Yeast Two-Hybrid, a Powerful Tool for Systems Biology

Abstract
:1. Interactomics Take Center Stage in Systems Biology
1.1. A central role for protein interactions
1.2. Systems bioenergetics
1.3. Interactomics tools
2. Screening Technologies for Protein-Protein Interactions
2.1. Yeast two-hybrid
2.2. Affinity purification/mass spectrometry
2.3. Comparison of Y2H- and MS-based methods
3. Aiming at in Vivo Interactions: The Yeast Two-Hybrid Approach
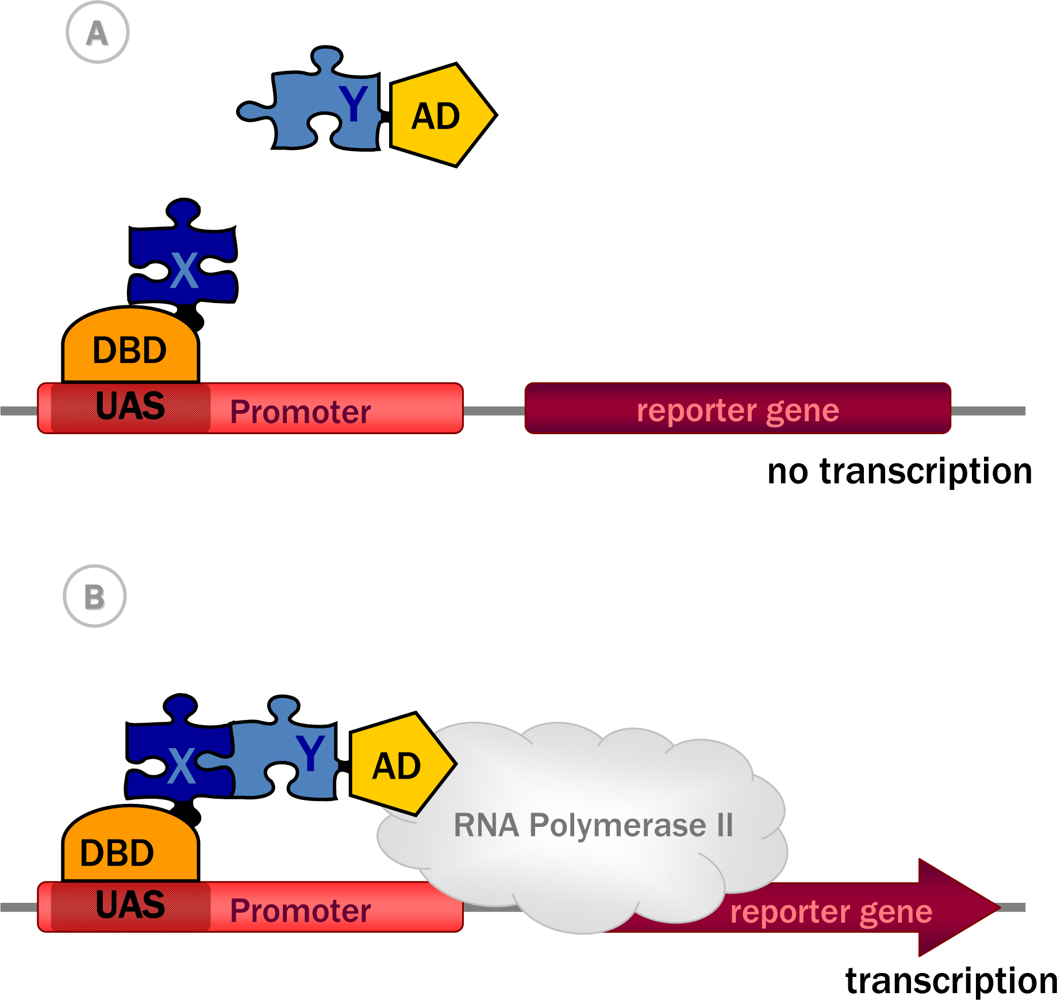
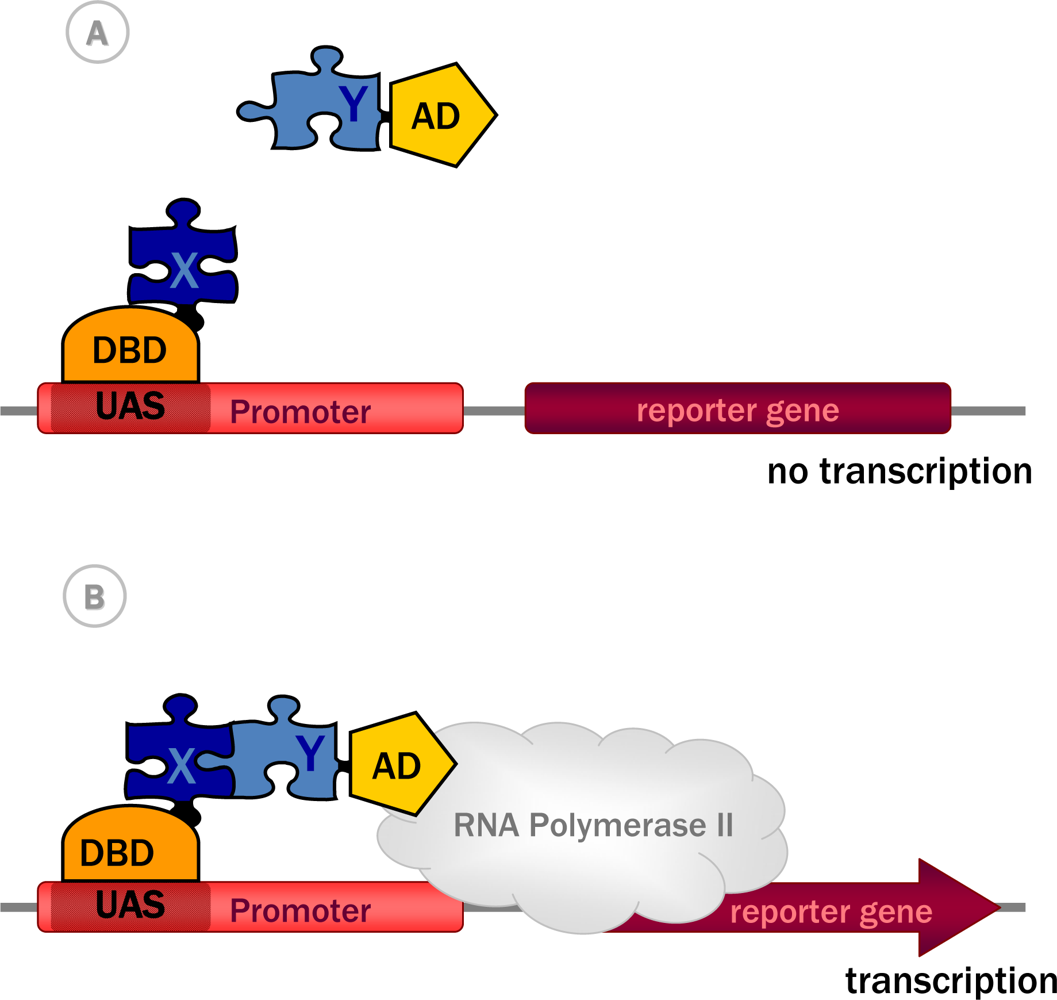
3.1. Historical perspective: The principles of the approach
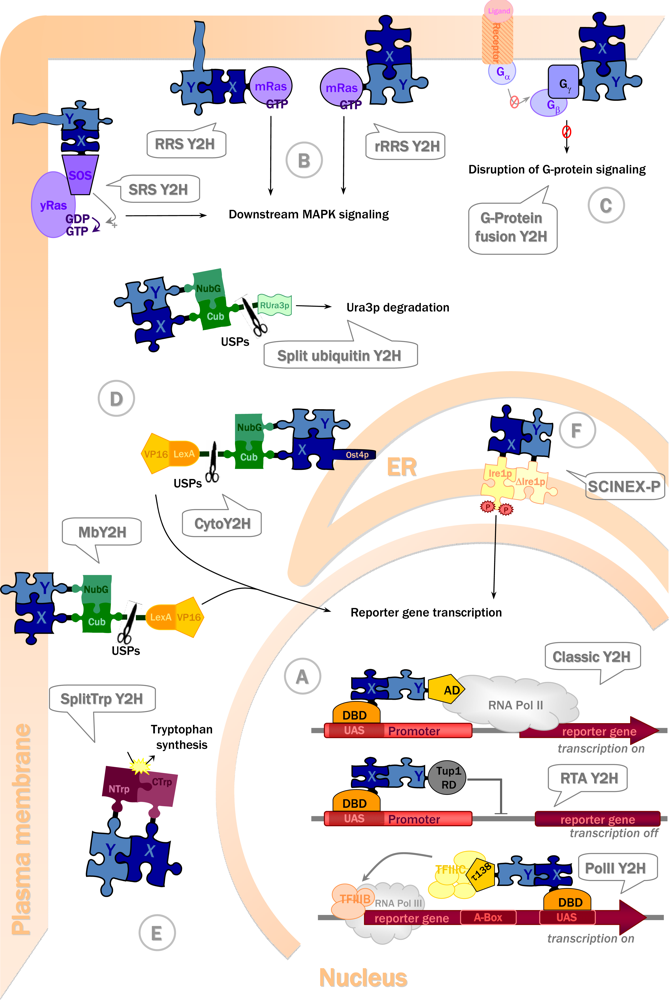
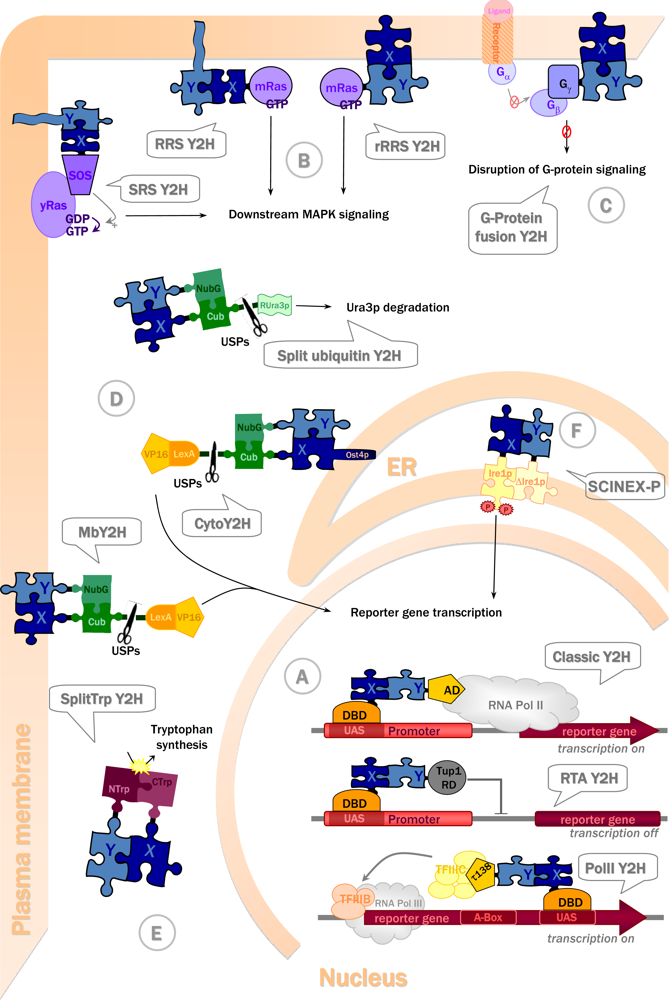
3.2. Choosing the right strategy: Available Y2H systems and their advantages
3.2.1. Y2H with transactivating proteins in the nucleus
3.2.2. Y2H with cytosolic and membrane proteins
3.2.3. Yeast two-hybrid with extracellular and transmembrane proteins
3.3. Dealing with doubt: Limitations of Y2H systems and methods for its validation
4. Further Confirmation: Protein-Protein Interactions within a Biological System
5. Conclusions
Acknowledgments
References and Notes
- Kiemer, L; Cesareni, G. Comparative interactomics: comparing apples and pears? Trends Biotechnol 2007, 25, 448–454. [Google Scholar]
- Kelly, W; Stumpf, M. Protein-protein interactions: from global to local analyses. Curr. Opin. Biotechnol 2008, 19, 396–403. [Google Scholar]
- Charbonnier, S; Gallego, O; Gavin, AC. The social network of a cell: recent advances in interactome mapping. Biotechnol. Annu. Rev 2008, 14, 1–28. [Google Scholar]
- Scheffler, IE. Mitochondria make a come back. Adv. Drug. Deliv. Rev 2001, 49, 3–26. [Google Scholar]
- Scheffler, IE. Mitochondria, 2nd ed; John Wiley & Sons: Hoboken, New Jersey, USA, 2007. [Google Scholar]
- Saks, V; Kaambre, T; Guzun, R; Anmann, T; Sikk, P; Schlattner, U; Wallimann, T; Aliev, M; Vendelin, M. The creatine kinase phosphotransfer network: thermodynamic and kinetic considerations, the impact of the mitochondrial outer membrane and modelling approaches. Subcell Biochem 2007, 46, 27–65. [Google Scholar]
- Bruggeman, FJ; Westerhoff, HV. The nature of systems biology. Trends Microbiol 2007, 15, 45–50. [Google Scholar]
- Srere, PA; Knull, HR. Location-location-location. Trends Biochem. Sci 1998, 23, 319–320. [Google Scholar]
- Beal, MF. Mitochondria take center stage in aging and neurodegeneration. Ann. Neurol 2005, 58, 495–505. [Google Scholar]
- Guigas, B; Detaille, D; Chauvin, C; Batandier, C; De Oliveira, F; Fontaine, E; Leverve, X. Metformin inhibits mitochondrial permeability transition and cell death: a pharmacological in vitro study. Biochem. J 2004, 382, 877–884. [Google Scholar]
- Ovadi, J; Srere, PA. Macromolecular compartmentation and channeling. Int. Rev. Cytol 2000, 192, 255–280. [Google Scholar]
- Schlattner, U; Gehring, F; Vernoux, N; Tokarska-Schlattner, M; Neumann, D; Marcillat, O; Vial, C; Wallimann, T. C-terminal lysines determine phospholipid interaction of sarcomeric mitochondrial creatine kinase. J. Biol. Chem 2004, 279, 24334–24342. [Google Scholar]
- Schlattner, U; Wallimann, T. Metabolite channeling: creatine kinase microcompartments. In Encyclopedia of Biological Chemistry; Lennarz, WJ, Lane, MD, Eds.; Academic Press: New York, NY, USA, 2004; pp. 646–651. [Google Scholar]
- Boireau, W; Rouleau, A; Lucchi, G; Ducoroy, P. Revisited BIA-MS combination: entire “on-a-chip” processing leading to the proteins identification at low femtomole to sub-femtomole levels. Biosens. Bioelectron 2009, 24, 1121–1127. [Google Scholar]
- Natsume, T; Nakayama, H; Jansson, O; Isobe, T; Takio, K; Mikoshiba, K. Combination of biomolecular interaction analysis and mass spectrometric amino acid sequencing. Anal. Chem 2000, 72, 4193–4198. [Google Scholar]
- Phizicky, EM; Fields, S. Protein-protein interactions: methods for detection and analysis. Microbiol. Rev 1995, 59, 94–123. [Google Scholar]
- Fields, S; Song, O. A novel genetic system to detect protein-protein interactions. Nature 1989, 340, 245–246. [Google Scholar]
- Bartel, PL; Roecklein, JA; SenGupta, D; Fields, S. A protein linkage map of Escherichia coli bacteriophage T7. Nat. Genet 1996, 12, 72–77. [Google Scholar]
- Ito, T; Chiba, T; Ozawa, R; Yoshida, M; Hattori, M; Sakaki, Y. A comprehensive two-hybrid analysis to explore the yeast protein interactome. Proc. Natl. Acad. Sci. USA 2001, 98, 4569–4574. [Google Scholar]
- Uetz, P; Giot, L; Cagney, G; Mansfield, TA; Judson, RS; Knight, JR; Lockshon, D; Narayan, V; Srinivasan, M; Pochart, P; Qureshi-Emili, A; Li, Y; Godwin, B; Conover, D; Kalbfleisch, T; Vijayadamodar, G; Yang, M; Johnston, M; Fields, S; Rothberg, JM. A comprehensive analysis of protein-protein interactions in Saccharomyces cerevisiae. Nature 2000, 403, 623–627. [Google Scholar]
- Formstecher, E; Aresta, S; Collura, V; Hamburger, A; Meil, A; Trehin, A; Reverdy, C; Betin, V; Maire, S; Brun, C; Jacq, B; Arpin, M; Bellaiche, Y; Bellusci, S; Benaroch, P; Bornens, M; Chanet, R; Chavrier, P; Delattre, O; Doye, V; Fehon, R; Faye, G; Galli, T; Girault, JA; Goud, B; de Gunzburg, J; Johannes, L; Junier, MP; Mirouse, V; Mukherjee, A; Papadopoulo, D; Perez, F; Plessis, A; Rosse, C; Saule, S; Stoppa-Lyonnet, D; Vincent, A; White, M; Legrain, P; Wojcik, J; Camonis, J; Daviet, L. Protein interaction mapping: a Drosophila case study. Genome Res 2005, 15, 376–384. [Google Scholar]
- Obrdlik, P; El-Bakkoury, M; Hamacher, T; Cappellaro, C; Vilarino, C; Fleischer, C; Ellerbrok, H; Kamuzinzi, R; Ledent, V; Blaudez, D; Sanders, D; Revuelta, JL; Boles, E; Andre, B; Frommer, WB. K+ channel interactions detected by a genetic system optimized for systematic studies of membrane protein interactions. Proc. Natl. Acad. Sci. USA 2004, 101, 12242–12247. [Google Scholar]
- Stelzl, U; Worm, U; Lalowski, M; Haenig, C; Brembeck, FH; Goehler, H; Stroedicke, M; Zenkner, M; Schoenherr, A; Koeppen, S; Timm, J; Mintzlaff, S; Abraham, C; Bock, N; Kietzmann, S; Goedde, A; Toksoz, E; Droege, A; Krobitsch, S; Korn, B; Birchmeier, W; Lehrach, H; Wanker, EE. A human protein-protein interaction network: a resource for annotating the proteome. Cell 2005, 122, 957–968. [Google Scholar]
- Rual, JF; Venkatesan, K; Hao, T; Hirozane-Kishikawa, T; Dricot, A; Li, N; Berriz, GF; Gibbons, FD; Dreze, M; Ayivi-Guedehoussou, N; Klitgord, N; Simon, C; Boxem, M; Milstein, S; Rosenberg, J; Goldberg, DS; Zhang, LV; Wong, SL; Franklin, G; Li, S; Albala, JS; Lim, J; Fraughton, C; Llamosas, E; Cevik, S; Bex, C; Lamesch, P; Sikorski, RS; Vandenhaute, J; Zoghbi, HY; Smolyar, A; Bosak, S; Sequerra, R; Doucette–Stamm, L; Cusick, ME; Hill, DE; Roth, FP; Vidal, M. Towards a proteome-scale map of the human protein-protein interaction network. Nature 2005, 437, 1173–1178. [Google Scholar]
- Rhodes, DR; Tomlins, SA; Varambally, S; Mahavisno, V; Barrette, T; Kalyana-Sundaram, S; Ghosh, D; Pandey, A; Chinnaiyan, AM. Probabilistic model of the human protein-protein interaction network. Nat. Biotechnol 2005, 23, 951–959. [Google Scholar]
- Gandhi, TK; Zhong, J; Mathivanan, S; Karthick, L; Chandrika, KN; Mohan, SS; Sharma, S; Pinkert, S; Nagaraju, S; Periaswamy, B; Mishra, G; Nandakumar, K; Shen, B; Deshpande, N; Nayak, R; Sarker, M; Boeke, JD; Parmigiani, G; Schultz, J; Bader, JS; Pandey, A. Analysis of the human protein interactome and comparison with yeast, worm and fly interaction datasets. Nat. Genet 2006, 38, 285–293. [Google Scholar]
- Lim, J; Hao, T; Shaw, C; Patel, AJ; Szabo, G; Rual, JF; Fisk, CJ; Li, N; Smolyar, A; Hill, DE; Barabasi, AL; Vidal, M; Zoghbi, HY. A protein-protein interaction network for human inherited ataxias and disorders of Purkinje cell degeneration. Cell 2006, 125, 801–814. [Google Scholar]
- Fromont-Racine, M; Mayes, AE; Brunet-Simon, A; Rain, JC; Colley, A; Dix, I; Decourty, L; Joly, N; Ricard, F; Beggs, JD; Legrain, P. Genome-wide protein interaction screens reveal functional networks involving Sm-like proteins. Yeast 2000, 17, 95–110. [Google Scholar]
- Auerbach, D; Thaminy, S; Hottiger, MO; Stagljar, I. The post-genomic era of interactive proteomics: facts and perspectives. Proteomics 2002, 2, 611–623. [Google Scholar]
- Fenn, JB; Mann, M; Meng, CK; Wong, SF; Whitehouse, CM. Electrospray ionization for mass spectrometry of large biomolecules. Science 1989, 246, 64–71. [Google Scholar]
- Tanaka, K; Waki, H; Ido, Y; Akita, S; Yoshida, Y; Yoshida, T. Protein and polymer analyses up to m/z 100 000 by laser ionization time-of-flight mass spectrometry. Rapid Commun. Mass Spectrom 1988, 2, 151–153. [Google Scholar]
- Karas, M; Hillenkamp, F. Laser desorption ionisation of proteins with molecular masses exceeding 10.000 daltons. Anal. Chem 1988, 60, 2299–2301. [Google Scholar]
- Nazabal, A; Wenzel, RJ; Zenobi, R. Immunoassays with direct mass spectrometric detection. Anal. Chem 2006, 78, 3562–3570. [Google Scholar]
- Pandey, A; Mann, M. Proteomics to study genes and genomes. Nature 2000, 405, 837–46. [Google Scholar]
- Ho, Y; Gruhler, A; Heilbut, A; Bader, GD; Moore, L; Adams, SL; Millar, A; Taylor, P; Bennett, K; Boutilier, K; Yang, L; Wolting, C; Donaldson, I; Schandorff, S; Shewnarane, J; Vo, M; Taggart, J; Goudreault, M; Muskat, B; Alfarano, C; Dewar, D; Lin, Z; Michalickova, K; Willems, AR; Sassi, H; Nielsen, PA; Rasmussen, KJ; Andersen, JR; Johansen, LE; Hansen, LH; Jespersen, H; Podtelejnikov, A; Nielsen, E; Crawford, J; Poulsen, V; Sorensen, BD; Matthiesen, J; Hendrickson, RC; Gleeson, F; Pawson, T; Moran, MF; Durocher, D; Mann, M; Hogue, CW; Figeys, D; Tyers, M. Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry. Nature 2002, 415, 180–183. [Google Scholar] [Green Version]
- Gavin, AC; Bosche, M; Krause, R; Grandi, P; Marzioch, M; Bauer, A; Schultz, J; Rick, JM; Michon, AM; Cruciat, CM; Remor, M; Hofert, C; Schelder, M; Brajenovic, M; Ruffner, H; Merino, A; Klein, K; Hudak, M; Dickson, D; Rudi, T; Gnau, V; Bauch, A; Bastuck, S; Huhse, B; Leutwein, C; Heurtier, MA; Copley, RR; Edelmann, A; Querfurth, E; Rybin, V; Drewes, G; Raida, M; Bouwmeester, T; Bork, P; Seraphin, B; Kuster, B; Neubauer, G; Superti-Furga, G. Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature 2002, 415, 141–147. [Google Scholar]
- Gavin, AC; Aloy, P; Grandi, P; Krause, R; Boesche, M; Marzioch, M; Rau, C; Jensen, LJ; Bastuck, S; Dumpelfeld, B; Edelmann, A; Heurtier, MA; Hoffman, V; Hoefert, C; Klein, K; Hudak, M; Michon, AM; Schelder, M; Schirle, M; Remor, M; Rudi, T; Hooper, S; Bauer, A; Bouwmeester, T; Casari, G; Drewes, G; Neubauer, G; Rick, JM; Kuster, B; Bork, P; Russell, RB; Superti-Furga, G. Proteome survey reveals modularity of the yeast cell machinery. Nature 2006, 440, 631–636. [Google Scholar]
- Krogan, NJ; Cagney, G; Yu, H; Zhong, G; Guo, X; Ignatchenko, A; Li, J; Pu, S; Datta, N; Tikuisis, AP; Punna, T; Peregrin-Alvarez, JM; Shales, M; Zhang, X; Davey, M; Robinson, MD; Paccanaro, A; Bray, JE; Sheung, A; Beattie, B; Richards, DP; Canadien, V; Lalev, A; Mena, F; Wong, P; Starostine, A; Canete, MM; Vlasblom, J; Wu, S; Orsi, C; Collins, SR; Chandran, S; Haw, R; Rilstone, JJ; Gandi, K; Thompson, NJ; Musso, G; St Onge, P; Ghanny, S; Lam, MH; Butland, G; Altaf-Ul, AM; Kanaya, S; Shilatifard, A; O'Shea, E; Weissman, JS; Ingles, CJ; Hughes, TR; Parkinson, J; Gerstein, M; Wodak, SJ; Emili, A; Greenblatt, JF. Global landscape of protein complexes in the yeast Saccharomyces cerevisiae. Nature 2006, 440, 637–643. [Google Scholar]
- Collins, MO; Choudhary, JS. Mapping multiprotein complexes by affinity purification and mass spectrometry. Curr. Opin. Biotechnol 2008, 19, 324–330. [Google Scholar]
- Puig, O; Caspary, F; Rigaut, G; Rutz, B; Bouveret, E; Bragado-Nilsson, E; Wilm, M; Seraphin, B. The tandem affinity purification (TAP) method: a general procedure of protein complex purification. Methods 2001, 24, 218–229. [Google Scholar]
- Uetz, P; Hughes, RE. Systematic and large-scale two-hybrid screens. Curr. Opin. Microbiol 2000, 3, 303–308. [Google Scholar]
- Hermjakob, H; Montecchi-Palazzi, L; Lewington, C; Mudali, S; Kerrien, S; Orchard, S; Vingron, M; Roechert, B; Roepstorff, P; Valencia, A; Margalit, H; Armstrong, J; Bairoch, A; Cesareni, G; Sherman, D; Apweiler, R. IntAct: an open source molecular interaction database. Nucleic Acids Res 2004, 32, D452–D455. [Google Scholar]
- Zanzoni, A; Montecchi-Palazzi, L; Quondam, M; Ausiello, G; Helmer-Citterich, M; Cesareni, G. MINT: a Molecular INTeraction database. FEBS Lett 2002, 513, 135–140. [Google Scholar]
- Glatter, T; Wepf, A; Aebersold, R; Gstaiger, M. An integrated workflow for charting the human interaction proteome: insights into the PP2A system. Mol. Syst. Biol 2009, 5, 237. [Google Scholar]
- Keegan, L; Gill, G; Ptashne, M. Separation of DNA binding from the transcription-activating function of a eukaryotic regulatory protein. Science 1986, 231, 699–704. [Google Scholar]
- Durfee, T; Becherer, K; Chen, PL; Yeh, SH; Yang, Y; Kilburn, AE; Lee, WH; Elledge, SJ. The retinoblastoma protein associates with the protein phosphatase type 1 catalytic subunit. Genes Dev 1993, 7, 555–569. [Google Scholar]
- Licitra, EJ; Liu, JO. A three-hybrid system for detecting small ligand-protein receptor interactions. Pro.c Natl. Acad. Sci. USA 1996, 93, 12817–12821. [Google Scholar]
- Huang, J; Schreiber, SL. A yeast genetic system for selecting small molecule inhibitors of protein-protein interactions in nanodroplets. Proc. Natl. Acad. Sci. USA 1997, 94, 13396–13401. [Google Scholar]
- Serebriiskii, I; Khazak, V; Golemis, EA. A two-hybrid dual bait system to discriminate specificity of protein interactions. J. Biol. Chem 1999, 274, 17080–17087. [Google Scholar]
- Serebriiskii, IG; Mitina, O; Pugacheva, EN; Benevolenskaya, E; Kotova, E; Toby, GG; Khazak, V; Kaelin, WG; Chernoff, J; Golemis, EA. Detection of peptides, proteins, and drugs that selectively interact with protein targets. Genome Res 2002, 12, 1785–1791. [Google Scholar]
- Aronheim, A; Engelberg, D; Li, N; al-Alawi, N; Schlessinger, J; Karin, M. Membrane targeting of the nucleotide exchange factor Sos is sufficient for activating the Ras signaling pathway. Cell 1994, 78, 949–961. [Google Scholar]
- Aronheim, A; Zandi, E; Hennemann, H; Elledge, SJ; Karin, M. Isolation of an AP-1 repressor by a novel method for detecting protein-protein interactions. Mol Cell Biol 1997, 17, 3094–3102. [Google Scholar]
- Johnsson, N; Varshavsky, A. Split ubiquitin as a sensor of protein interactions in vivo. Proc. Natl. Acad. Sci. USA 1994, 91, 10340–10344. [Google Scholar]
- Dirnberger, D; Messerschmid, M; Baumeister, R. An optimized split-ubiquitin cDNA-library screening system to identify novel interactors of the human Frizzled 1 receptor. Nucleic Acids Res 2008, 36, e37. [Google Scholar]
- Stagljar, I; Korostensky, C; Johnsson, N; te Heesen, S. A genetic system based on split-ubiquitin for the analysis of interactions between membrane proteins in vivo. Proc. Natl. Acad. Sci. USA 1998, 95, 5187–5192. [Google Scholar]
- Hooker, BS; Bigelow, DJ; Lin, CT. Methods for mapping of interaction networks involving membrane proteins. Biochem. Biophys. Res. Commun 2007, 363, 457–461. [Google Scholar]
- Broder, YC; Katz, S; Aronheim, A. The ras recruitment system, a novel approach to the study of protein-protein interactions. Curr. Biol 1998, 8, 1121–1124. [Google Scholar]
- Ehrhard, KN; Jacoby, JJ; Fu, XY; Jahn, R; Dohlman, HG. Use of G-protein fusions to monitor integral membrane protein-protein interactions in yeast. Nat. Biotechnol 2000, 18, 1075–1079. [Google Scholar]
- Petrascheck, M; Castagna, F; Barberis, A. Two-hybrid selection assay to identify proteins interacting with polymerase II transcription factors and regulators. Biotechniques 2001, 30, 296–298. [Google Scholar]
- Hirst, M; Ho, C; Sabourin, L; Rudnicki, M; Penn, L; Sadowski, I. A two-hybrid system for transactivator bait proteins. Proc. Natl. Acad. Sci. USA 2001, 98, 8726–8731. [Google Scholar]
- Huang, A; Ho, CS; Ponzielli, R; Barsyte-Lovejoy, D; Bouffet, E; Picard, D; Hawkins, CE; Penn, LZ. Identification of a novel c-Myc protein interactor, JPO2, with transforming activity in medulloblastoma cells. Cancer Res 2005, 65, 5607–5619. [Google Scholar]
- Wafa, LA; Cheng, H; Rao, MA; Nelson, CC; Cox, M; Hirst, M; Sadowski, I; Rennie, PS. Isolation and identification of L-dopa decarboxylase as a protein that binds to and enhances transcriptional activity of the androgen receptor using the repressed transactivator yeast two-hybrid system. Biochem. J 2003, 375, 373–383. [Google Scholar]
- Hubsman, M; Yudkovsky, G; Aronheim, A. A novel approach for the identification of protein-protein interaction with integral membrane proteins. Nucleic Acids Res 2001, 29, e18. [Google Scholar]
- Urech, DM; Lichtlen, P; Barberis, A. Cell growth selection system to detect extracellular and transmembrane protein interactions. Biochim. Biophys. Acta 2003, 1622, 117–127. [Google Scholar]
- Tafelmeyer, P; Johnsson, N; Johnsson, K. Transforming a (beta/alpha)8--barrel enzyme into a split-protein sensor through directed evolution. Chem. Biol 2004, 11, 681–689. [Google Scholar]
- Mockli, N; Deplazes, A; Hassa, PO; Zhang, Z; Peter, M; Hottiger, MO; Stagljar, I; Auerbach, D. Yeast split-ubiquitin-based cytosolic screening system to detect interactions between transcriptionally active proteins. Biotechniques 2007, 42, 725–730. [Google Scholar]
- Joshi, PB; Hirst, M; Malcolm, T; Parent, J; Mitchell, D; Lund, K; Sadowski, I. Identification of protein interaction antagonists using the repressed transactivator two-hybrid system. Biotechniques 2007, 42, 635–644. [Google Scholar]
- Marsolier, MC; Prioleau, MN; Sentenac, A. A RNA polymerase III-based two-hybrid system to study RNA polymerase II transcriptional regulators. J. Mol. Biol 1997, 268, 243–249. [Google Scholar]
- Borg, JP; Marchetto, S; Le Bivic, A; Ollendorff, V; Jaulin-Bastard, F; Saito, H; Fournier, E; Adelaide, J; Margolis, B; Birnbaum, D. ERBIN: a basolateral PDZ protein that interacts with the mammalian ERBB2/HER2 receptor. Nat. Cell Biol 2000, 2, 407–414. [Google Scholar]
- Sugita, S; Hata, Y; Sudhof, TC. Distinct Ca(2+)-dependent properties of the first and second C2-domains of synaptotagmin I. J. Biol. Chem 1996, 271, 1262–1265. [Google Scholar]
- Niethammer, M; Kim, E; Sheng, M. Interaction between the C terminus of NMDA receptor subunits and multiple members of the PSD-95 family of membrane-associated guanylate kinases. J. Neurosci 1996, 16, 2157–2163. [Google Scholar]
- Wittke, S; Lewke, N; Muller, S; Johnsson, N. Probing the molecular environment of membrane proteins in vivo. Mol. Biol. Cell 1999, 10, 2519–2530. [Google Scholar]
- Varshavsky, A. The N-end rule pathway of protein degradation. Genes Cells 1997, 2, 13–28. [Google Scholar]
- Laser, H; Bongards, C; Schuller, J; Heck, S; Johnsson, N; Lehming, N. A new screen for protein interactions reveals that the Saccharomyces cerevisiae high mobility group proteins Nhp6A/B are involved in the regulation of the GAL1 promoter. Proc. Natl. Acad. Sci. USA 2000, 97, 13732–13737. [Google Scholar]
- Kerkmann, K; Lehming, N. Genome-wide expression analysis of a Saccharomyces cerevisiae strain deleted for the Tup1p-interacting protein Cdc73p. Curr. Genet 2001, 39, 284–290. [Google Scholar]
- Eckert, JH; Johnsson, N. Pex10p links the ubiquitin conjugating enzyme Pex4p to the protein import machinery of the peroxisome. J. Cell. Sci 2003, 116, 3623–3634. [Google Scholar]
- Thaminy, S; Auerbach, D; Arnoldo, A; Stagljar, I. Identification of novel ErbB3-interacting factors using the split-ubiquitin membrane yeast two-hybrid system. Genome Res 2003, 13, 1744–1753. [Google Scholar]
- Wang, B; Pelletier, J; Massaad, MJ; Herscovics, A; Shore, GC. The yeast split-ubiquitin membrane protein two-hybrid screen identifies BAP31 as a regulator of the turnover of endoplasmic reticulum-associated protein tyrosine phosphatase-like B. Mol. Cell. Biol 2004, 24, 2767–2778. [Google Scholar]
- Matsuda, S; Giliberto, L; Matsuda, Y; Davies, P; McGowan, E; Pickford, F; Ghiso, J; Frangione, B; D'Adamio, L. The familial dementia BRI2 gene binds the Alzheimer gene amyloid-beta precursor protein and inhibits amyloid-beta production. J. Biol. Chem 2005, 280, 28912–28916. [Google Scholar]
- Felkl, M; Leube, RE. Interaction assays in yeast and cultured cells confirm known and identify novel partners of the synaptic vesicle protein synaptophysin. Neuroscience 2008, 156, 344–352. [Google Scholar]
- Pasch, JC; Nickelsen, J; Schunemann, D. The yeast split-ubiquitin system to study chloroplast membrane protein interactions. Appl. Microbiol. Biotechnol 2005, 69, 440–447. [Google Scholar]
- Miller, JP; Lo, RS; Ben-Hur, A; Desmarais, C; Stagljar, I; Noble, WS; Fields, S. Large-scale identification of yeast integral membrane protein interactions. Proc. Natl. Acad. Sci. USA 2005, 102, 12123–12128. [Google Scholar]
- Pollock, S; Kozlov, G; Pelletier, MF; Trempe, JF; Jansen, G; Sitnikov, D; Bergeron, JJ; Gehring, K; Ekiel, I; Thomas, DY. Specific interaction of ERp57 and calnexin determined by NMR spectroscopy and an ER two-hybrid system. Embo. J 2004, 23, 1020–1029. [Google Scholar]
- Osborne, MA; Zenner, G; Lubinus, M; Zhang, X; Songyang, Z; Cantley, LC; Majerus, P; Burn, P; Kochan, JP. The inositol 5′-phosphatase SHIP binds to immunoreceptor signaling motifs and responds to high affinity IgE receptor aggregation. J. Biol. Chem 1996, 271, 29271–29278. [Google Scholar]
- Fukada, M; Kawachi, H; Fujikawa, A; Noda, M. Yeast substrate-trapping system for isolating substrates of protein tyrosine phosphatases: Isolation of substrates for protein tyrosine phosphatase receptor type z. Methods 2005, 35, 54–63. [Google Scholar]
- Guo, D; Hazbun, TR; Xu, XJ; Ng, SL; Fields, S; Kuo, MH. A tethered catalysis, two-hybrid system to identify protein-protein interactions requiring post-translational modifications. Nat. Biotechnol 2004, 22, 888–892. [Google Scholar]
- Hamilton, SR; Gerngross, TU. Glycosylation engineering in yeast: the advent of fully humanized yeast. Curr. Opin. Biotechnol 2007, 18, 387–392. [Google Scholar]
- von Mering, C; Krause, R; Snel, B; Cornell, M; Oliver, SG; Fields, S; Bork, P. Comparative assessment of large-scale data sets of protein-protein interactions. Nature 2002, 417, 399–403. [Google Scholar]
- Jackson, M; Song, W; Liu, MY; Jin, L; Dykes-Hoberg, M; Lin, CI; Bowers, WJ; Federoff, HJ; Sternweis, PC; Rothstein, JD. Modulation of the neuronal glutamate transporter EAAT4 by two interacting proteins. Nature 2001, 410, 89–93. [Google Scholar]
- Tanaka, H; Katoh, H; Negishi, M. Pragmin, a novel effector of Rnd2 GTPase, stimulates RhoA activity. J. Biol. Chem 2006, 281, 10355–10364. [Google Scholar]
- Burklen, TS; Hirschy, A; Wallimann, T. Brain-type creatine kinase BB-CK interacts with the Golgi Matrix Protein GM130 in early prophase. Mol. Cell Biochem 2007, 297, 53–64. [Google Scholar]
- Dye, DE; Karlen, S; Rohrbach, B; Staub, O; Braathen, LR; Eidne, KA; Coombe, DR. hShroom1 links a membrane bound protein to the actin cytoskeleton. Cell. Mol. Life Sci 2009, 66, 681–696. [Google Scholar]
- Hornemann, T; Kempa, S; Himmel, M; Hayess, K; Furst, DO; Wallimann, T. Muscle-type creatine kinase interacts with central domains of the M-band proteins myomesin and M-protein. J. Mol. Biol 2003, 332, 877–887. [Google Scholar]
- Fetchko, M; Stagljar, I. Application of the split-ubiquitin membrane yeast two-hybrid system to investigate membrane protein interactions. Methods 2004, 32, 349–362. [Google Scholar]
- Day, RN. Visualization of Pit-1 transcription factor interactions in the living cell nucleus by fluorescence resonance energy transfer microscopy. Mol. Endocrinol 1998, 12, 1410–1419. [Google Scholar]
- Deane, CM; Salwinski, L; Xenarios, I; Eisenberg, D. Protein interactions: two methods for assessment of the reliability of high throughput observations. Mol. Cell Proteomics 2002, 1, 349–356. [Google Scholar]
- Patil, A; Nakamura, H. Filtering high-throughput protein-protein interaction data using a combination of genomic features. BMC Bioinformatics 2005, 6, 100. [Google Scholar]
- Giot, L; Bader, JS; Brouwer, C; Chaudhuri, A; Kuang, B; Li, Y; Hao, YL; Ooi, CE; Godwin, B; Vitols, E; Vijayadamodar, G; Pochart, P; Machineni, H; Welsh, M; Kong, Y; Zerhusen, B; Malcolm, R; Varrone, Z; Collis, A; Minto, M; Burgess, S; McDaniel, L; Stimpson, E; Spriggs, F; Williams, J; Neurath, K; Ioime, N; Agee, M; Voss, E; Furtak, K; Renzulli, R; Aanensen, N; Carrolla, S; Bickelhaupt, E; Lazovatsky, Y; DaSilva, A; Zhong, J; Stanyon, CA; Finley, RL, Jr; White, KP; Braverman, M; Jarvie, T; Gold, S; Leach, M; Knight, J; Shimkets, RA; McKenna, MP; Chant, J; Rothberg, JM. A protein interaction map of Drosophila melanogaster. Science 2003, 302, 1727–1736. [Google Scholar]

{kind=link}
{kind=link}
| Year | Y2H method | Possible baits | Response | Cellular compartment * | Screen compatibility # |
|---|---|---|---|---|---|
| 1989 | Classic Y2H system [17] | Non-transactivating proteins capable of entering nucleus | Transcriptional activation | Nucleus | Yes [17] |
| 1994 | SOS recruitment system (SRS) [51] | Transactivating, cytosolic proteins | Ras signalling | Membrane periphery | Yes [52] |
| 1994 | Split-ubiquitin system [53] | Nuclear, membrane and cytosolic proteins | Uracil auxotrophy and 5-FoA resistance | Cytosol | Yes [54] |
| 1998 | Membrane split-ubiquitin system (MbY2H) [55] | Membrane proteins | Transcriptional activation | Membrane periphery | Yes [56] |
| 1998 | Ras recruitment system (RRS) [57] | Transactivating, cytosolic proteins | Ras signalling | Membrane periphery | Yes [57] |
| 1999 | Dual bait system [49] | Two non-transactivating proteins capable of entering nucleus | Transcriptional activation | Nucleus | Yes [49] |
| 2000 | G-protein fusion system [58] | Membrane proteins | Inhibition of protein G signalling | Membrane periphery | No |
| 2001 | RNA polymerase III based two-hybrid (Pol III) [59] | Transactivating proteins (in the RNA polymerase II pathway) | Transcriptional activation | Nucleus | Yes [59] |
| 2001 | Repressed transactivator system (RTA) [60] | Transactivating proteins capable of entering nucleus | Inhibition of transcriptional activation | Nucleus | Yes [60–62] |
| 2001 | Reverse Ras recruitment system (rRRS) [63] | Membrane proteins | Ras signalling | Membrane periphery | Yes [63] |
| 2003 | SCINEX-P system [64] | Extracellular and transmembrane proteins | Downstream signalling & transcriptional activation | Endoplasmic reticulum (ER) | No |
| 2004 | Split-Trp system [65] | Cytosolic, membrane proteins | Trp1p activity | Cytosol | Yes (Lentze & Auerbach, unpubl.) |
| 2007 | Cytosolic split-ubiquitin system (cytoY2H) [66] | Transactivating, cytosolic proteins | Transcriptional activation | ER membrane periphery | Yes [66] |
| Method | Type | Description |
|---|---|---|
| Pull-down assay [89–91] | in vitro | Tagged bait (mostly expressed in E.coli) is immobilized on a resin and subsequently “pulls down” target protein (prey) from lysates (of eukaryotic cells or of E.coli expressing proteins of interest). After washing steps, prey is detected by SDS-PAGE/immunoblot or MS. |
| Coimmunoprecipitation [80,89,90,92] | ex vivo | A specific antibody is used to precipitate the bait from cell lysates (see above). After washing steps, coimmunoprecipitated prey is detected as above. |
| Surface plasmon resonance (Biacore) [93] | in vitro | Bait immobilized on the surface of a sensor chip is probed by injection of prey onto the surface. Protein interaction is detected online via a biophysical principle (using the change in refractive index at the sensor surface in case of protein interaction). Protein is eluted and analyzed by MS. |
| In situ hybridization [90] | in situ | Hybridization of a labelled complementary DNA or RNA strand (i.e. probe) to a specific DNA or RNA sequence in a tissue section. Visualizes expression of specific genes to evaluate potential coexpression of proteins of interest in the same cell of a given tissue. |
| Immunohistochemistry, immunocytochemistry [80,89,90] | in situ | Proteins in fixed cells or tissue sections are detected by immune-labelling with fluorescently tagged antibodies, e.g. using confocal microscopy. Visualizes coexpression of proteins of interest in the same cell and potential subcellular colocalization. |
| Fluorescent detection in live cells [91] | in vivo | Proteins in living cells are detected with fluorescently tagged antibodies as above (using permeabilized cells) or after expression of fluorescently tagged protein variants. Visualizes colocalization of proteins of interest. |
| Fluorescence resonance energy transfer (FRET) [80] | in vivo | Bait and prey are fused to two different fluorescent tags with overlapping emission/excitation spectra. If both proteins are in close proximity, excitation of the first fluorophore (donor) leads to energy transfer to the second fluorophore (acceptor). Acceptor fluorescence can be observed in vitro (fluorimeter) or in living cells (confocal microscopy). |
| Bioluminescencer resonance energy transfer (BRET) [92] | in vivo | Similar to FRET (see above), but with bait fused to bioluminescent luciferase, thus avoiding the external excitation step susceptible to generate background. Detection as with FRET. |
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Brückner, A.; Polge, C.; Lentze, N.; Auerbach, D.; Schlattner, U. Yeast Two-Hybrid, a Powerful Tool for Systems Biology. Int. J. Mol. Sci. 2009, 10, 2763-2788. https://doi.org/10.3390/ijms10062763
Brückner A, Polge C, Lentze N, Auerbach D, Schlattner U. Yeast Two-Hybrid, a Powerful Tool for Systems Biology. International Journal of Molecular Sciences. 2009; 10(6):2763-2788. https://doi.org/10.3390/ijms10062763
Chicago/Turabian StyleBrückner, Anna, Cécile Polge, Nicolas Lentze, Daniel Auerbach, and Uwe Schlattner. 2009. "Yeast Two-Hybrid, a Powerful Tool for Systems Biology" International Journal of Molecular Sciences 10, no. 6: 2763-2788. https://doi.org/10.3390/ijms10062763
APA StyleBrückner, A., Polge, C., Lentze, N., Auerbach, D., & Schlattner, U. (2009). Yeast Two-Hybrid, a Powerful Tool for Systems Biology. International Journal of Molecular Sciences, 10(6), 2763-2788. https://doi.org/10.3390/ijms10062763