Abstract
The Cu-catalyzed intramolecular CH insertion of phenyliodonium ylide 5b has been investigated at 0° C in the presence of several chiral ligands. Enantioselectivities vary in the range of 38–72 %, and are higher than those resulting from reaction of the diazo compound 5c at 65° C. The results are consistent with a carbenoid mechanism for Cu-catalyzed decomposition of phenyliodonium ylides.
Introduction
Phenyliodonium ylides are of interest as substitutes for diazo compounds in transition metalcatalyzed carbenoid reactions. Their decomposition under photochemical [1,2] or thermal conditions [1,3], or in the presence of transition metal catalysts [2,3,4] affords products typical for carbene or metal carbenoid intermediates. However, the experimental evidence supporting these mechanistic hypotheses is scarce. The intermediacy of metal carbenoids upon diazo decomposition by transition metal-catalysts is well established. These reactions proceed with almost perfect enantioselectivity with the appropriate chiral, non-racemic catalysts [5]. In contrast, the mechanism of the transition metal-catalyzed decomposition of phenyliodonium ylides is controversial, and enantioselective reactions are limited. Some years ago we presented evidence for metal carbenoid pathways in Rh(II)-catalyzed cyclopropanations and CH insertions of phenyliodonium ylides [6].
The Cu(I)-catalyzed decomposition of phenyliodonium ylides in the presence of olefins affords cyclopropanes. A mechanism involving electrophilic addition of the iodonium center to the double bond followed by reductive elimination of PhI, as shown in Scheme 1 has been proposed for this transformation by Moriarty. A carbene or metal carbenoid mechanism was specifically ruled out [7].
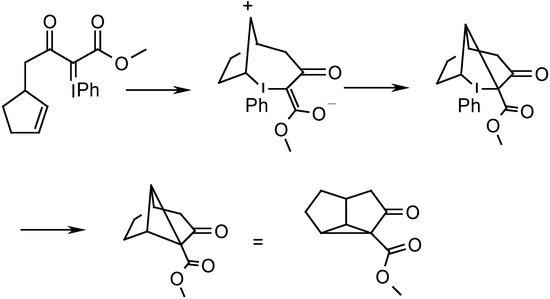
Scheme 1.
Scheme 1.
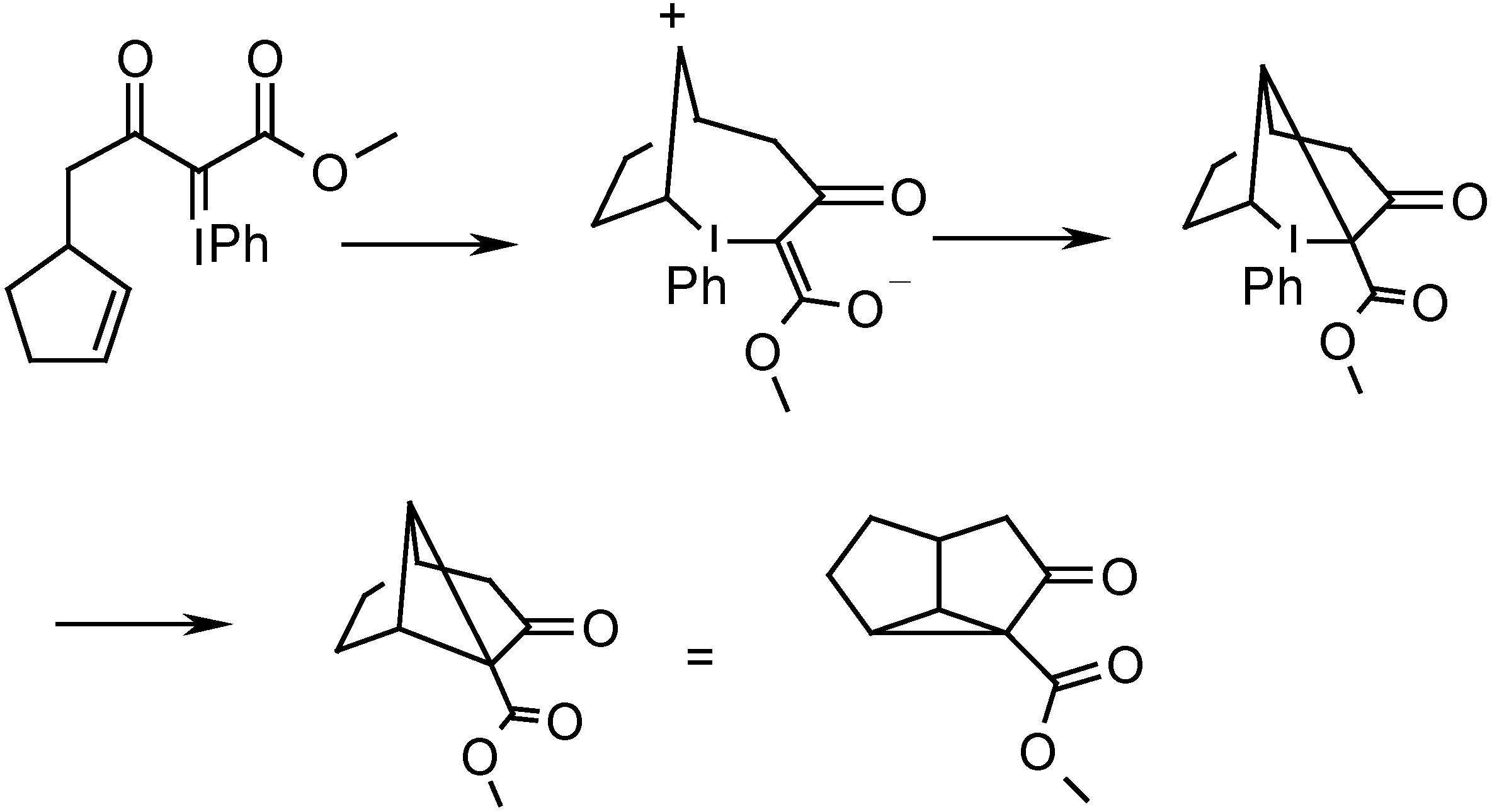
Recently we reported the intramolecular cyclopropanation of phenyliodonium ylides derived from acetoacetates and malonates with [Cu(OTf)2] in the presence of chiral ligands. Thus, the reaction of ylide 1a afforded 2 with the binaphthalene derived oxazoline A as ligand in 48 % yield and with 68 % ee (Scheme 2) [8].
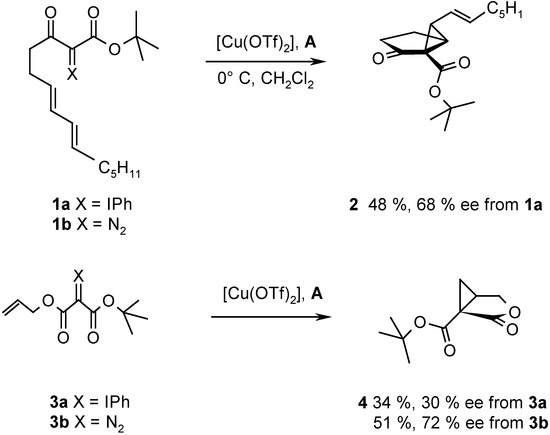
Scheme 2.
Scheme 2.
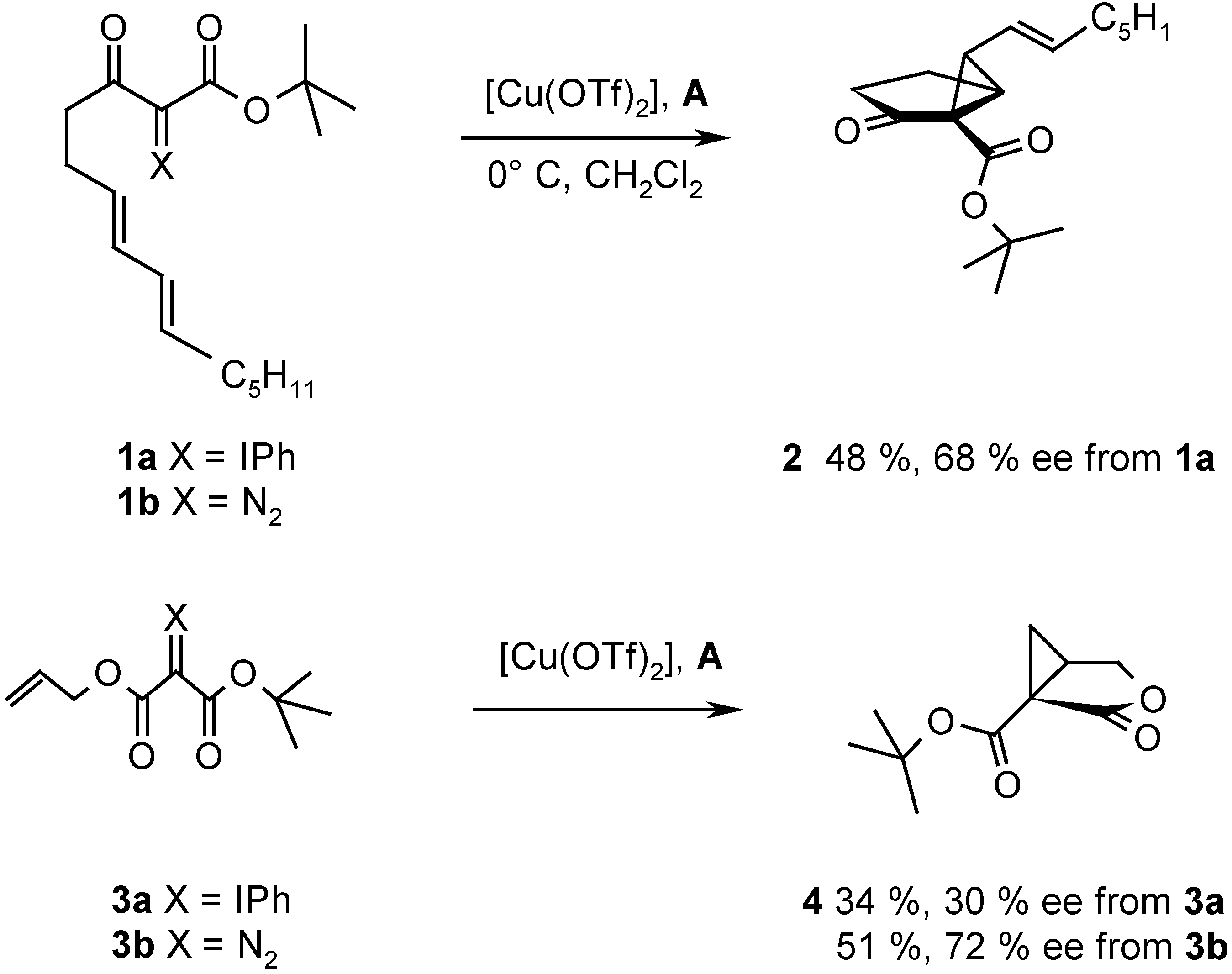
This result is inconsistent with the mechanism for cyclopropanation as shown in Scheme 1. If the reaction proceeds via electrophilic attack of the iodonium center on the double bond, the metal is not involved in the reaction and, therefore, no asymmetric induction should occur. On the other hand, conclusive evidence for metal carbenoid intermediates upon Cu-catalyzed decomposition of diazo compounds has been reported [6]. If a carbenoid mechanism applies to the cyclopropanation with phenyliodonium ylides, the enantioselectivity resulting upon decomposition of 1a should be identical to that obtained with the corresponding diazo compound 1b, provided the same chiral ligand is used. Unfortunately, the conventionally used chiral Cu-catalysts proved to be insufficiently reactive for decomposition of 1b or of similar diazo compounds derived from β-ketoesters and other 1,3-dicarbonyl derivatives under conditions suitable for reaction of phenyliodonium ylides. Typically, the ylide 3a underwent intramolecular cyclopropanation with [Cu(OTf)2] and ligand A in CH2Cl2 at 0° C to provide 4 in 34 % yield and 30 % ee, but decomposition of the corresponding diazo compound 3b required heating in trifluorotoluene at 100° C. It is known that enantioselectivity increases generally with decreasing temperature and, therefore, a higher enantioselectivity for the reaction of 3a was expected. Surprisingly, however, the product 4 resulting from diazo decomposition of 3b had a higher enantiomeric excess (72 % ee) than that resulting from 3a (30 % ee). The intramolecular cyclopropanation of 3a and 3b in the presence of other chiral ligands revealed remarkable inconsistencies. With some ligands the enantioselectivity observed upon reaction of the ylide was higher than that of the diazo decomposition, and with others the trend was inversed. These discrepancies suggest that the Cu-catalyzed cyclopropanation with phenyliodonium ylides may not entirely proceed via a metal carbenoid, but also via some other, uncatalyzed pathway as suggested by Moriarty. Indeed, uncatalyzed intramolecular cyclopropanations of phenyliodonium ylides have been observed previously [5,7].
The mechanism of Moriarty for uncatalyzed cyclopropanations with phenyliodonium ylides is plausible, but this mechanism should not apply to uncatalyzed CH bond insertions. Indeed, no insertion products upon uncatalyzed decomposition of phenyliodonium ylides at ambient temperatures have ever been observed or reported. Although the phenyliodonium ylide derived from diethyl malonate does insert into the CH bonds of cyclohexane, this latter reaction, which is believed to proceed via a free carbene, requires a temperature of 100° C, much higher than the 0 °C used for the metal-catalyzed cyclopropanation [1]. We reasoned that comparison of enantioselectivities in CH bond insertions resulting from Cu-catalyzed decompositions of phenyliodonium ylides and of the corresponding diazo compounds, respectively, would not be affected by the irregularities occurring in cyclopropanations, and would, therefore, provide unambiguous evidence in support of the carbenoid pathway [9].
Results and Discussion
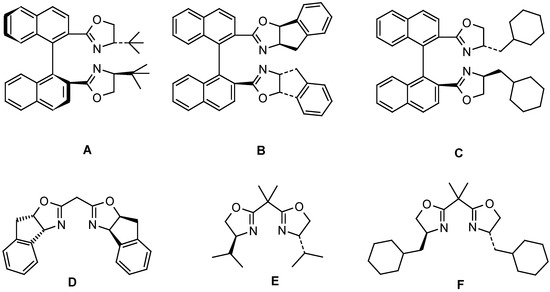
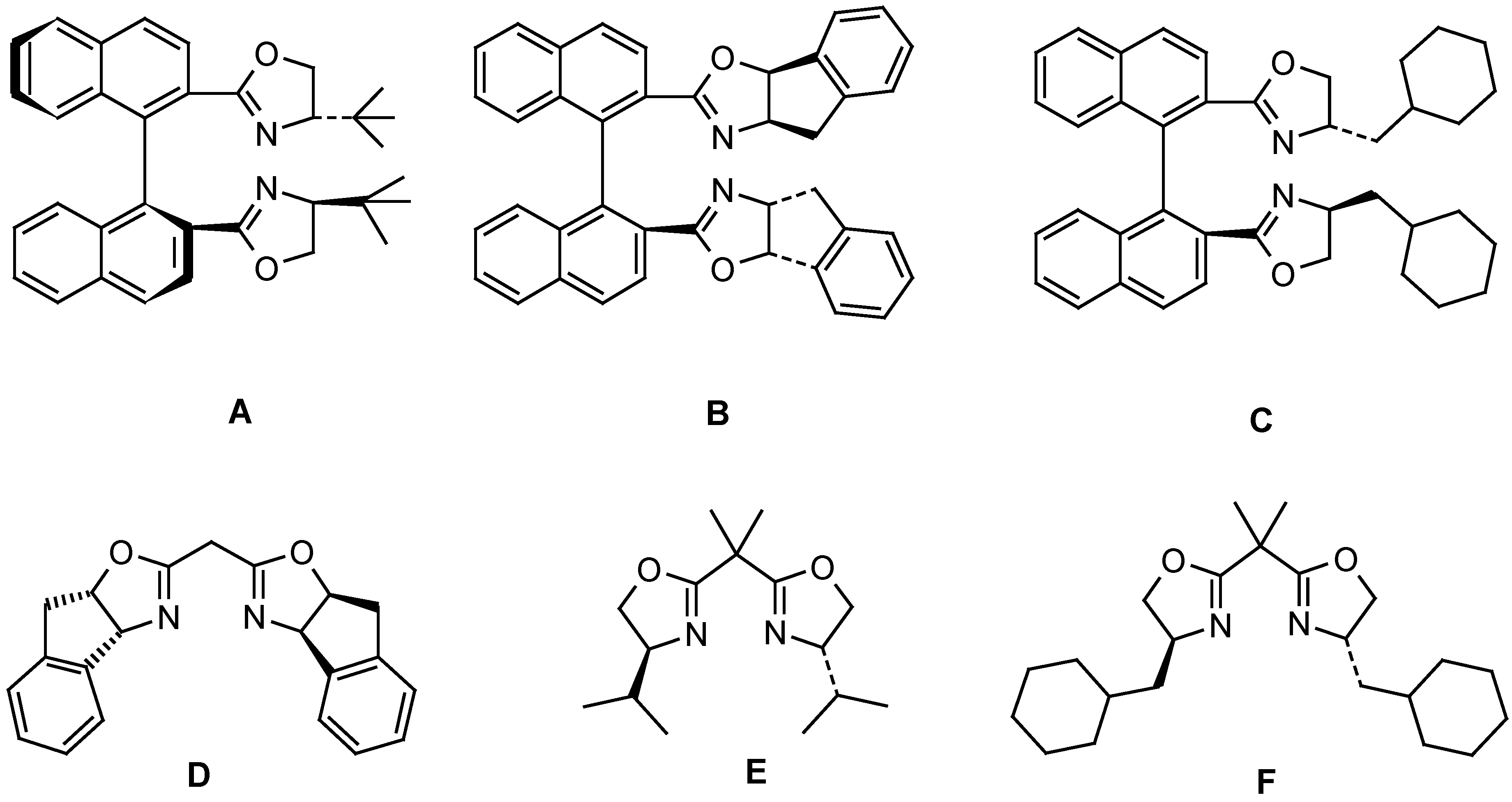
The phenyliodonium ylide 5b was synthesized by reaction of the hydrocarbon 5a [10] with PhI(OAc)2 [11]. Exposure of 5b to [Cu(OTf)2] in CH2Cl2 at 0° C in the presence of chiral ligands A – E resulted in intramolecular CH insertion and afforded the cyclopentanone carboxylate 6. The enantioselectivity of the reaction was established on the ketone 7 (g. c., DAICEL, Lipodex B), which was obtained via ester hydrolysis of 6 (HBr / EtOH) and subsequent decarboxylation of the intermediate β-ketoacid. Reactions with the diazo compound 5c were carried out in 1,2-dichloroethane at 65° C. The results are summarized in Table 1.
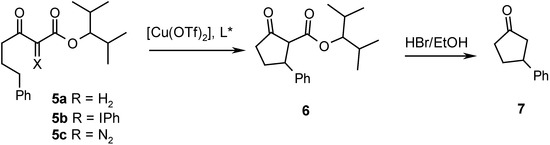
Scheme 3.
Scheme 3.
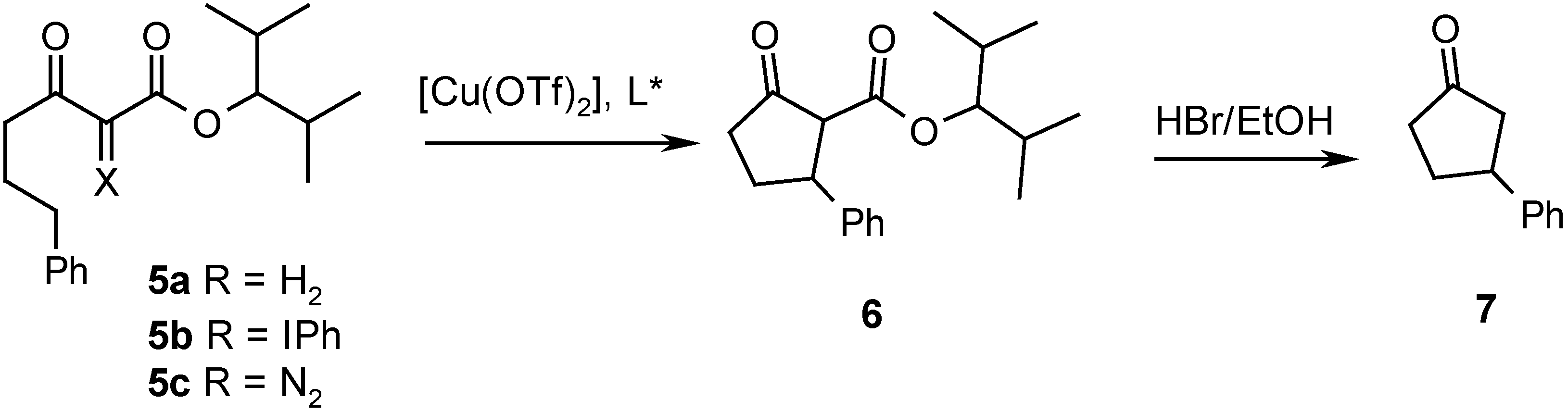

Table 1.
Yields and enantioselectivities in intramolecular CH insertions of phenyliodonium ylide 5b and diazo ketoester 5c
| Entry | Ligand | Yield from 5b (%)a) | ee from 5b (%) | Yield from 5c (%)b) | ee from 5c (%) |
| 1 | A | 55 | 22 | -- | -- |
| 2 | B | 49 | 70 | 17 | 51 |
| 3 | C | 46 | 59 | 35 | 60 |
| 4 | D | 52 | 72 | 14 | 31 |
| 5 | E | 51 | 42 | 38 | 15 |
| 6 | F | 49 | 38 | 32 | 18 |
(a) in CH2Cl2, 0° C. (b) in DCE, 65° C.
In general, we find that the Cu-catalyzed insertions proceed with acceptable yields from the ylide. The occurrence of CH insertions upon catalysis with Cu is remarkable in itself, since it is well known, that Cu-catalysts are the catalysts of choice for cyclopropanations and are less suitable for CH insertions. However, this preference is only significant when cyclopropanation and insertion pathways are competitive, and this is not the case with 5b and 5c. Other Cu-catalyzed CH insertions of diazocompounds have been reported [12]. The yields of insertion product 6 resulting from ylide decomposition are generally higher than those of the diazo decomposition. This is a consequence of the notorious low reactivity of diazo esters and diazo ketones derived from β-dicarbonyl compounds, which require temperatures of up to 80° C with Cu-catalysts and with dirhodium(II)-carboxamidates [13]. Phenyliodonium ylides are significantly more reactive and may be decomposed already at 0° C with these catalysts. This enhanced reactivity in comparison of that of diazo compounds constitutes the main interest of phenyliodonium ylides in the context of metal carbenoid reactions.
The enantioselectivity resulting from ylide decomposition is with all ligands higher than that from diazo decomposition, except with ligand C, where it is essentially equal within the limits of experimental error. This trend is mainly due to the different temperatures of the reactions. In addition, catalyst stability becomes a problem at elevated temperatures, and the low ee’s observed in some of the diazo decompositions may be due to partial degradation of the catalyst. The intriguing irregularities in the enantioselectivities of Cu-catalyzed cyclopropanations of phenyliodonium ylides and diazo compounds do clearly not occur in the CH insertions. These observations are not only of mechanistic interest; they also extend the synthetic potential of phenyliodonium ylides.
Ligands.
Ligands.
Conclusions
To our knowledge, these are the first enantioselective CH insertions observed upon Cu-catalyzed decompositions of phenyliodonium ylides. The results show clearly that the reactions proceed in the intimate vicinity of the chiral catalyst, and that the mechanism proposed by Moriarty for cyclopropanations cannot apply to the CH insertions. A carbenoid mechanism is generally accepted for CH insertions resulting from transition metal-catalyzed diazo decomposition, and the same mechanism should apply to the reaction of phenyliodonium ylides. This mechanism requires retention of configuration at the center undergoing insertion. Verification of the stereochemistry of the Cu-catalyzed CH insertion of phenyliodonium ylides is currently in progress in this laboratory.
Experimental
General
For general experimental details and instrumentation see [14]. The abbreviation FC refers to flash chromatography.
Synthesis of phenyliodonium ylide 5b and diazo keto ester 5c: see refs [5a] and [10].
Intramolecular CH-insertion with 5b. General procedure: The appropriate ligand (13.0 μmol) and [Cu(OTf)2] (3.9 mg, 11 μmol) were stirred in CH2Cl2 (6.0 mL) during 1 h. The ylide 5d (273 mg, 0.54 mmol) in CH2Cl2 (6.0 mL) was added at 0° C and the mixture was stirred overnight at 0° C. After evaporation of the solvent, the residue was purified by FC (SiO2, pentane/AcOEt 97:3) to give 6. For yields and enantioselectivity: see Table 1. For analytical data of 6 and 7, see ref. [5a].
Intramolecular CH insertion with 5c. Same procedure, but in DCE at 65° C.
Preparation of catalysts: The following ligands were synthesized according to published procedures: A: ref. [15]; D: ref. [16]; E: ref. [17]. The synthesis of ligands B and C will be reported elsewhere [18].
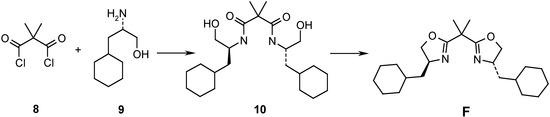
Scheme 4.
Scheme 4.
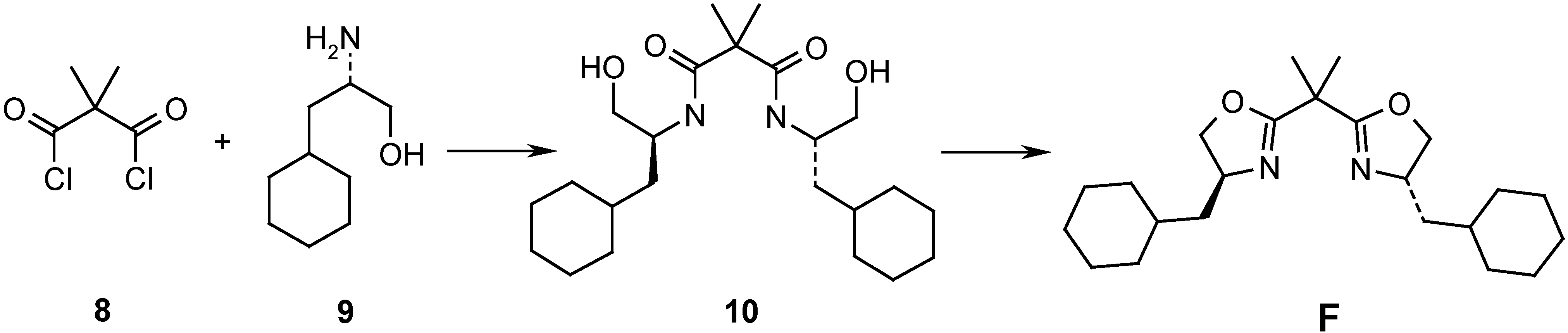
N,N’-Bis[(1S)-2-cyclohexyl-1-hydroxymethylethyl]-2,2-dimethylpropane-1,3-diamide (10). Dimethyl-malonyl dichloride (8, 500 mg, 2.96 mmol) in CH2Cl2 (3.0 mL) was added slowly, at 0oC, to (S)-3-cyclohexyl-2-aminopropanol hydrochloride (9, 6.2 mmol) and Et3N (2.5 ml, 17.7 mmol). The mixture was stirred overnight at r.t.. It was diluted with CH2Cl2 (10 mL) and washed with 1N HCl (10 mL). After usual work-up, the crude product was recrystallized (hexane/AcOEt) to afford 10 (1.20 g, 99 %), m.p. 139 C. [α]D20 = -9.6 (c = 1.15, CHCl3). IR (CHCl3): 3420m, 3030s, 2926s, 1654s, 1509s, 1230s, 1202s. 1H- NMR (400 MHz, CDCl3): 0.79-1.02 (m, 4H); 1.08-1.43 (m, 12H); 1.46 (s, 6H); 1.61-1.79 (m, 10H); 3.30-3.45 (m, 2H); 3.71 (dd, J=2.8, 11.4, 2H); 3.64-3.92 (s, br., 2H); 4.05-4.18 (m, 2H); 6.39 (d, J=8.6, 2H). 13C NMR (100 MHz): 23.4 (q); 26.0 (t); 26.1 (t); 26.3 (t); 32.6 (t); 33.6 (t); 34.3 (d); 38.1 (t); 49.2 (d); 49.9 (s); 65.7 (t); 174.2 (s). MS: 380 (M+-CH2O, 41), 379 (89), 362 (10), 361 (29), 255 (13), 254 (79), 227 (31), 209 (31), 208 (12), 198 (20), 184 (27), 141 (20), 140 (20), 126 (53), 123 (23), 114 (11), 95 (18), 88 (13), 86 (13), 84 (19), 83 (30), 82 (10), 81 (57), 71 (32), 70 (45), 69 (48), 67 (35), 60 (13), 58 (24), 57 (13), 56 (13), 55 (100), 53 (11). HRMS: 380.2999 (C22H40N2O3+ (M+-CH2O); calc. 380.3039).
2,2-Dimethyl-1,3-bis[(4S)-4-cyclohexylmethyloxazolin-2-yl]-propane (F). SOCl2 (1.90 mL, 7.0 mmol) was added dropwise to 10 (0.70 mmol) in CH2Cl2 (5.0 mL). The mixture was refluxed for 4 h, then poured on ice. The organic layer was separated, washed with satd. NaCl and 0.1M K2CO3. The combined organic phases were dried (Na2SO4) and evaporated. The residue was purified by FC (pentane/AcOEt 70:30) to afford an oil, which was heated to reflux (2 h) after addition of MeOH (4.0 mL), H2O (4.0 mL) and solid NaOH (169 mg, 6 equiv.). The mixture was then extracted with CH2Cl2, the organic phase was dried (Na2SO4) and evaporated. The residue was purified by FC (pentane/AcOEt 70:30) and afforded F as viscous oil, [α]D20 = -68.6 (c=1.04, CHCl3). IR (film): 2921s, 2851s, 1659s, 1448s, 1351w, 1147m, 981s. 1H-NMR (400 MHz, CDCl3): 0.86-1.01 (m, 4H); 1.10-1.43 (m, 10H); 1.51 (s, 6H); 1.60-1.83 (m, 12H); 3.83-3.90 (m, 2H); 4.13-4.22 (m, 2H); 4.32 (dd, J=8.1, 9.4, 2H). 13C-NMR (100 MHz): 24.3 (q); 26.0 (t); 26.1 (t); 26.4 (t); 33.1 (t); 33.8 (t); 34.7 (d); 38.4 (s); 43.8 (t); 64.0 (d); 73.3 (t); 168.6 (s). MS: 374 (M+, 1), 362 (21), 361 (62), 277 (27), 253 (12), 210 (16), 209 (100), 208 (27), 196 (23), 166 (12), 127 (16), 126 (74), 112 (13), 88 (14), 83 (17), 82 (11), 81 (27), 71 (19), 69 (39), 67 (20), 57 (21), 56 (12), 55 (61). HRMS: 374.2920 (C23H38N2O2+; calc. 374.2933).
Acknowledgments
This work was supported by the Swiss National Science Foundation (Grant Nos. 20-52581.97 and 2027-048156) and by the European Commission for Science, Research and Development (COST Action D12).
References and Notes
- Camacho, M. B.; Clark, A. E.; Liebrecht, T. A.; DeLuca, J. P. A phenyliodonium ylide as a precursor for dicarboethoxycarbenes: Demonstration of a strategy for carbene generation. J. Am. Chem. Soc. 2000, 122, 5210–5211. [Google Scholar] [CrossRef]
- Hayasi, Y.; Okada, T.; Kawanisi, M. Cyclic diacylcarbenes generated from iodonium ylides and diazodiketones. Bull. Chem. Soc. Jpn. 1970, 43, 2506–2511. [Google Scholar] [CrossRef]
- Hadjiarapoglou, L.; Varvoglis, A.; Alcock, N. W.; Pike, G. A. Reactivity of Phenyliodonium bis(arylsulphonyl)methylides towards alkenes and alkynes: Crystal structure of 9-phenylsulfonyl-1,2,3,4,4a,9a-hexahydro-1,4-methanofluorene. J. Chem. Soc. Perkin Trans. 1 1988, 2839–2846. [Google Scholar] [CrossRef] Hadjiarapoglou, L.; Spyroudis, S.; Varvoglis, A. Phenyliodonium bis(phenylsulfonyl)methylide: A new hypervalent iodonium ylide. J. Am. Chem. Soc. 1985, 107, 7178–7179. [Google Scholar] [CrossRef] Hadjiarapoglou, L. The chemistry of photolytically and thermally generated α-ketocarbenes from iodonium ylides of β−diketones. Tetrahedron Lett. 1987, 28, 4449–4450. [Google Scholar] [CrossRef]
- Hood, J. N. C.; Lloyd, D.; McDonald, W. A.; Shepherd, T. M. The effects of different copper (and some other) catalysts on the conversion of triphenyl- and tetraphenyl-diazocyclopentadienes and of some phenyliodonium α,α’-dicarbonylylides into arsonium and other ylides. Tetrahedron 1982, 38, 3355–3358. [Google Scholar] Saito, T.; Kikuchi, K.; Kondo, A. A new and simple synthesis of heterocycle-fused [c]thiophenes: Reaction of heteroaromatic thioketones with bis(arylsulfonyl)diazomethanes and phenyliodonium bis(phenylsulfonyl)-methylide. Synthesis 1995, 87–91. [Google Scholar] [CrossRef] Saito, T.; Gon, S.; Kikuchi, H.; Motoki, S. 2, 223–235. Fairfax, D. J.; Austin, D. J.; Xu, S. L.; Padwa, A. Alternatives to α-diazo ketones for tandem cyclization-cycloaddition and carbenoid-alkyne metathesis strategies. Novel cyclic enol ether formation via carbonyl ylide rearrangement reactions. J. Chem. Soc. Perkin Trans. 1 1992, 2837–2844. [Google Scholar] [CrossRef]
- Müller, P.; Fernandez, D. Carbenoid reactions in rhodium(II)-catalyzed decomposition of iodonium ylides. Helv. Chim. Acta 1995, 78, 947–958. [Google Scholar] [CrossRef] Müller, P.; Fernandez, D.; Nury, P.; Rossier, J.-C. Metal-catalyzed carbenoid reactions with iodonium and sulfonium ylides. J. Phys. Org. Chem. 1998, 11, 321–333. [Google Scholar] [CrossRef]
- Doyle, M. P.; McKervey, M. A.; Ye, T. Modern Catalytic Methods for Organic Synthesis with Diazo Compounds: From Cyclopropanes to Ylides; Wiley: New York, 1998; Chapter 2. [Google Scholar] Doyle, M. P.; Forbes, D. C. Recent advances in asymmetric catalytic metal carbene transformations. Chem. Rev. 1998, 98, 911–935. [Google Scholar] [CrossRef] [PubMed]
- Moriarty, R. M.; Prakash, O.; Vaid, R. K.; Zhao, L. A novel intramolecular cyclopropanation using iodonium ylides. J. Am. Chem. Soc. 1989, 111, 6443–6444. [Google Scholar] [CrossRef] Moriarty, R. M.; Kim, J.; Guo, L. Intramolecular cyclopropanation using iodonium ylides: The 3,5-cyclovitamin D ring A synthon. Tetrahedron Lett. 1993, 34, 4129–4132. [Google Scholar] [CrossRef] Moriarty, R. M.; May, E. J.; Prakash, O. Intramolecular cyclization of aryl substituted iodonium ylides with copper(I) chloride. Tetrahedron Lett. 1997, 38, 4333–4336. [Google Scholar] [CrossRef] Gallos, J. K.; Koftis, T. V.; Koumbis, A. E. Synthesis of enantiomerically pure bicyclo[3.1.0]hexanes from D-ribose by intramolecular cyclopropanation. J. Chem. Soc. Perkin Trans. 1 1994, 611–612. [Google Scholar] [CrossRef]
- Müller, P.; Boléa, C. Asymmetric induction in Cu-catalyzed intramolecular cyclopropanations of phenyliodonium ylides. Synlett 2000, 826–828. [Google Scholar]
- Preliminary communication: Müller, P.; Boléa, C. Enantioselective intramolecular CH-insertions upon Cu-catalyzed decomposition of phenyliodonium ylides 4th electronic Conference on Synthetic Organic Chemistry, ecsoc.-4:. http://www.mdpi.org/ecsoc-4a0015/0015.htm.
- Hashimoto, S.; Watanabe, N.; Sato, T.; Shiro, M.; Ikegami, S. Enhancement of enantioselectivity in intramolecular CH-insertion reactions of α-diazo β-keto esters catalyzed by chiral dirhodium(II) carboxylates. Tetrahedron Lett. 1993, 32, 5109–5112. [Google Scholar] [CrossRef] Hashimoto, S.; Watanabe, N.; Ikegami, S. Enantioselective intramolecular CH insertion reactions of α-diazo β-keto esters catalyzed by dirhodium(II) tetrakis[N-phthaloyl-(S)-phenylalaninate]: The effect of the substituent at the insertion site on enantioselectivity. Synlett 1994, 353–355. [Google Scholar] [CrossRef] Hashimoto, S.; Watanabe, N.; Kawano, K.; Ikegami, S. Double asymmetric induction in intramolecular CH insertion reactions of α-diazo β-keto esters. Synth. Commun. 1994, 24, 3277–3287. [Google Scholar] [CrossRef]
- Schank, K.; Lick, C. Ozonolytic fragmentation of phenyliodonium β-diketonates, a convenient synthesis of unsolvated vic-triketones. Synthesis 1983, 392–395. [Google Scholar] [CrossRef]
- Müller, P.; Maîtrejean, E. Rhodium(II)- and copper(I)-catalyzed intramolecular carbon-hydrogen bond insertions with metal carbenoids derived from diazo ketones. Collect. Czech. Chem. Commun. 1999, 64, 1807–1825. [Google Scholar] [CrossRef]
- Doyle, M. P.; Davies, S. B.; Hu, W. Dirhodium(II) tetrakis[methyl 2-oxaazetidine-4-carboxylate]: A chiral dirhodium(II) carboxamidate of exceptional reactivity and selectivity. Org. Lett. 2000, 2, 1145–1147. [Google Scholar] [CrossRef] [PubMed]
- Doyle, M. P.; Winchester, W. R.; Protopopova, M. N.; Müller, P.; Bernardinelli, G.; Ene, D.; Motallebi, S. Tetrakis[(4S)-4-phenyloxazolidin-2-one]dirhodium(II) and its catalytic applications for metal carbene transformations. Helv. Chim. Acta 1993, 76, 2227–2235. [Google Scholar] [CrossRef] Müller, P.; Nury, P. Desymmetrization of meso-N-sulfonated aziridines with chiral nonracemic nucleophiles and bases. Helv. Chim. Acta 2000. submitted. [Google Scholar]
- Andrus, M. B.; Asgari, D.; Sclafani, J. A. Efficient synthesis of 1,1'-binaphthyl and 2,2'-bi-o-tolyl-2,2'-bis(oxazoline)s and preliminary use for the catalytic asymmetric allylic oxidation of cyclohexene. J. Org. Chem. 1997, 62, 9365–9368. [Google Scholar] [CrossRef]
- Ghosh, A. K.; Mathivanan, P.; Cappiello, J. Conformationally constrained bis(oxazoline) derived chiral catalysts: A highly effective enantioselective Diels-Alder Reaction. Tetrahedron Lett. 1996, 37, 3815–1818. [Google Scholar] [CrossRef]
- Evans, D. A.; Woerpel, K. A.; Hinman, M. M.; Faul, M. M. Bis(oxazolines) as chiral ligands in metal-catalyzed asymmetric reactions. Catalytic asymmetric cyclopropanation of olefins. J. Am. Chem. Soc. 1991, 113, 726–728. [Google Scholar] [CrossRef] Evans, D. A.; Peterson, G. S.; Johnson, J. S.; Barnes, D. M.; Campos, K. R.; Woerpel, K. A. An improved procedure for the preparation of 2,2-bis[2-[4(S)-tert-butyl-1,3-oxazolinyl]]propane [(S,S)-tert-butyl bis(oxazoline)] and derived copper(II) complexes. J. Org. Chem. 1998, 63, 4541–4544. [Google Scholar] [CrossRef]
- Müller, P.; Boléa, C. Carbenoid pathways in Cu-catalyzed intramolecular cyclopropanations of phenyliodonium ylides. Helv. Chim. Acta 2001. in preparation. [Google Scholar]
- Sample Availability:. Samples not available.
© 2001 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes