Implication of Tumor Microenvironment in Chemoresistance: Tumor-Associated Stromal Cells Protect Tumor Cells from Cell Death

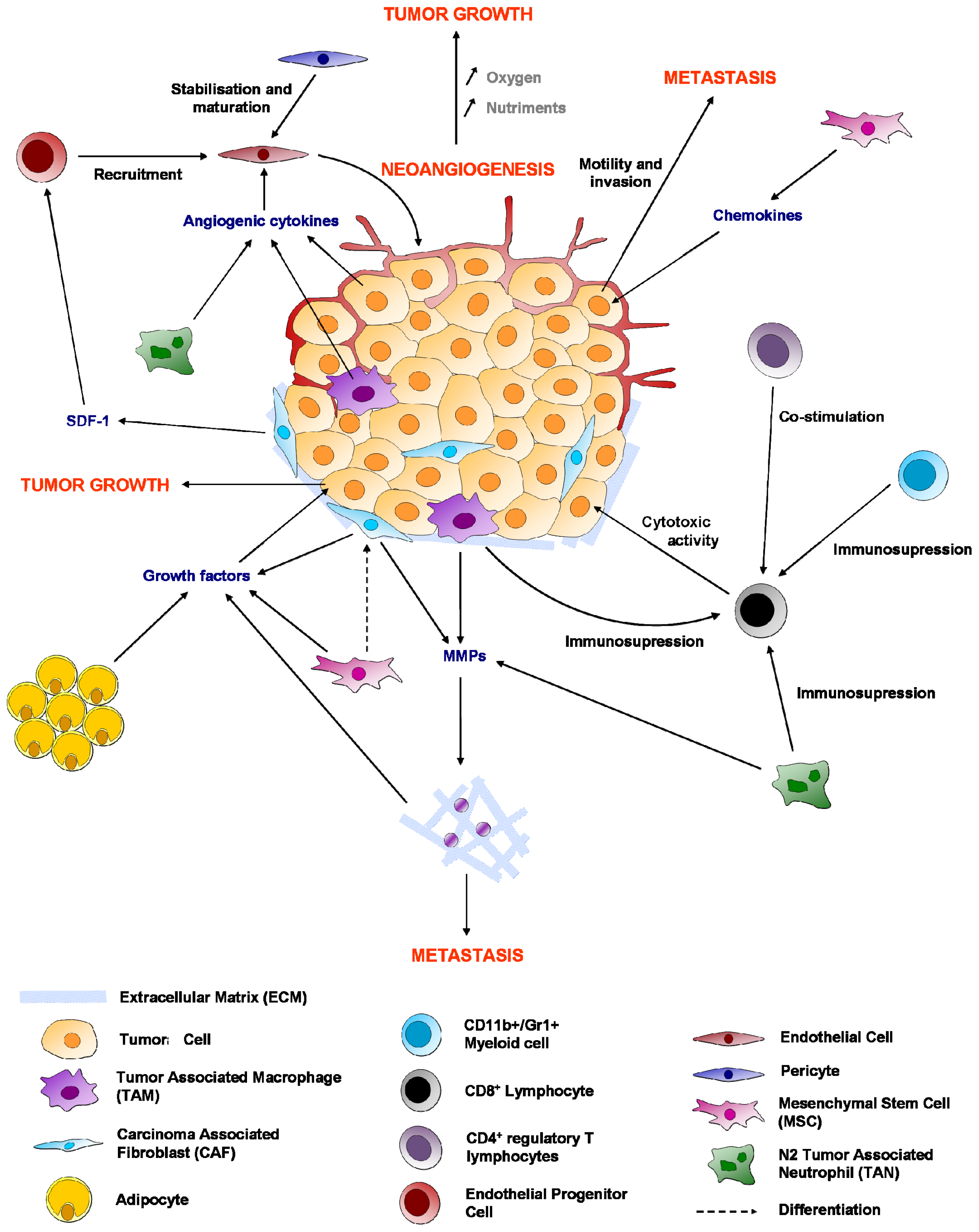{kind=link}
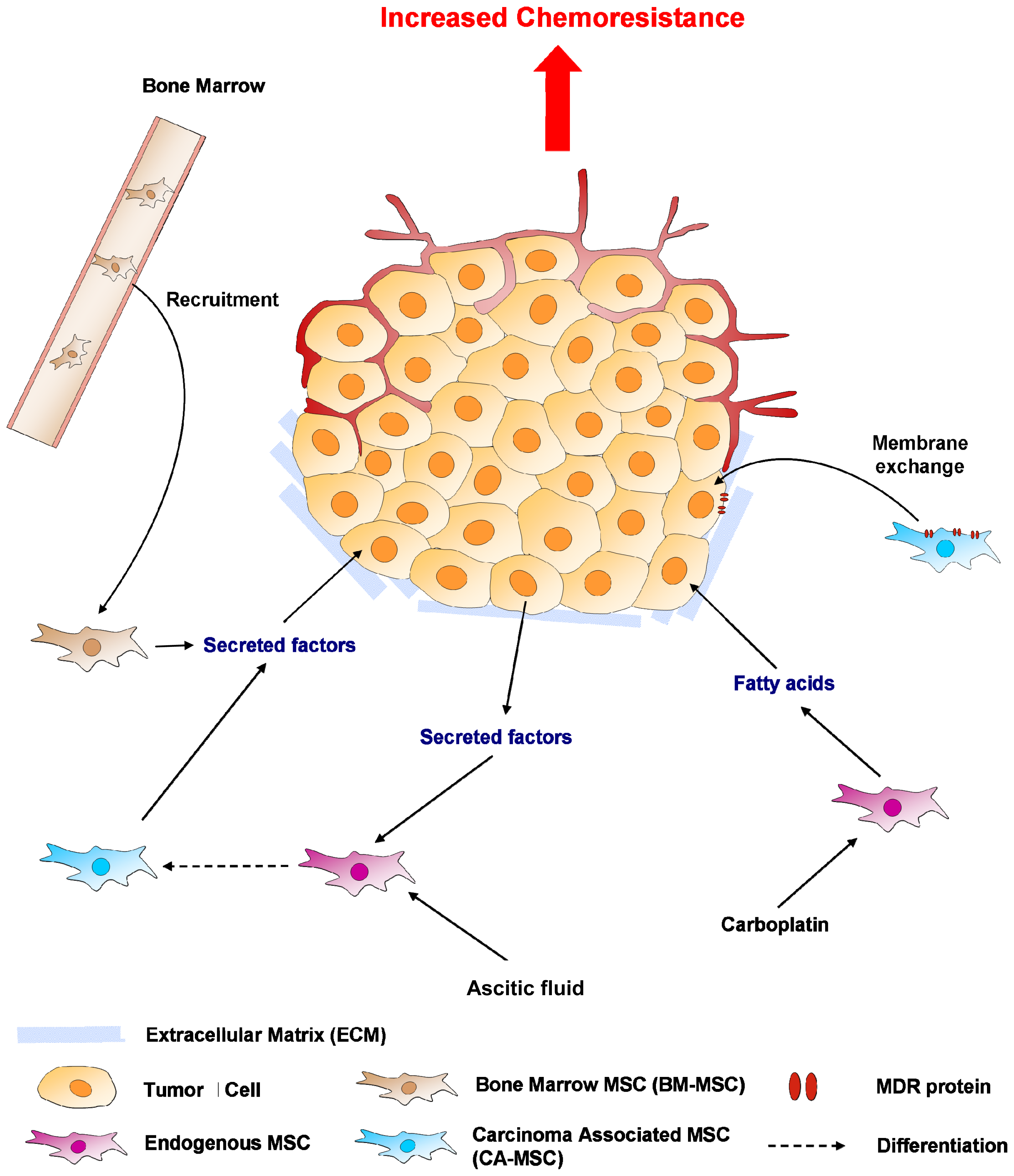{kind=link}
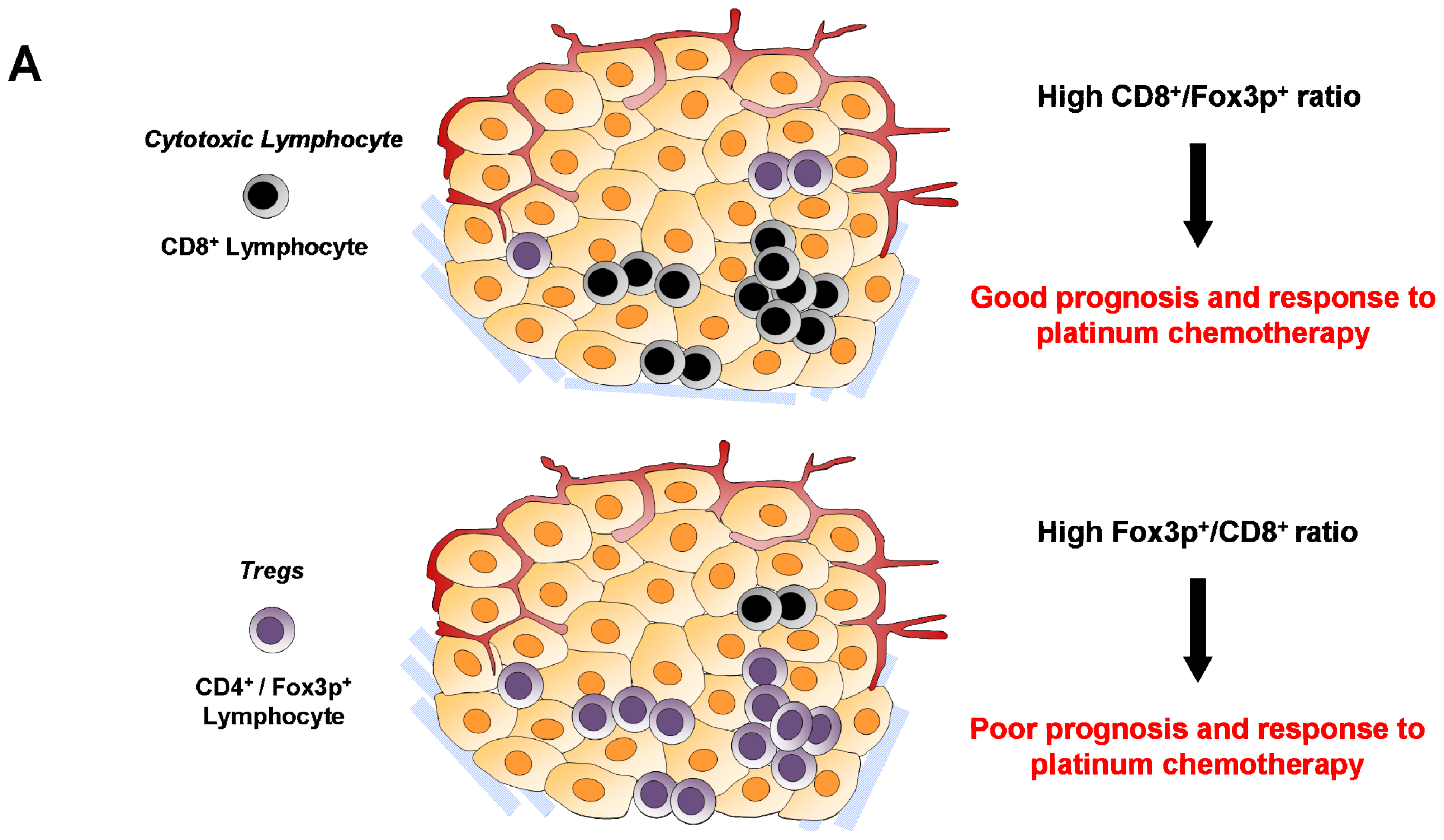{kind=link}
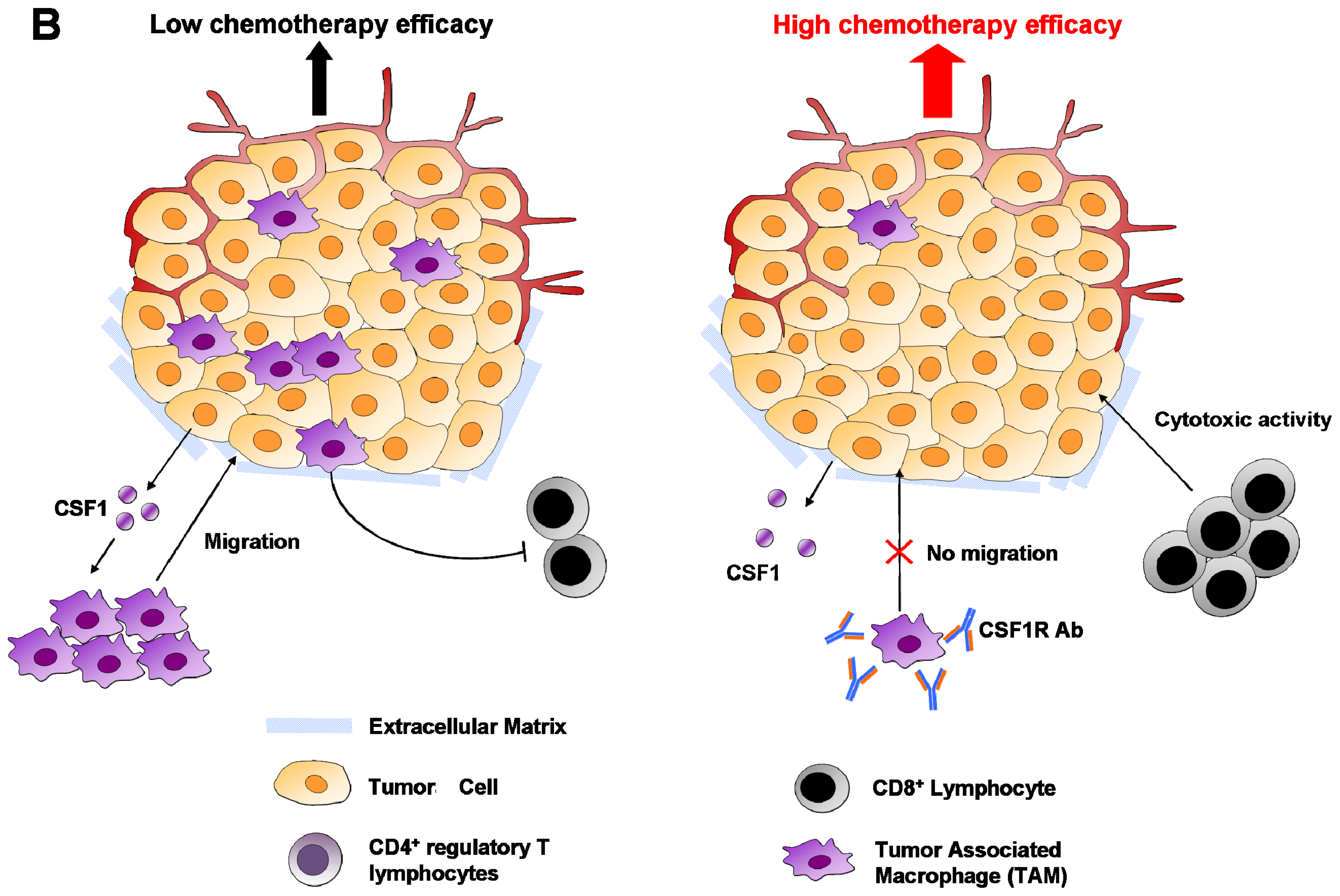{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Cell Adhesion and Chemoresistance
3. Endothelial Cells
3.1. Growth Factors Involved in Angiogenesis Are Pro Survival
3.2. Endothelial Cell Protection against Apoptosis
3.3. Treatments
3.4. From Hypoxia to Angiogenesis
4. Fibroblasts
4.1. Non Activated Fibroblasts
4.2. Myofibroblasts or Carcinoma-Associated Fibroblasts (CAFs)
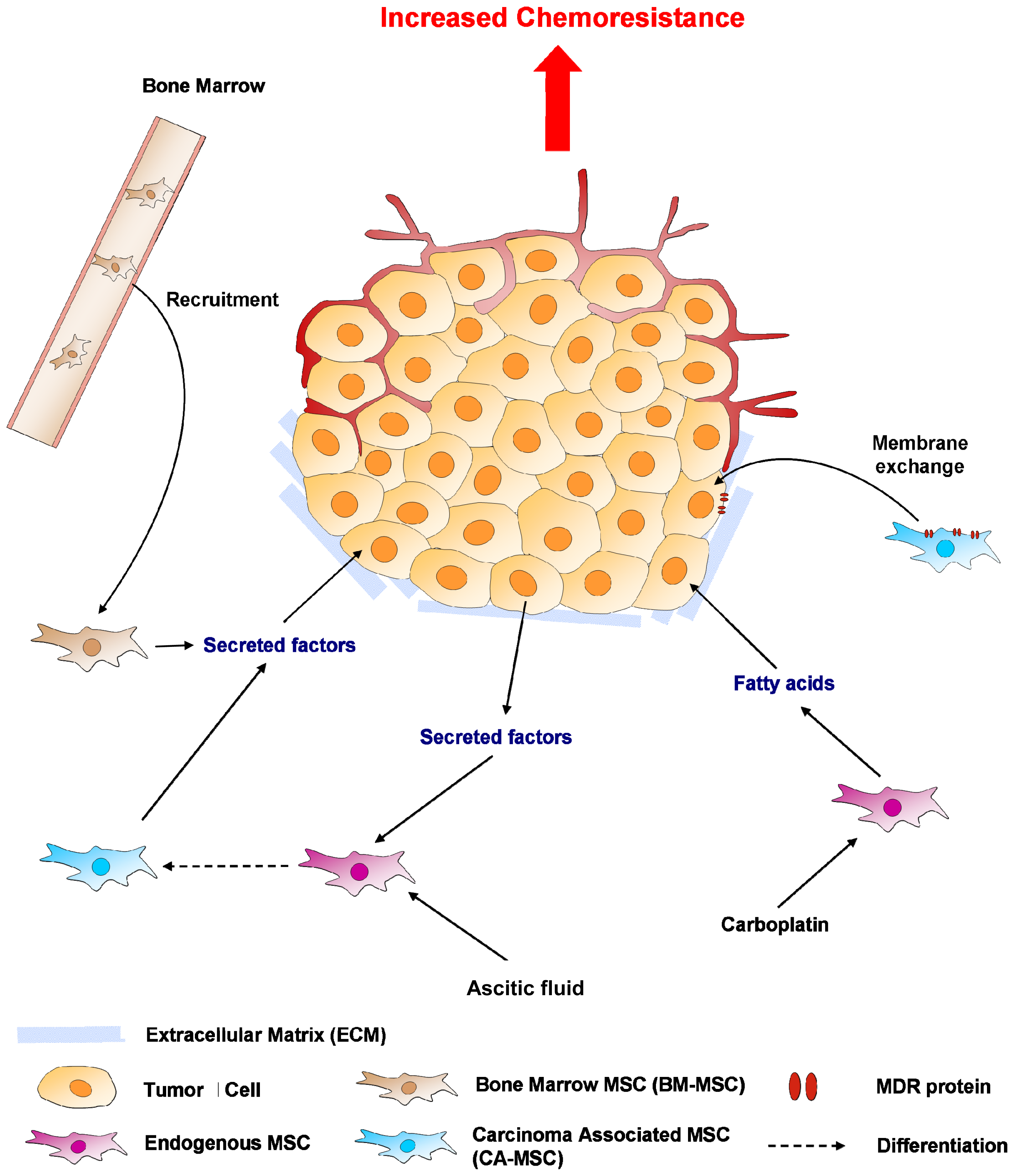
5. Mesenchymal Stem Cells (MSCs) or MSC-Like Cells (CA-MSCs)
5.1. Cell-Cell Contact
5.2. Soluble Factors Released Locally
5.3. Hypoxia
5.4. Conversion of Mesenchymal Cells to Cancer-Initiating Cells
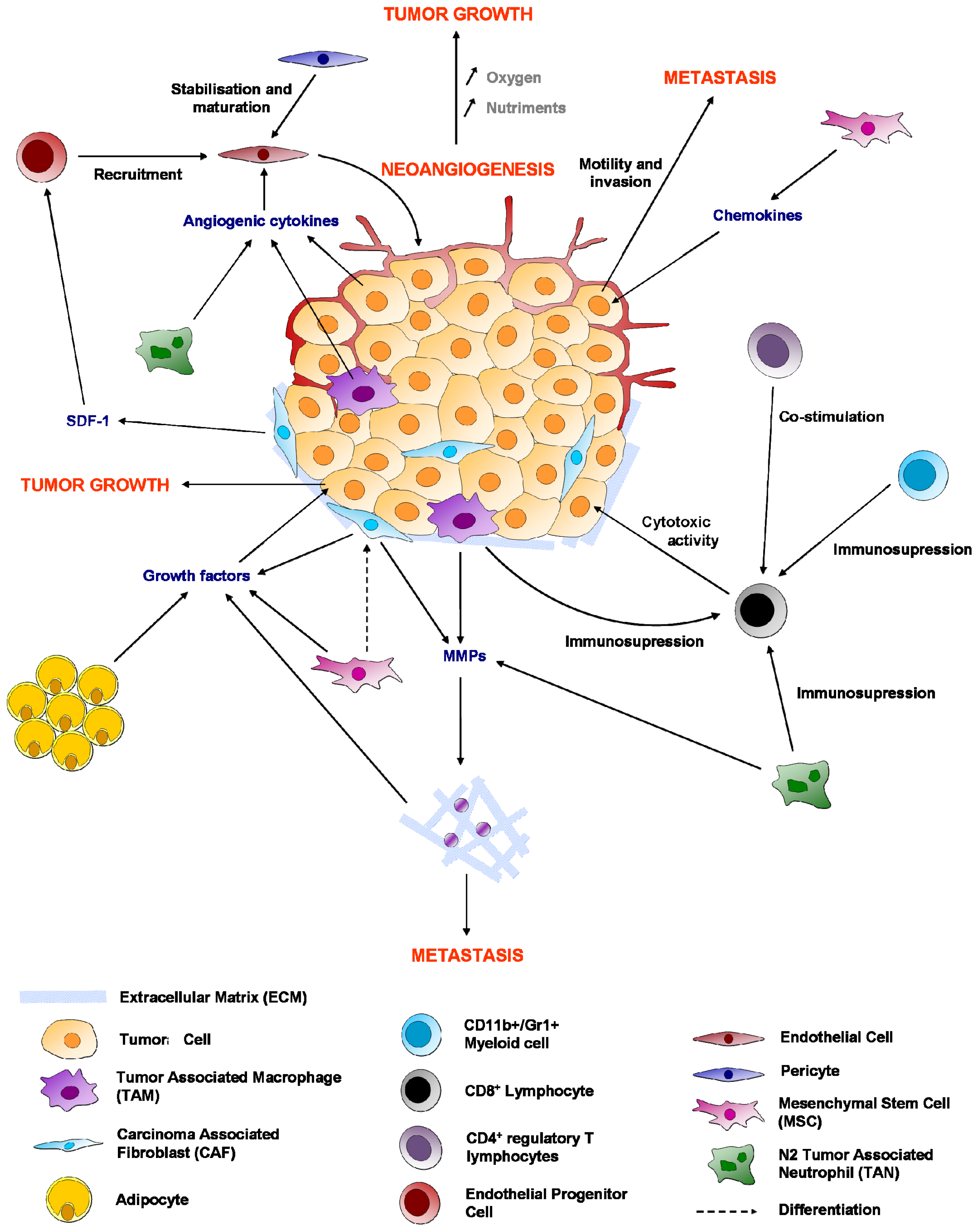
6. Immune System
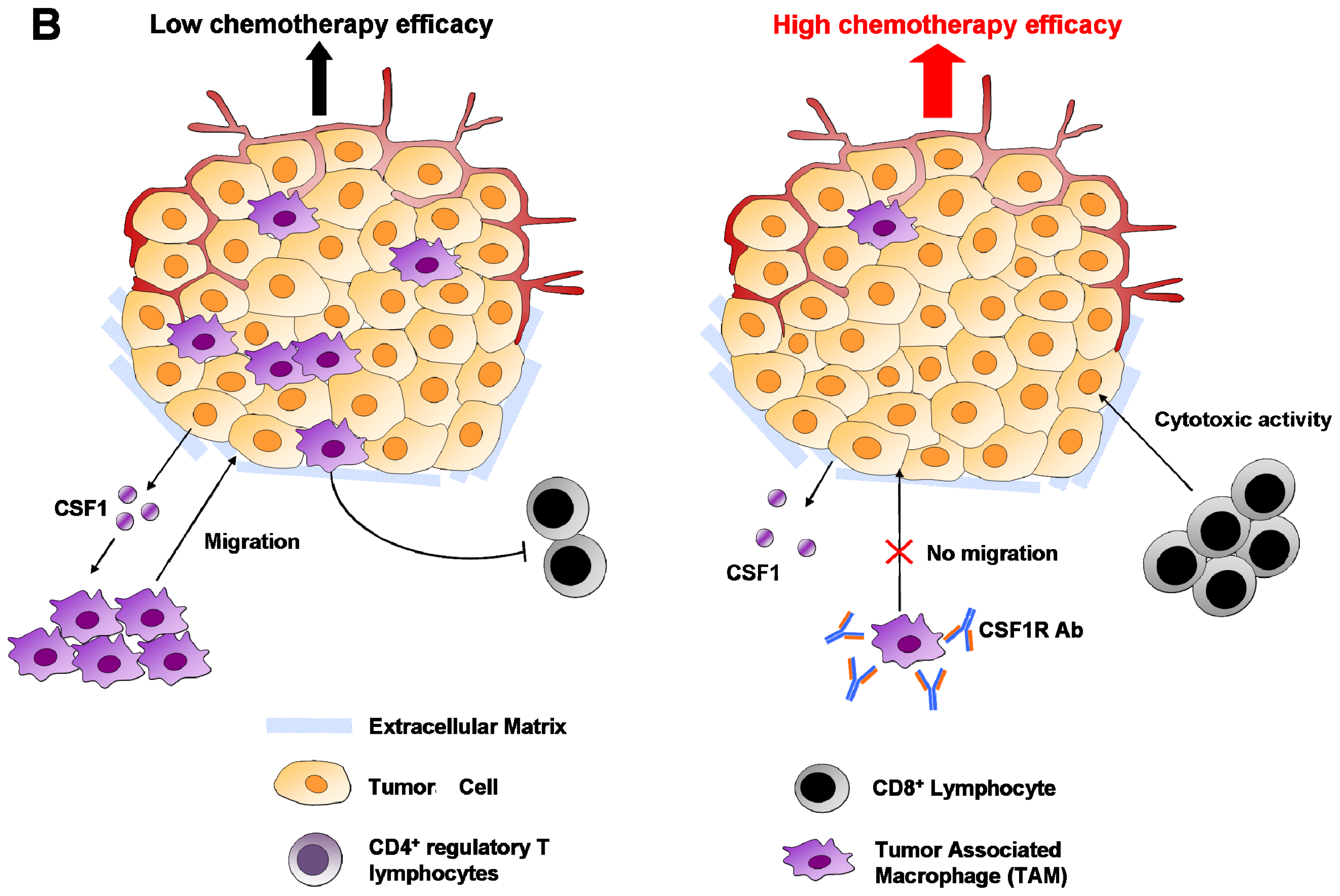
6.1. Macrophages
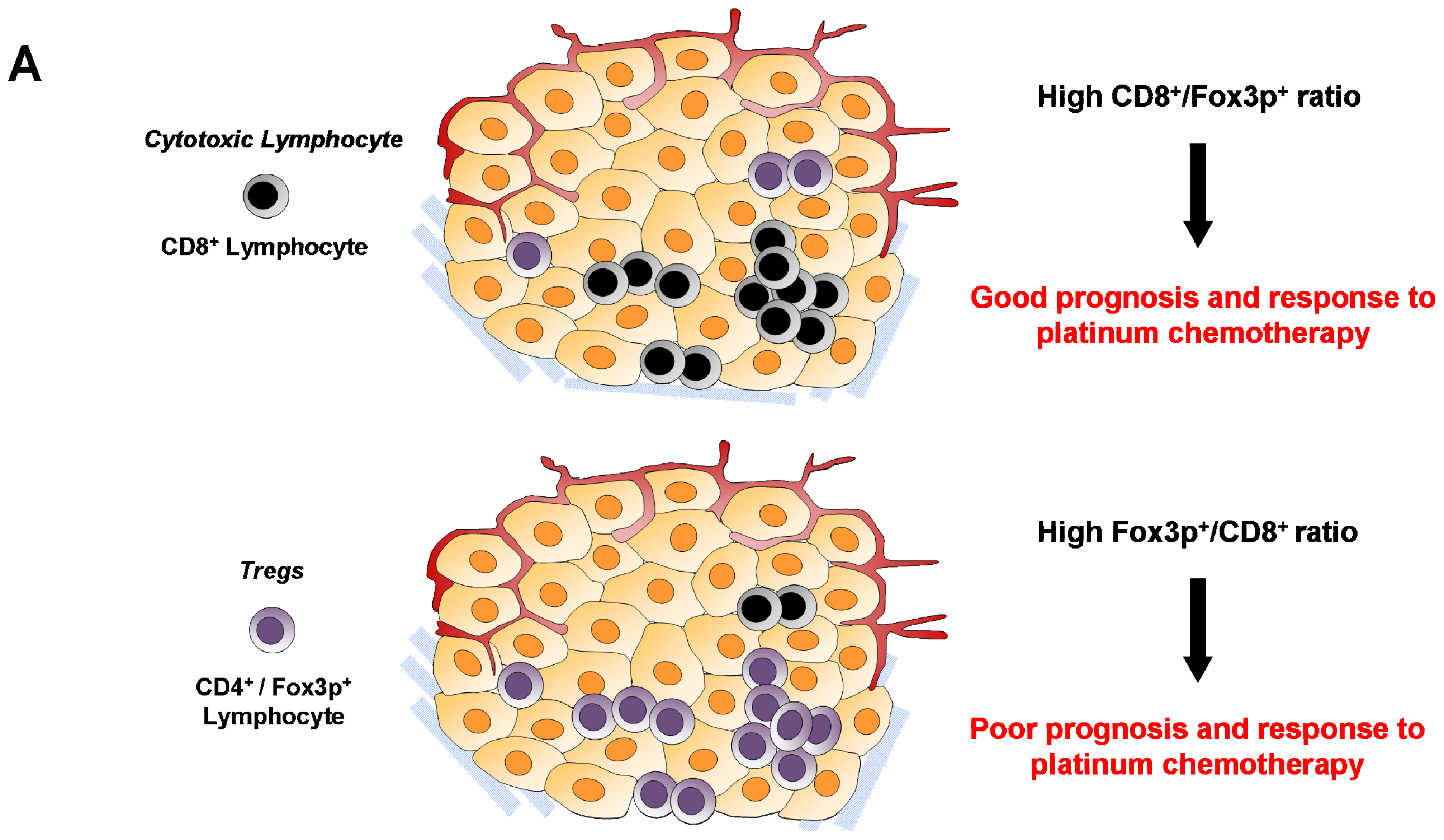
6.2. Cytotoxic CD8+ T Lymphocytes
6.3. γδ T Lymphocytes
6.4. Regulatory T Cells (Treg)
6.5. T Helpers (Th) Cells
6.6. Mast Cells
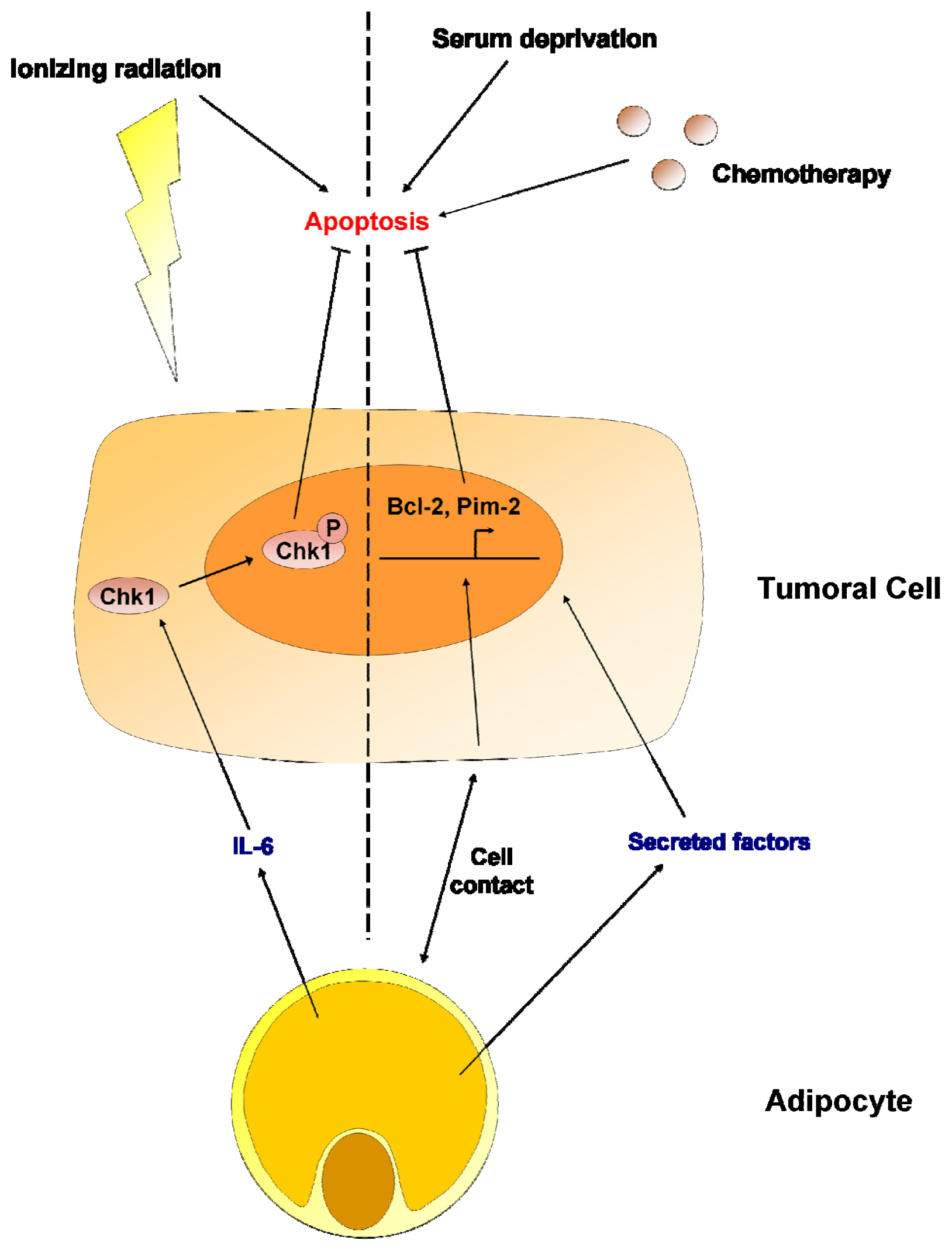
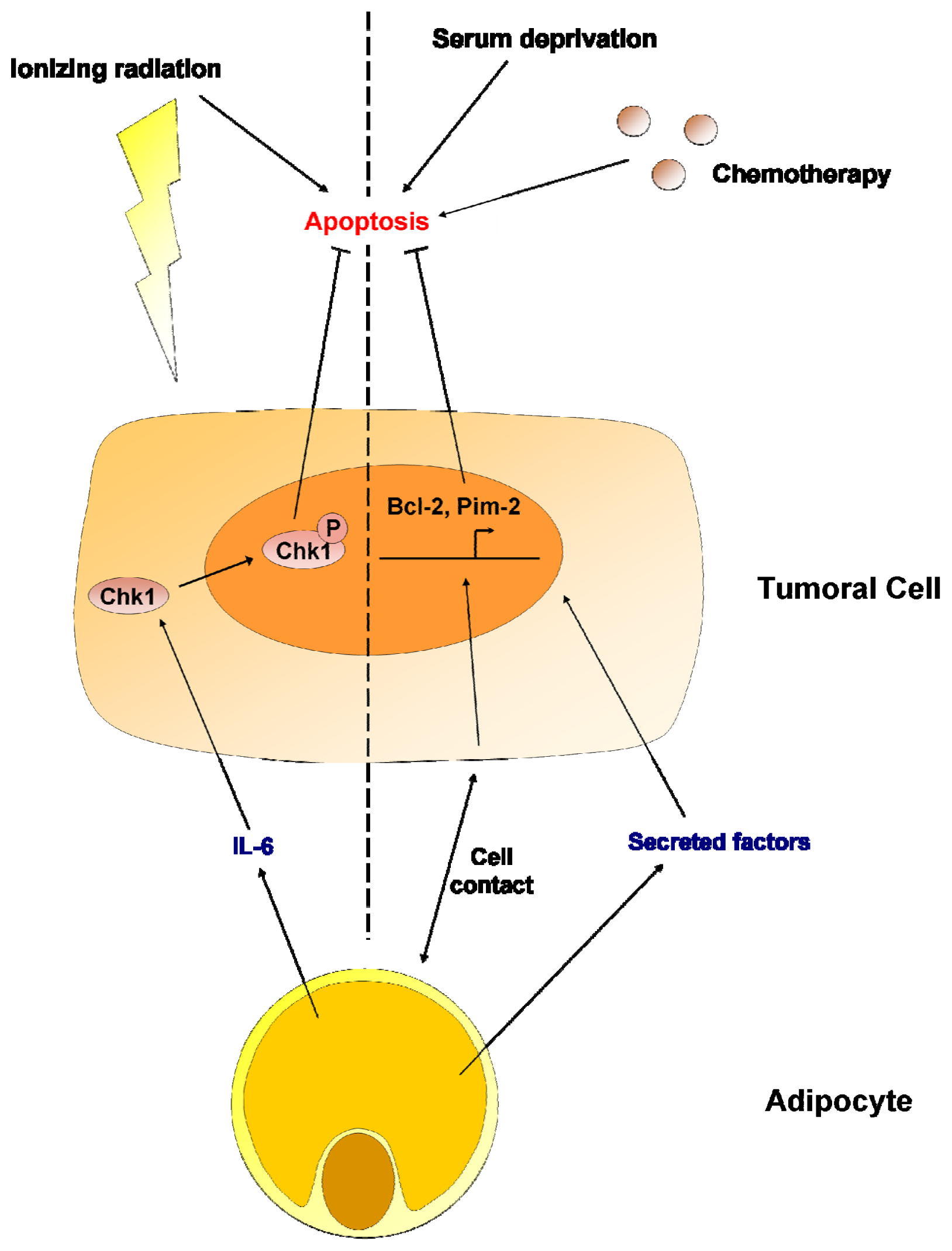
7. Adipocytes
8. miRNAs
9. Conclusions
Acknowledgments
References
- Meads, M.B.; Gatenby, R.A.; Dalton, W.S. Environment-mediated drug resistance: A major contributor to minimal residual disease. Nat. Rev. Cancer 2009, 9, 665–674. [Google Scholar]
- Basak, G.W.; Srivastava, A.S.; Malhotra, R.; Carrier, E. Multiple myeloma bone marrow niche. Curr. Pharm. Biotechnol 2009, 3, 345–346. [Google Scholar]
- Roodhart, J.M.; Daenen, L.G.; Stigter, E.C.; Prins, H.J.; Gerrits, J.; Houthuijzen, J.M.; Gerritsen, M.G.; Schipper, H.S.; Backer, M.J.; van Amersfoort, M.; et al. Mesenchymal stem cells induce resistance to chemotherapy through the release of platinum-induced fatty acids. Cancer Cell 2011, 3, 370–383. [Google Scholar]
- Castells, M.; Thibault, B.; Mery, E.; Golzio, M.; Pasquet, M.; Hennebelle, I.; Bourin, P.; Mirshahi, M.; Delord, J.P.; Querleu, D.; et al. Ovarian ascites-derived Hospicells promote angiogenesis via activation of macrophages. Cancer Lett 2012, in press. [Google Scholar]
- Lis, R.; Touboul, C.; Mirshahi, P.; Ali, F.; Mathew, S.; Nolan, D.J.; Maleki, M.; Abdalla, S.A.; Raynaud, C.M.; Querleu, D.; et al. Tumor associated mesenchymal stem cells protects ovarian cancer cells from hyperthermia through CXCL12. Int. J. Cancer 2011, 3, 715–725. [Google Scholar]
- Scherzed, A.; Hackenberg, S.; Froelich, K.; Kessler, M.; Koehler, C.; Hagen, R.; Radeloff, A.; Friehs, G.; Kleinsasser, N. BMSC enhance the survival of paclitaxel treated squamous cell carcinoma cells in vitro. Cancer Biol. Ther 2011, 3, 349–357. [Google Scholar]
- Rafii, A.; Mirshahi, P.; Poupot, M.; Faussat, A.M.; Simon, A.; Ducros, E.; Mery, E.; Couderc, B.; Lis, R.; Capdet, J.; et al. Oncologic trogocytosis of an original stromal cells induces chemoresistance of ovarian tumours. PLoS. One 2008, 12, e3894. [Google Scholar]
- Rohwer, N.; Cramer, T. Hypoxia-mediated drug resistance: Novel insights on the functional interaction of HIFs and cell death pathways. Drug Resist. Updat 2011, 3, 191–201. [Google Scholar]
- Tredan, O.; Galmarini, C.M.; Patel, K.; Tannock, I.F. Drug resistance and the solid tumor microenvironment. J. Natl. Cancer Inst 2007, 19, 1441–1454. [Google Scholar]
- Teng, I.W.; Hou, P.C.; Lee, K.D.; Chu, P.Y.; Yeh, K.T.; Jin, V.X.; Tseng, M.J.; Tsai, S.J.; Chang, Y.S.; Wu, C.S.; et al. Targeted methylation of two tumor suppressor genes is sufficient to transform mesenchymal stem cells into cancer stem/initiating cells. Cancer Res 2011, 13, 4653–4663. [Google Scholar]
- Correia, A.L.; Bissell, M.J. The tumor microenvironment is a dominant force in multidrug resistance. Drug Resist. Updat 2012, 15, 39–49. [Google Scholar]
- Sutherland, R.M.; Eddy, H.A.; Bareham, B.; Reich, K.; Vanantwerp, D. Resistance to adriamycin in multicellular spheroids. Int. J. Radiat. Oncol. Biol. Phys 1979, 8, 1225–1230. [Google Scholar]
- Stoeck, A.; Gast, D.; Sanderson, M.P.; Issa, Y.; Gutwein, P.; Altevogt, P. L1-CAM in a membrane-bound or soluble form augments protection from apoptosis in ovarian carcinoma cells. Gynecol. Oncol 2007, 2, 461–469. [Google Scholar]
- Schafer, H.; Dieckmann, C.; Korniienko, O.; Moldenhauer, G.; Kiefel, H.; Salnikov, A.; Kruger, A.; Altevogt, P.; Sebens, S. Combined treatment of L1CAM antibodies and cytostatic drugs improve the therapeutic response of pancreatic and ovarian carcinoma. Cancer Lett 2012, 1, 66–82. [Google Scholar]
- Eberle, K.E.; Sansing, H.A.; Szaniszlo, P.; Resto, V.A.; Berrier, A.L. Carcinoma matrix controls resistance to cisplatin through talin regulation of NF-kB. PLoS One 2011, 6, e21496. [Google Scholar]
- Hazlehurst, L.A.; Enkemann, S.A.; Beam, C.A.; Argilagos, R.F.; Painter, J.; Shain, K.H.; Saporta, S.; Boulware, D.; Moscinski, L.; Alsina, M.; et al. Genotypic and phenotypic comparisons of de novo and acquired melphalan resistance in an isogenic multiple myeloma cell line model. Cancer Res 2003, 22, 7900–7906. [Google Scholar]
- Holmgren, L.; O’Reilly, M.S.; Folkman, J. Dormancy of micrometastases: Balanced proliferation and apoptosis in the presence of angiogenesis suppression. Nat. Med 1995, 2, 149–153. [Google Scholar]
- Carmeliet, P.; Jain, R.K. Molecular mechanisms and clinical applications of angiogenesis. Nature 2011, 7347, 298–307. [Google Scholar]
- Ferrara, N.; Gerber, H.P.; LeCouter, J. The biology of VEGF and its receptors. Nat. Med 2003, 6, 669–676. [Google Scholar]
- Hicklin, D.J.; Ellis, L.M. Role of the vascular endothelial growth factor pathway in tumor growth and angiogenesis. J. Clin. Oncol 2005, 5, 1011–1027. [Google Scholar]
- Schwartz, J.D.; Rowinsky, E.K.; Youssoufian, H.; Pytowski, B.; Wu, Y. Vascular endothelial growth factor receptor-1 in human cancer: Concise review and rationale for development of IMC-18F1 (Human antibody targeting vascular endothelial growth factor receptor-1). Cancer 2010, 4, S1027–S1032. [Google Scholar]
- Dias, S.; Hattori, K.; Zhu, Z.; Heissig, B.; Choy, M.; Lane, W.; Wu, Y.; Chadburn, A.; Hyjek, E.; Gill, M.; et al. Autocrine stimulation of VEGFR-2 activates human leukemic cell growth and migration. J. Clin. Invest 2000, 4, 511–521. [Google Scholar]
- Harmey, J.H.; Bouchier-Hayes, D. Vascular endothelial growth factor (VEGF), a survival factor for tumour cells: Implications for anti-angiogenic therapy. Bioessays 2002, 3, 280–283. [Google Scholar]
- Zhang, L.; Hannay, J.A.; Liu, J.; Das, P.; Zhan, M.; Nguyen, T.; Hicklin, D.J.; Yu, D.; Pollock, R.E.; Lev, D. Vascular endothelial growth factor overexpression by soft tissue sarcoma cells: Implications for tumor growth, metastasis, and chemoresistance. Cancer Res 2006, 17, 8770–8778. [Google Scholar]
- Yang, F.; Tang, X.; Riquelme, E.; Behrens, C.; Nilsson, M.B.; Giri, U.; Varella-Garcia, M.; Byers, L.A.; Lin, H.Y.; Wang, J.; et al. Increased VEGFR-2 gene copy is associated with chemoresistance and shorter survival in patients with non-small-cell lung carcinoma who receive adjuvant chemotherapy. Cancer Res 2011, 16, 5512–5521. [Google Scholar]
- Tran, J.; Master, Z.; Yu, J.L.; Rak, J.; Dumont, D.J.; Kerbel, R.S. A role for survivin in chemoresistance of endothelial cells mediated by VEGF. Proc. Natl Acad Sci. USA 2002, 7, 4349–4354. [Google Scholar]
- Fukuda, S.; Pelus, L.M. Survivin, a cancer target with an emerging role in normal adult tissues. Mol. Cancer Ther 2006, 5, 1087–1098. [Google Scholar]
- Nor, J.E.; Christensen, J.; Mooney, D.J.; Polverini, P.J. Vascular endothelial growth factor (VEGF)-mediated angiogenesis is associated with enhanced endothelial cell survival and induction of Bcl-2 expression. Am. J. Pathol 1999, 2, 375–384. [Google Scholar]
- Samuel, S.; Fan, F.; Dang, L.H.; Xia, L.; Gaur, P.; Ellis, L.M. Intracrine vascular endothelial growth factor signaling in survival and chemoresistance of human colorectal cancer cells. Oncogene 2011, 10, 1205–1212. [Google Scholar]
- Dias, S.; Shmelkov, S.V.; Lam, G.; Rafii, S. VEGF(165) promotes survival of leukemic cells by Hsp90-mediated induction of Bcl-2 expression and apoptosis inhibition. Blood 2002, 7, 2532–2540. [Google Scholar]
- Fujio, Y.; Walsh, K. Akt mediates cytoprotection of endothelial cells by vascular endothelial growth factor in an anchorage-dependent manner. J. Biol. Chem 1999, 23, 16349–16354. [Google Scholar]
- Wang, L.; Chen, L.; Benincosa, J.; Fortney, J.; Gibson, L.F. VEGF-induced phosphorylation of Bcl-2 influences B lineage leukemic cell response to apoptotic stimuli. Leukemia 2005, 3, 344–353. [Google Scholar]
- Pidgeon, G.P.; Barr, M.P.; Harmey, J.H.; Foley, D.A.; Bouchier-Hayes, D.J. Vascular endothelial growth factor (VEGF) upregulates BCL-2 and inhibits apoptosis in human and murine mammary adenocarcinoma cells. Br. J. Cancer 2001, 2, 273–278. [Google Scholar]
- Lee, Y.K.; Shanafelt, T.D.; Bone, N.D.; Strege, A.K.; Jelinek, D.F.; Kay, N.E. VEGF receptors on chronic lymphocytic leukemia (CLL) B cells interact with STAT 1 and 3: Implication for apoptosis resistance. Leukemia 2005, 4, 513–523. [Google Scholar]
- Gerhardt, H.; Betsholtz, C. Endothelial-pericyte interactions in angiogenesis. Cell Tissue Res 2003, 1, 15–23. [Google Scholar]
- Franco, M.; Roswall, P.; Cortez, E.; Hanahan, D.; Pietras, K. Pericytes promote endothelial cell survival through induction of autocrine VEGF-A signaling and Bcl-w expression. Blood 2011, 10, 2906–2917. [Google Scholar]
- Ferrari, G.; Pintucci, G.; Seghezzi, G.; Hyman, K.; Galloway, A.C.; Mignatti, P. VEGF, a prosurvival factor, acts in concert with TGF-beta1 to induce endothelial cell apoptosis. Proc. Natl. Acad Sci. USA 2006, 46, 17260–17265. [Google Scholar]
- Michaelis, M.; Klassert, D.; Barth, S.; Suhan, T.; Breitling, R.; Mayer, B.; Hinsch, N.; Doerr, H.W.; Cinatl, J.; Cinatl, J., Jr. Chemoresistance acquisition induces a global shift of expression of aniogenesis-associated genes and increased pro-angogenic activity in neuroblastoma cells. Mol. Cancer 2009. [Google Scholar] [CrossRef]
- Biroccio, A.; Candiloro, A.; Mottolese, M.; Sapora, O.; Albini, A.; Zupi, G.; del Bufalo, D. Bcl-2 overexpression and hypoxia synergistically act to modulate vascular endothelial growth factor expression and in vivo angiogenesis in a breast carcinoma line. FASEB J 2000, 5, 652–660. [Google Scholar]
- Anai, S.; Goodison, S.; Shiverick, K.; Hirao, Y.; Brown, B.D.; Rosser, C.J. Knock-down of Bcl-2 by antisense oligodeoxynucleotides induces radiosensitization and inhibition of angiogenesis in human PC-3 prostate tumor xenografts. Mol. Cancer Ther 2007, 1, 101–111. [Google Scholar]
- Tu, S.P.; Cui, J.T.; Liston, P.; Huajiang, X.; Xu, R.; Lin, M.C.; Zhu, Y.B.; Zou, B.; Ng, S.S.; Jiang, S.H.; et al. Gene therapy for colon cancer by adeno-associated viral vector-mediated transfer of survivin Cys84Ala mutant. Gastroenterology 2005, 2, 361–375. [Google Scholar]
- Fuks, Z.; Persaud, R.S.; Alfieri, A.; McLoughlin, M.; Ehleiter, D.; Schwartz, J.L.; Seddon, A.P.; Cordon-Cardo, C.; Haimovitz-Friedman, A. Basic fibroblast growth factor protects endothelial cells against radiation-induced programmed cell death in vitro and in vivo. Cancer Res 1994, 10, 2582–2590. [Google Scholar]
- Karsan, A.; Yee, E.; Poirier, G.G.; Zhou, P.; Craig, R.; Harlan, J.M. Fibroblast growth factor-2 inhibits endothelial cell apoptosis by Bcl-2-dependent and independent mechanisms. Am. J. Pathol 1997, 6, 1775–1784. [Google Scholar]
- O’Connor, D.S.; Schechner, J.S.; Adida, C.; Mesri, M.; Rothermel, A.L.; Li, F.; Nath, A.K.; Pober, J.S.; Altieri, D.C. Control of apoptosis during angiogenesis by survivin expression in endothelial cells. Am. J. Pathol 2000, 2, 393–398. [Google Scholar]
- Carmeliet, P.; Lampugnani, M.G.; Moons, L.; Breviario, F.; Compernolle, V.; Bono, F.; Balconi, G.; Spagnuolo, R.; Oosthuyse, B.; Dewerchin, M.; et al. Targeted deficiency or cytosolic truncation of the VE-cadherin gene in mice impairs VEGF-mediated endothelial survival and angiogenesis. Cell 1999, 2, 147–157. [Google Scholar]
- Chavakis, E.; Dimmeler, S. Regulation of endothelial cell survival and apoptosis during angiogenesis. Arterioscler. Thromb. Vasc. Biol 2002, 6, 887–893. [Google Scholar]
- Brown, C.K.; Khodarev, N.N.; Yu, J.; Moo-Young, T.; Labay, E.; Darga, T.E.; Posner, M.C.; Weichselbaum, R.R.; Mauceri, H.J. Glioblastoma cells block radiation-induced programmed cell death of endothelial cells. FEBS Lett 2004, 1–3, 167–170. [Google Scholar]
- Fonsato, V.; Buttiglieri, S.; Deregibus, M.C.; Puntorieri, V.; Bussolati, B.; Camussi, G. Expression of Pax2 in human renal tumor-derived endothelial cells sustains apoptosis resistance and angiogenesis. Am. J. Pathol 2006, 2, 706–713. [Google Scholar]
- Virrey, J.J.; Guan, S.; Li, W.; Schonthal, A.H.; Chen, T.C.; Hofman, F.M. Increased survivin expression confers chemoresistance to tumor-associated endothelial cells. Am. J. Pathol 2008, 2, 575–585. [Google Scholar]
- Meng, F.; Henson, R.; Patel, T. Chemotherapeutic stress selectively activates NF-kappa B-dependent AKT and VEGF expression in liver cancer-derived endothelial cells. Am. J. Physiol. Cell Physiol 2007, 2, C749–C760. [Google Scholar]
- Kim, K.J.; Li, B.; Winer, J.; Armanini, M.; Gillett, N.; Phillips, H.S.; Ferrara, N. Inhibition of vascular endothelial growth factor-induced angiogenesis suppresses tumour growth in vivo. Nature 1993, 6423, 841–844. [Google Scholar]
- Lee, C.G.; Heijn, M.; di Tomaso, E.; Griffon-Etienne, G.; Ancukiewicz, M.; Koike, C.; Park, K.R.; Ferrara, N.; Jain, R.K.; Suit, H.D.; Boucher, Y. Anti-Vascular endothelial growth factor treatment augments tumor radiation response under normoxic or hypoxic conditions. Cancer Res 2000, 19, 5565–5570. [Google Scholar]
- Tong, R.T.; Boucher, Y.; Kozin, S.V.; Winkler, F.; Hicklin, D.J.; Jain, R.K. Vascular normalization by vascular endothelial growth factor receptor 2 blockade induces a pressure gradient across the vasculature and improves drug penetration in tumors. Cancer Res 2004, 11, 3731–3736. [Google Scholar]
- O’Reilly, M.S.; Holmgren, L.; Chen, C.; Folkman, J. Angiostatin induces and sustains dormancy of human primary tumors in mice. Nat. Med 1996, 6, 689–692. [Google Scholar]
- Carmeliet, P.; Jain, R.K. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat. Rev. Drug Discov 2011, 6, 417–427. [Google Scholar]
- Willett, C.G.; Boucher, Y.; di Tomaso, E.; Duda, D.G.; Munn, L.L.; Tong, R.T.; Chung, D.C.; Sahani, D.V.; Kalva, S.P.; Kozin, S.V.; et al. Direct evidence that the VEGF-specific antibody bevacizumab has antivascular effects in human rectal cancer. Nat. Med 2004, 2, 145–147. [Google Scholar]
- Xu, T.; Chen, J.; Lu, Y.; Wolff, J.E. Effects of bevacizumab plus irinotecan on response and survival in patients with recurrent malignant glioma: A systematic review and survival-gain analysis. BMC Cancer 2010. [Google Scholar] [CrossRef]
- Mayer, R.J. Two steps forward in the treatment of colorectal cancer. N. Engl. J. Med 2004, 23, 2406–2408. [Google Scholar]
- Jain, R.K. Normalization of tumor vasculature: An emerging concept in antiangiogenic therapy. Science 2005, 5706, 58–62. [Google Scholar]
- De Bock, K.; Mazzone, M.; Carmeliet, P. Antiangiogenic therapy, hypoxia, and metastasis: Risky liaisons, or not? Nat. Rev. Clin. Oncol 2011, 7, 393–404. [Google Scholar]
- Brahimi-Horn, M.C.; Bellot, G.; Pouyssegur, J. Hypoxia and energetic tumour metabolism. Curr. Opin. Genet. Dev 2011, 1, 67–72. [Google Scholar]
- Bottaro, D.P.; Liotta, L.A. Cancer: Out of air is not out of action. Nature 2003, 6940, 593–595. [Google Scholar]
- Semenza, G.L. Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 2003, 10, 721–732. [Google Scholar]
- Liu, Y.; Cox, S.R.; Morita, T.; Kourembanas, S. Hypoxia regulates vascular endothelial growth factor gene expression in endothelial cells. Identification of a 5′ enhancer. Circ. Res 1995, 3, 638–643. [Google Scholar]
- Eltzschig, H.K.; Carmeliet, P. Hypoxia and inflammation. N. Engl. J. Med 2011, 7, 656–665. [Google Scholar]
- Blagosklonny, M.V. Antiangiogenic therapy and tumor progression. Cancer Cell 2004, 1, 13–17. [Google Scholar]
- Ebos, J.M.; Lee, C.R.; Cruz-Munoz, W.; Bjarnason, G.A.; Christensen, J.G.; Kerbel, R.S. Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell 2009, 3, 232–239. [Google Scholar]
- Paez-Ribes, M.; Allen, E.; Hudock, J.; Takeda, T.; Okuyama, H.; Vinals, F.; Inoue, M.; Bergers, G.; Hanahan, D.; Casanovas, O. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell 2009, 3, 220–231. [Google Scholar]
- Miles, D.W.; Chan, A.; Dirix, L.Y.; Cortes, J.; Pivot, X.; Tomczak, P.; Delozier, T.; Sohn, J.H.; Provencher, L.; Puglisi, F.; et al. Phase III study of bevacizumab plus docetaxel compared with placebo plus docetaxel for the first-line treatment of human epidermal growth factor receptor 2-negative metastatic breast cancer. J. Clin. Oncol 2010, 20, 3239–3247. [Google Scholar]
- Padera, T.P.; Kuo, A.H.; Hoshida, T.; Liao, S.; Lobo, J.; Kozak, K.R.; Fukumura, D.; Jain, R.K. Differential response of primary tumor versus lymphatic metastasis to VEGFR-2 and VEGFR-3 kinase inhibitors cediranib and vandetanib. Mol. Cancer Ther 2008, 8, 2272–2279. [Google Scholar]
- Thomas, X.; Anglaret, B.; Magaud, J.P.; Epstein, J.; Archimbaud, E. Interdependence between cytokines and cell adhesion molecules to induce interleukin-6 production by stromal cells in myeloma. Leuk. Lymphoma 1998, 32, 107–119. [Google Scholar]
- Shain, K.H.; Landowski, T.H.; Dalton, W.S. The tumor microenvironment as a determinant of cancer cell survival: A possible mechanism for de novo drug resistance. Curr. Opin. Oncol 2000, 6, 557–563. [Google Scholar]
- Xing, F.; Saidou, J.; Watabe, K. Cancer associated fibroblasts (CAFs) in tumor microenvironment. Front Biosci 2010, 15, 166–179. [Google Scholar]
- Sirica, A.E. The role of cancer-associated myofibroblasts in intrahepatic cholangiocarcinoma. Nat. Rev. Gastroenterol. Hepatol 2012, 1, 44–54. [Google Scholar]
- Fingas, C.D.; Bronk, S.F.; Werneburg, N.W.; Mott, J.L.; Guicciardi, M.E.; Cazanave, S.C.; Mertens, J.C.; Sirica, A.E.; Gores, G.J. Myofibroblast-derived PDGF-BB promotes Hedgehog survival signaling in cholangiocarcinoma cells. Hepatology 2011, 6, 2076–2088. [Google Scholar]
- Ruan, K.; Bao, S.; Ouyang, G. The multifaceted role of periostin in tumorigenesis. Cell Mol. Life Sci 2009, 14, 2219–2230. [Google Scholar]
- Park, J.E.; Lenter, M.C.; Zimmermann, R.N.; Garin-Chesa, P.; Old, L.J.; Rettig, W.J. Fibroblast activation protein, a dual specificity serine protease expressed in reactive human tumor stromal fibroblasts. J. Biol. Chem 1999, 51, 36505–36512. [Google Scholar]
- Heldin, C.H.; Rubin, K.; Pietras, K.; Ostman, A. High interstitial fluid pressure-an obstacle in cancer therapy. Nat. Rev. Cancer 2004, 10, 806–813. [Google Scholar]
- Serebriiskii, I.; Castello-Cros, R.; Lamb, A.; Golemis, E.A.; Cukierman, E. Fibroblast-derived 3D matrix differentially regulates the growth and drug-responsiveness of human cancer cells. Matrix. Biol 2008, 6, 573–585. [Google Scholar]
- Muerkoster, S.S.; Werbing, V.; Koch, D.; Sipos, B.; Ammerpohl, O.; Kalthoff, H.; Tsao, M.S.; Folsch, U.R.; Schafer, H. Role of myofibroblasts in innate chemoresistance of pancreatic carcinoma--epigenetic downregulation of caspases. Int. J. Cancer 2008, 8, 1751–1760. [Google Scholar]
- Reardon, D.A.; Akabani, G.; Coleman, R.E.; Friedman, A.H.; Friedman, H.S.; Herndon, J.E.; McLendon, R.E.; Pegram, C.N.; Provenzale, J.M.; Quinn, J.A.; et al. Salvage radioimmunotherapy with murine iodine-131-labeled antitenascin monoclonal antibody 81C6 for patients with recurrent primary and metastatic malignant brain tumors: Phase II study results. J. Clin. Oncol 2006, 1, 115–122. [Google Scholar]
- Loeffler, M.; Kruger, J.A.; Niethammer, A.G.; Reisfeld, R.A. Targeting tumor-associated fibroblasts improves cancer chemotherapy by increasing intratumoral drug uptake. J. Clin. Invest 2006, 7, 1955–1962. [Google Scholar]
- Ringe, J.; Strassburg, S.; Neumann, K.; Endres, M.; Notter, M.; Burmester, G.R.; Kaps, C.; Sittinger, M. Towards in situ tissue repair: Human mesenchymal stem cells express chemokine receptors CXCR1, CXCR2 and CCR2, and migrate upon stimulation with CXCL8 but not CCL2. J. Cell Biochem 2007, 1, 135–146. [Google Scholar]
- Pasquet, M.; Golzio, M.; Mery, E.; Rafii, A.; Benabbou, N.; Mirshahi, P.; Hennebelle, I.; Bourin, P.; Allal, B.; Teissie, J.; et al. Hospicells (ascites-derived stromal cells) promote tumorigenicity and angiogenesis. Int. J. Cancer 2010, 9, 2090–2101. [Google Scholar]
- Rhodes, L.V.; Muir, S.E.; Elliott, S.; Guillot, L.M.; Antoon, J.W.; Penfornis, P.; Tilghman, S.L.; Salvo, V.A.; Fonseca, J.P.; Lacey, M.R.; et al. Adult human mesenchymal stem cells enhance breast tumorigenesis and promote hormone independence. Breast Cancer Res. Treat 2010, 2, 293–300. [Google Scholar]
- Karnoub, A.E.; Dash, A.B.; Vo, A.P.; Sullivan, A.; Brooks, M.W.; Bell, G.W.; Richardson, A.L.; Polyak, K.; Tubo, R.; Weinberg, R.A. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature 2007, 7162, 557–563. [Google Scholar]
- Xu, Y.; Tabe, Y.; Jin, L.; Watt, J.; McQueen, T.; Ohsaka, A.; Andreeff, M.; Konopleva, M. TGF-beta receptor kinase inhibitor LY2109761 reverses the anti-apoptotic effects of TGF-beta1 in myelo-monocytic leukaemic cells co-cultured with stromal cells. Br. J. Haematol 2008, 2, 192–201. [Google Scholar]
- Hao, M.; Zhang, L.; An, G.; Meng, H.; Han, Y.; Xie, Z.; Xu, Y.; Li, C.; Yu, Z.; Chang, H.; Qiu, L. Bone marrow stromal cells protect myeloma cells from bortezomib induced apoptosis by suppressing microRNA-15a expression. Leuk. Lymphoma 2011, 9, 1787–1794. [Google Scholar]
- Jin, L.; Tabe, Y.; Konoplev, S.; Xu, Y.; Leysath, C.E.; Lu, H.; Kimura, S.; Ohsaka, A.; Rios, M.B.; Calvert, L.; et al. CXCR4 up-regulation by imatinib induces chronic myelogenous leukemia (CML) cell migration to bone marrow stroma and promotes survival of quiescent CML cells. Mol. Cancer Ther 2008, 1, 48–58. [Google Scholar]
- Benito, J.; Shi, Y.; Szymanska, B.; Carol, H.; Boehm, I.; Lu, H.; Konoplev, S.; Fang, W.; Zweidler-McKay, P.A.; Campana, D.; et al. Pronounced hypoxia in models of murine and human leukemia: High efficacy of hypoxia-activated prodrug PR-104. PLoS One 2011, 8, e23108. [Google Scholar]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 5, 646–674. [Google Scholar]
- Igney, F.H.; Krammer, P.H. Immune escape of tumors: Apoptosis resistance and tumor counterattack. J. Leukoc. Biol 2002, 6, 907–920. [Google Scholar]
- Shojaei, F.; Wu, X.; Malik, A.K.; Zhong, C.; Baldwin, M.E.; Schanz, S.; Fuh, G.; Gerber, H.P.; Ferrara, N. Tumor refractoriness to anti-VEGF treatment is mediated by CD11b+Gr1+ myeloid cells. Nat. Biotechnol 2007, 8, 911–920. [Google Scholar]
- Fischer, C.; Jonckx, B.; Mazzone, M.; Zacchigna, S.; Loges, S.; Pattarini, L.; Chorianopoulos, E.; Liesenborghs, L.; Koch, M.; de Mol, M.; et al. Anti-PlGF inhibits growth of VEGF(R)-inhibitor-resistant tumors without affecting healthy vessels. Cell 2007, 3, 463–475. [Google Scholar]
- Biswas, S.K.; Sica, A.; Lewis, C.E. Plasticity of macrophage function during tumor progression: Regulation by distinct molecular mechanisms. J. Immunol 2008, 4, 2011–2017. [Google Scholar]
- Denardo, D.G.; Brennan, D.J.; Rexhepaj, E.; Ruffell, B.; Shiao, S.L.; Madden, S.F.; Gallagher, W.M.; Wadhwani, N.; Keil, S.D.; Junaid, S.A.; et al. Leukocyte Complexity Predicts Breast Cancer Survival and Functionally Regulates Response to Chemotherapy. Cancer Discov 2011. [Google Scholar] [CrossRef]
- Shree, T.; Olson, O.C.; Elie, B.T.; Kester, J.C.; Garfall, A.L.; Simpson, K.; Bell-McGuinn, K.M.; Zabor, E.C.; Brogi, E.; Joyce, J.A. Macrophages and cathepsin proteases blunt chemotherapeutic response in breast cancer. Genes Dev 2011, 23, 2465–2479. [Google Scholar]
- Angst, E.; Reber, H.A.; Hines, O.J.; Eibl, G. Mononuclear cell-derived interleukin-1 beta confers chemoresistance in pancreatic cancer cells by upregulation of cyclooxygenase-2. Surgery 2008, 1, 57–65. [Google Scholar]
- Halama, N.; Michel, S.; Kloor, M.; Zoernig, I.; Pommerencke, T.; von Knebel Doeberitz, M.; Schirmacher, P.; Weitz, J.; Grabe, N.; Jager, D. The localization and density of immune cells in primary tumors of human metastatic colorectal cancer shows an association with response to chemotherapy. Cancer Immun 2009, 9, 1. [Google Scholar]
- Halama, N.; Michel, S.; Kloor, M.; Zoernig, I.; Benner, A.; Spille, A.; Pommerencke, T.; von Knebel, D.M.; Folprecht, G.; Luber, B.; et al. Localization and density of immune cells in the invasive margin of human colorectal cancer liver metastases are prognostic for response to chemotherapy. Cancer Res 2011, 17, 5670–5677. [Google Scholar]
- Denkert, C.; Loibl, S.; Noske, A.; Roller, M.; Muller, B.M.; Komor, M.; Budczies, J.; Darb-Esfahani, S.; Kronenwett, R.; Hanusch, C.; et al. Tumor-associated lymphocytes as an independent predictor of response to neoadjuvant chemotherapy in breast cancer. J. Clin. Oncol 2010, 1, 105–113. [Google Scholar]
- Van der Most, R.G.; Currie, A.J.; Cleaver, A.L.; Salmons, J.; Nowak, A.K.; Mahendran, S.; Larma, I.; Prosser, A.; Robinson, B.W.; Smyth, M.J.; et al. Cyclophosphamide chemotherapy sensitizes tumor cells to TRAIL-dependent CD8 T cell-mediated immune attack resulting in suppression of tumor growth. PLoS One 2009, 9, e6982. [Google Scholar]
- Ramakrishnan, R.; Assudani, D.; Nagaraj, S.; Hunter, T.; Cho, H.I.; Antonia, S.; Altiok, S.; Celis, E.; Gabrilovich, D.I. Chemotherapy enhances tumor cell susceptibility to CTL-mediated killing during cancer immunotherapy in mice. J. Clin. Invest 2010, 4, 1111–1124. [Google Scholar]
- Mattarollo, S.R.; Loi, S.; Duret, H.; Ma, Y.; Zitvogel, L.; Smyth, M.J. Pivotal role of innate and adaptive immunity in anthracycline chemotherapy of established tumors. Cancer Res 2011, 14, 4809–4820. [Google Scholar]
- Ma, Y.; Aymeric, L.; Locher, C.; Mattarollo, S.R.; Delahaye, N.F.; Pereira, P.; Boucontet, L.; Apetoh, L.; Ghiringhelli, F.; Casares, N.; et al. Contribution of IL-17-producing gamma delta T cells to the efficacy of anticancer chemotherapy. J. Exp. Med 2011, 3, 491–503. [Google Scholar]
- Liu, H.; Zhang, T.; Ye, J.; Li, H.; Huang, J.; Li, X.; Wu, B.; Huang, X.; Hou, J. Tumor-infiltrating lymphocytes predict response to chemotherapy in patients with advance non-small cell lung cancer. Cancer Immunol. Immunother 2012. [Google Scholar] [CrossRef]
- Gobert, M.; Treilleux, I.; Bendriss-Vermare, N.; Bachelot, T.; Goddard-Leon, S.; Arfi, V.; Biota, C.; Doffin, A.C.; Durand, I.; Olive, D.; et al. Regulatory T cells recruited through CCL22/CCR4 are selectively activated in lymphoid infiltrates surrounding primary breast tumors and lead to an adverse clinical outcome. Cancer Res 2009, 5, 2000–2009. [Google Scholar]
- Bates, G.J.; Fox, S.B.; Han, C.; Leek, R.D.; Garcia, J.F.; Harris, A.L.; Banham, A.H. Quantification of regulatory T cells enables the identification of high-risk breast cancer patients and those at risk of late relapse. J. Clin. Oncol 2006, 34, 5373–5380. [Google Scholar]
- Tosolini, M.; Kirilovsky, A.; Mlecnik, B.; Fredriksen, T.; Mauger, S.; Bindea, G.; Berger, A.; Bruneval, P.; Fridman, W.H.; Pages, F.; et al. Clinical impact of different classes of infiltrating T cytotoxic and helper cells (Th1, th2, treg, th17) in patients with colorectal cancer. Cancer Res 2011, 4, 1263–1271. [Google Scholar]
- Denardo, D.G.; Barreto, J.B.; Andreu, P.; Vasquez, L.; Tawfik, D.; Kolhatkar, N.; Coussens, L.M. CD4(+) T cells regulate pulmonary metastasis of mammary carcinomas by enhancing protumor properties of macrophages. Cancer Cell 2009, 2, 91–102. [Google Scholar]
- Stassi, G.; Todaro, M.; Zerilli, M.; Ricci-Vitiani, L.; di Liberto, D.; Patti, M.; Florena, A.; di Gaudio, F.; di Gesu, G.; de Maria, R. Thyroid cancer resistance to chemotherapeutic drugs via autocrine production of interleukin-4 and interleukin-10. Cancer Res 2003, 20, 6784–6790. [Google Scholar]
- Zou, W.; Restifo, N.P. T(H)17 cells in tumour immunity and immunotherapy. Nat. Rev. Immunol 2010, 4, 248–256. [Google Scholar]
- Ding, Z.C.; Zhou, G. Cytotoxic chemotherapy and CD4+ effector T cells: An emerging alliance for durable antitumor effects. Clin. Dev. Immunol 2012. [Google Scholar] [CrossRef]
- Pittoni, P.; Piconese, S.; Tripodo, C.; Colombo, M.P. Tumor-intrinsic and -extrinsic roles of c-Kit: Mast cells as the primary off-target of tyrosine kinase inhibitors. Oncogene 2011, 7, 757–769. [Google Scholar]
- Johansson, A.; Rudolfsson, S.; Hammarsten, P.; Halin, S.; Pietras, K.; Jones, J.; Stattin, P.; Egevad, L.; Granfors, T.; Wikstrom, P.; et al. Mast cells are novel independent prognostic markers in prostate cancer and represent a target for therapy. Am. J. Pathol 2010, 2, 1031–1041. [Google Scholar]
- Pittoni, P.; Colombo, M.P. The dark side of mast cell-targeted therapy in prostate cancer. Cancer Res 2012, 4, 831–835. [Google Scholar]
- Iyengar, P.; Combs, T.P.; Shah, S.J.; Gouon-Evans, V.; Pollard, J.W.; Albanese, C.; Flanagan, L.; Tenniswood, M.P.; Guha, C.; Lisanti, M.P.; et al. Adipocyte-secreted factors synergistically promote mammary tumorigenesis through induction of anti-apoptotic transcriptional programs and proto-oncogene stabilization. Oncogene 2003, 41, 6408–6423. [Google Scholar]
- Behan, J.W.; Yun, J.P.; Proektor, M.P.; Ehsanipour, E.A.; Arutyunyan, A.; Moses, A.S.; Avramis, V.I.; Louie, S.G.; Butturini, A.; Heisterkamp, N.; et al. Adipocytes impair leukemia treatment in mice. Cancer Res 2009, 19, 7867–7874. [Google Scholar]
- Bochet, L.; Meulle, A.; Imbert, S.; Salles, B.; Valet, P.; Muller, C. Cancer-associated adipocytes promotes breast tumor radioresistance. Biochem. Biophys. Res. Commun 2011, 1, 102–106. [Google Scholar]
- Ma, J.; Dong, C.; Ji, C. MicroRNA and drug resistance. Cancer Gene Ther 2010, 8, 523–531. [Google Scholar]
- Sorrentino, A.; Liu, C.G.; Addario, A.; Peschle, C.; Scambia, G.; Ferlini, C. Role of microRNAs in drug-resistant ovarian cancer cells. Gynecol. Oncol 2008, 3, 478–486. [Google Scholar]
- Wang, H.; Tan, G.; Dong, L.; Cheng, L.; Li, K.; Wang, Z.; Luo, H. Circulating MiR-125b as a marker predicting chemoresistance in breast cancer. PLoS One 2012, 4, e34210. [Google Scholar]
- Zhao, R.; Wu, J.; Jia, W.; Gong, C.; Yu, F.; Ren, Z.; Chen, K.; He, J.; Su, F. Plasma miR-221 as a predictive biomarker for chemoresistance in breast cancer patients who previously received neoadjuvant chemotherapy. Onkologie 2011, 12, 675–680. [Google Scholar]
- Rao, E.; Jiang, C.; Ji, M.; Huang, X.; Iqbal, J.; Lenz, G.; Wright, G.; Staudt, L.M.; Zhao, Y.; McKeithan, T.W.; et al. The miRNA-17 approximately 92 cluster mediates chemoresistance and enhances tumor growth in mantle cell lymphoma via PI3K/AKT pathway activation. Leukemia 2012, 5, 1064–1072. [Google Scholar]
- Hamano, R.; Miyata, H.; Yamasaki, M.; Kurokawa, Y.; Hara, J.; Moon, J.H.; Nakajima, K.; Takiguchi, S.; Fujiwara, Y.; Mori, M.; et al. Overexpression of miR-200c induces chemoresistance in esophageal cancers mediated through activation of the Akt signaling pathway. Clin. Cancer Res 2011, 9, 3029–3038. [Google Scholar]
- Kong, W.; He, L.; Coppola, M.; Guo, J.; Esposito, N.N.; Coppola, D.; Cheng, J.Q. MicroRNA-155 regulates cell survival, growth, and chemosensitivity by targeting FOXO3a in breast cancer. J. Biol. Chem 2010, 23, 17869–17879. [Google Scholar]
- Bourguignon, L.Y.; Earle, C.; Wong, G.; Spevak, C.C.; Krueger, K. Stem cell marker (Nanog) and Stat-3 signaling promote MicroRNA-21 expression and chemoresistance in hyaluronan/CD44-activated head and neck squamous cell carcinoma cells. Oncogene 2012, 2, 149–160. [Google Scholar]
- Loscalzo, J. The cellular response to hypoxia: Tuning the system with microRNAs. J. Clin. Invest 2010, 11, 3815–3817. [Google Scholar]
- Hebert, C.; Norris, K.; Scheper, M.A.; Nikitakis, N.; Sauk, J.J. High mobility group A2 is a target for miRNA-98 in head and neck squamous cell carcinoma. Mol. Cancer 2007, 6. [Google Scholar] [CrossRef]
- Hanahan, D.; Coussens, L.M. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 2012, 3, 309–322. [Google Scholar]
- Margolin, D.A.; Silinsky, J.; Grimes, C.; Spencer, N.; Aycock, M.; Green, H.; Cordova, J.; Davis, N.K.; Driscoll, T.; Li, L. Lymph node stromal cells enhance drug-resistant colon cancer cell tumor formation through SDF-1alpha/CXCR4 paracrine signaling. Neoplasia 2011, 9, 874–886. [Google Scholar]
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Castells, M.; Thibault, B.; Delord, J.-P.; Couderc, B. Implication of Tumor Microenvironment in Chemoresistance: Tumor-Associated Stromal Cells Protect Tumor Cells from Cell Death. Int. J. Mol. Sci. 2012, 13, 9545-9571. https://doi.org/10.3390/ijms13089545
Castells M, Thibault B, Delord J-P, Couderc B. Implication of Tumor Microenvironment in Chemoresistance: Tumor-Associated Stromal Cells Protect Tumor Cells from Cell Death. International Journal of Molecular Sciences. 2012; 13(8):9545-9571. https://doi.org/10.3390/ijms13089545
Chicago/Turabian StyleCastells, Magali, Benoît Thibault, Jean-Pierre Delord, and Bettina Couderc. 2012. "Implication of Tumor Microenvironment in Chemoresistance: Tumor-Associated Stromal Cells Protect Tumor Cells from Cell Death" International Journal of Molecular Sciences 13, no. 8: 9545-9571. https://doi.org/10.3390/ijms13089545
APA StyleCastells, M., Thibault, B., Delord, J.-P., & Couderc, B. (2012). Implication of Tumor Microenvironment in Chemoresistance: Tumor-Associated Stromal Cells Protect Tumor Cells from Cell Death. International Journal of Molecular Sciences, 13(8), 9545-9571. https://doi.org/10.3390/ijms13089545