers, S.K.; Criswell, D.; Lawler, J.; Ji, L.L.; Martin, D.; Herb, R.A.; Dudley, G. Influence of exercise and fiber type on antioxidant enzyme activity in rat skeletal muscle. Am. J. Physiol 1994, 266, R375–R380. [Google Scholar]
- Criswell, D.; Powers, S.; Dodd, S.; Lawler, J.; Edwards, W.; Renshler, K.; Grinton, S. High intensity training-induced changes in skeletal muscle antioxidant enzyme activity. Med. Sci. Sports Exerc 1993, 25, 1135–1140. [Google Scholar]
- Lawler, J.M.; Kwak, H.B.; Song, W.; Parker, J.L. Exercise training reverses downregulation of HSP70 and antioxidant enzymes in porcine skeletal muscle after chronic coronary artery occlusion. Am. J. Physiol. Regul. Integr. Comp. Physiol 2006, 291, R1756–R1763. [Google Scholar]
- Bjornstedt, M.; Xue, J.; Huang, W.; Akesson, B.; Holmgren, A. The thioredoxin and glutaredoxin systems are efficient electron donors to human plasma glutathione peroxidase. J. Biol. Chem 1994, 269, 29382–29384. [Google Scholar]
- Bjornstedt, M.; Kumar, S.; Bjorkhem, L.; Spyrou, G.; Holmgren, A. Selenium and the thioredoxin and glutaredoxin systems. Biomed. Environ. Sci 1997, 10, 271–279. [Google Scholar]
- Lawler, J.M.; Powers, S.K.; Van, D.H.; Visser, T.; Kordus, M.J.; Ji, L.L. Metabolic and antioxidant enzyme activities in the diaphragm: Effects of acute exercise. Respir. Physiol 1994, 96, 139–149. [Google Scholar]
- Karanth, J.; Kumar, R.; Jeevaratnam, K. Response of antioxidant system in rats to dietary fat and physical activity. Indian J. Physiol. Pharmacol 2004, 48, 446–452. [Google Scholar]
- Lambertucci, R.H.; Levada-Pires, A.C.; Rossoni, L.V.; Curi, R.; Pithon-Curi, T.C. Effects of aerobic exercise training on antioxidant enzyme activities and mRNA levels in soleus muscle from young and aged rats. Mech. Ageing Dev 2007, 128, 267–275. [Google Scholar]
- Leeuwenburgh, C.; Fiebig, R.; Chandwaney, R.; Ji, L.L. Aging and exercise training in skeletal muscle: Responses of glutathione and antioxidant enzyme systems. Am. J. Physiol 1994, 267, R439–R445. [Google Scholar]
- Vincent, H.K.; Powers, S.K.; Stewart, D.J.; Demirel, H.A.; Shanely, R.A.; Naito, H. Short-term exercise training improves diaphragm antioxidant capacity and endurance. Eur. J. Appl. Physiol 2000, 81, 67–74. [Google Scholar]
- Tibballs, J. The role of nitric oxide (formerly endothelium-derived relaxing factor-EDRF) in vasodilatation and vasodilator therapy. Anaesth. Intensive Care 1993, 21, 759–773. [Google Scholar]
- Hirai, D.M.; Copp, S.W.; Ferguson, S.K.; Holdsworth, C.T.; McCullough, D.J.; Behnke, B.J.; Musch, T.I.; Poole, D.C. Exercise training and muscle microvascular oxygenation: Functional role of nitric oxide. J. Appl. Physiol 2012, 113, 557–565. [Google Scholar]
- Ferguson, S.K.; Hirai, D.M.; Copp, S.W.; Holdsworth, C.T.; Allen, J.D.; Jones, A.M.; Musch, T.I.; Poole, D.C. Impact of dietary nitrate supplementation via beetroot juice on exercising muscle vascular control in rats. J. Physiol 2013, 591, 547–557. [Google Scholar]
- Wolosker, H.; Panizzutti, R.; Engelender, S. Inhibition of creatine kinase by S-nitrosoglutathione. FEBS Lett 1996, 392, 274–276. [Google Scholar]
- Gross, W.L.; Bak, M.I.; Ingwall, J.S.; Arstall, M.A.; Smith, T.W.; Balligand, J.L.; Kelly, R.A. Nitric oxide inhibits creatine kinase and regulates rat heart contractile reserve. Proc. Natl. Acad. Sci. USA 1996, 93, 5604–5609. [Google Scholar]
- Wilson, C.M.; Cushman, S.W. Insulin stimulation of glucose transport activity in rat skeletal muscle: Increase in cell surface GLUT4 as assessed by photolabelling. Biochem. J 1994, 299, 755–759. [Google Scholar]
- Youn, J.H.; Gulve, E.A.; Holloszy, J.O. Calcium stimulates glucose transport in skeletal muscle by a pathway independent of contraction. Am. J. Physiol 1991, 260, C555–C561. [Google Scholar]
- Lund, S.; Holman, G.D.; Schmitz, O.; Pedersen, O. Contraction stimulates translocation of glucose transporter GLUT4 in skeletal muscle through a mechanism distinct from that of insulin. Proc. Natl. Acad. Sci. USA 1995, 92, 5817–5821. [Google Scholar]
- Roy, D.; Marette, A. Exercise induces the translocation of GLUT4 to transverse tubules from an intracellular pool in rat skeletal muscle. Biochem. Biophys. Res. Commun 1996, 223, 147–152. [Google Scholar]
- Lemieux, K.; Konrad, D.; Klip, A.; Marette, A. The AMP-activated protein kinase activator AICAR does not induce GLUT4 translocation to transverse tubules but stimulates glucose uptake and p38 mitogen-activated protein kinases alpha and beta in skeletal muscle. FASEB J 2003, 17, 1658–1665. [Google Scholar]
- Etgen, G.J., Jr; Fryburg, D.A.; Gibbs, E.M. Nitric oxide stimulates skeletal muscle glucose transport through a calcium/contraction- and phosphatidylinositol-3-kinase-independent pathway. Diabetes 1997, 46, 1915–1919. [Google Scholar]
- Mohr, S.; Stamler, J.S.; Brune, B. Posttranslational modification of glyceraldehyde-3-phosphate dehydrogenase by S-nitrosylation and subsequent NADH attachment. J. Biol. Chem 1996, 271, 4209–4214. [Google Scholar]
- Brown, G.C. Nitric oxide regulates mitochondrial respiration and cell functions by inhibiting cytochrome oxidase. FEBS Lett 1995, 369, 136–139. [Google Scholar]
- Palacios-Callender, M.; Hollis, V.; Frakich, N.; Mateo, J.; Moncada, S. Cytochrome c oxidase maintains mitochondrial respiration during partial inhibition by nitric oxide. J. Cell Sci 2007, 120, 160–165. [Google Scholar]
- Glass, D.J. Signalling pathways that mediate skeletal muscle hypertrophy and atrophy. Nat. Cell Biol 2003, 5, 87–90. [Google Scholar]
- Sellman, J.E.; DeRuisseau, K.C.; Betters, J.L.; Lira, V.A.; Soltow, Q.A.; Selsby, J.T.; Criswell, D.S. In vivo inhibition of nitric oxide synthase impairs upregulation of contractile protein mRNA in overloaded plantaris muscle. J. Appl. Physiol 2006, 100, 258–265. [Google Scholar]
- Leiter, J.R.; Upadhaya, R.; Anderson, J.E. Nitric oxide and voluntary exercise together promote quadriceps hypertrophy and increase vascular density in female 18-mo-old mice. Am. J. Physiol. Cell Physiol 2012, 302, C1306–C1315. [Google Scholar]
- Ito, N.; Ruegg, U.T.; Kudo, A.; Miyagoe-Suzuki, Y.; Takeda, S. Activation of calcium signaling through Trpv1 by nNOS and peroxynitrite as a key trigger of skeletal muscle hypertrophy. Nat. Med 2013, 19, 101–106. [Google Scholar]
- Samengo, G.; Avik, A.; Fedor, B.; Whittaker, D.; Myung, K.H.; Wehling-Henricks, M.; Tidball, J.G. Age-related loss of nitric oxide synthase in skeletal muscle causes reductions in calpain S-nitrosylation that increase myofibril degradation and sarcopenia. Aging Cell 2012, 11, 1036–1045. [Google Scholar]
- Gundersen, K.; Bruusgaard, J.C. Nuclear domains during muscle atrophy: Nuclei lost or paradigm lost? J. Physiol 2008, 586, 2675–2681. [Google Scholar]
- Qaisar, R.; Renaud, G.; Morine, K.; Barton, E.R.; Sweeney, H.L.; Larsson, L. Is functional hypertrophy and specific force coupled with the addition of myonuclei at the single muscle fiber level? FASEB J 2012, 26, 1077–1085. [Google Scholar]
- Walker, D.K.; Fry, C.S.; Drummond, M.J.; Dickinson, J.M.; Timmerman, K.L.; Gundermann, D.M.; Jennings, K.; Volpi, E.; Rasmussen, B.B. PAX7+ satellite cells in young and older adults following resistance exercise. Muscle Nerve 2012, 46, 51–59. [Google Scholar]
- Mackey, A.L.; Esmarck, B.; Kadi, F.; Koskinen, S.O.; Kongsgaard, M.; Sylvestersen, A.; Hansen, J.J.; Larsen, G.; Kjaer, M. Enhanced satellite cell proliferation with resistance training in elderly men and women. Scand. J. Med. Sci. Sports 2007, 17, 34–42. [Google Scholar]
- Martins, K.J.; St-Louis, M.; Murdoch, G.K.; MacLean, I.M.; McDonald, P.; Dixon, W.T.; Putman, C.T.; Michel, R.N. Nitric oxide synthase inhibition prevents activity-induced calcineurin-NFATc1 signalling and fast-to-slow skeletal muscle fibre type conversions. J. Physiol 2012, 590, 1427–1442. [Google Scholar]
- Zammit, P.S.; Partridge, T.A.; Yablonka-Reuveni, Z. The skeletal muscle satellite cell: The stem cell that came in from the cold. J. Histochem. Cytochem 2006, 54, 1177–1191. [Google Scholar]
- Friday, B.B.; Pavlath, G.K. A calcineurin- and NFAT-dependent pathway regulates Myf5 gene expression in skeletal muscle reserve cells. J. Cell Sci 2001, 114, 303–310. [Google Scholar]
- Tengan, C.H.; Rodrigues, G.S.; Godinho, R.O. Nitric oxide in skeletal muscle: Role on mitochondrial biogenesis and function. Int. J. Mol. Sci 2012, 13, 17160–17184. [Google Scholar]
- Cerqueira, F.M.; Laurindo, F.R.; Kowaltowski, A.J. Mild mitochondrial uncoupling and calorie restriction increase fasting eNOS, akt and mitochondrial biogenesis. PLoS One 2011, 6, e18433. [Google Scholar]
- Hood, D.A. Mechanisms of exercise-induced mitochondrial biogenesis in skeletal muscle. Appl. Physiol. Nutr. Metab 2009, 34, 465–472. [Google Scholar]
- Hood, D.A. Invited review: Contractile activity-induced mitochondrial biogenesis in skeletal muscle. J. Appl. Physiol 2001, 90, 1137–1157. [Google Scholar]
- Gomes, E.C.; Silva, A.N.; de Oliveira, M.R. Oxidants, antioxidants, and the beneficial roles of exercise-induced production of reactive species. Oxid. Med. Cell Longev 2012, 2012, 756132. [Google Scholar]
- Jorgensen, S.B.; Richter, E.A.; Wojtaszewski, J.F. Role of AMPK in skeletal muscle metabolic regulation and adaptation in relation to exercise. J. Physiol 2006, 574, 17–31. [Google Scholar]
- Kulisz, A.; Chen, N.; Chandel, N.S.; Shao, Z.; Schumacker, P.T. Mitochondrial ROS initiate phosphorylation of p38 MAP kinase during hypoxia in cardiomyocytes. Am. J. Physiol. Lung Cell Mol. Physiol 2002, 282, L1324–L1329. [Google Scholar]
- Liu, C.; Lin, J.D. PGC-1 coactivators in the control of energy metabolism. Acta Biochim. Biophys. Sin. (Shanghai) 2011, 43, 248–257. [Google Scholar]
- Sriwijitkamol, A.; Coletta, D.K.; Wajcberg, E.; Balbontin, G.B.; Reyna, S.M.; Barrientes, J.; Eagan, P.A.; Jenkinson, C.P.; Cersosimo, E.; DeFronzo, R.A.; et al. Effect of acute exercise on AMPK signaling in skeletal muscle of subjects with type 2 diabetes: A time-course and dose-response study. Diabetes 2007, 56, 836–848. [Google Scholar]
- McConell, G.K.; Ng, G.P.; Phillips, M.; Ruan, Z.; Macaulay, S.L.; Wadley, G.D. Central role of nitric oxide synthase in AICAR and caffeine-induced mitochondrial biogenesis in L6 myocytes. J. Appl. Physiol 2010, 108, 589–595. [Google Scholar]
- Wadley, G.D.; Choate, J.; McConell, G.K. NOS isoform-specific regulation of basal but not exercise-induced mitochondrial biogenesis in mouse skeletal muscle. J. Physiol 2007, 585, 253–262. [Google Scholar]
- Clementi, E.; Nisoli, E. Nitric oxide and mitochondrial biogenesis: A key to long-term regulation of cellular metabolism. Comp. Biochem. Physiol. A 2005, 142, 102–110. [Google Scholar]
- Mortensen, O.H.; Frandsen, L.; Schjerling, P.; Nishimura, E.; Grunnet, N. PGC-1alpha and PGC-1beta have both similar and distinct effects on myofiber switching toward an oxidative phenotype. Am. J. Physiol. Endocrinol. Metab 2006, 291, E807–E816. [Google Scholar]
- Gehlert, S.; Weber, S.; Weidmann, B.; Gutsche, K.; Platen, P.; Graf, C.; Kappes-Horn, K.; Bloch, W. Cycling exercise-induced myofiber transitions in skeletal muscle depend on basal fiber type distribution. Eur. J. Appl. Physiol 2011, 112, 2393–2402. [Google Scholar]
- Murgia, M.; Serrano, A.L.; Calabria, E.; Pallafacchina, G.; Lomo, T.; Schiaffino, S. Ras is involved in nerve-activity-dependent regulation of muscle genes. Nat. Cell Biol 2000, 2, 142–147. [Google Scholar]
- Calabria, E.; Ciciliot, S.; Moretti, I.; Garcia, M.; Picard, A.; Dyar, K.A.; Pallafacchina, G.; Tothova, J.; Schiaffino, S.; Murgia, M. NFAT isoforms control activity-dependent muscle fiber type specification. Proc. Natl. Acad. Sci. USA 2009, 106, 13335–13340. [Google Scholar]
- Drenning, J.A.; Lira, V.A.; Simmons, C.G.; Soltow, Q.A.; Sellman, J.E.; Criswell, D.S. Nitric oxide facilitates NFAT-dependent transcription in mouse myotubes. Am. J. Physiol. Cell Physiol 2008, 294, C1088–C1095. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Suhr, F.; Gehlert, S.; Grau, M.; Bloch, W. Skeletal Muscle Function during Exercise—Fine-Tuning of Diverse Subsystems by Nitric Oxide. Int. J. Mol. Sci. 2013, 14, 7109-7139. https://doi.org/10.3390/ijms14047109
Suhr F, Gehlert S, Grau M, Bloch W. Skeletal Muscle Function during Exercise—Fine-Tuning of Diverse Subsystems by Nitric Oxide. International Journal of Molecular Sciences. 2013; 14(4):7109-7139. https://doi.org/10.3390/ijms14047109
Chicago/Turabian StyleSuhr, Frank, Sebastian Gehlert, Marijke Grau, and Wilhelm Bloch. 2013. "Skeletal Muscle Function during Exercise—Fine-Tuning of Diverse Subsystems by Nitric Oxide" International Journal of Molecular Sciences 14, no. 4: 7109-7139. https://doi.org/10.3390/ijms14047109
APA StyleSuhr, F., Gehlert, S., Grau, M., & Bloch, W. (2013). Skeletal Muscle Function during Exercise—Fine-Tuning of Diverse Subsystems by Nitric Oxide. International Journal of Molecular Sciences, 14(4), 7109-7139. https://doi.org/10.3390/ijms14047109