Enantioselective Intramolecular CH-Insertions upon Cu-Catalyzed Decomposition of Phenyliodonium Ylides

Abstract
:Introduction
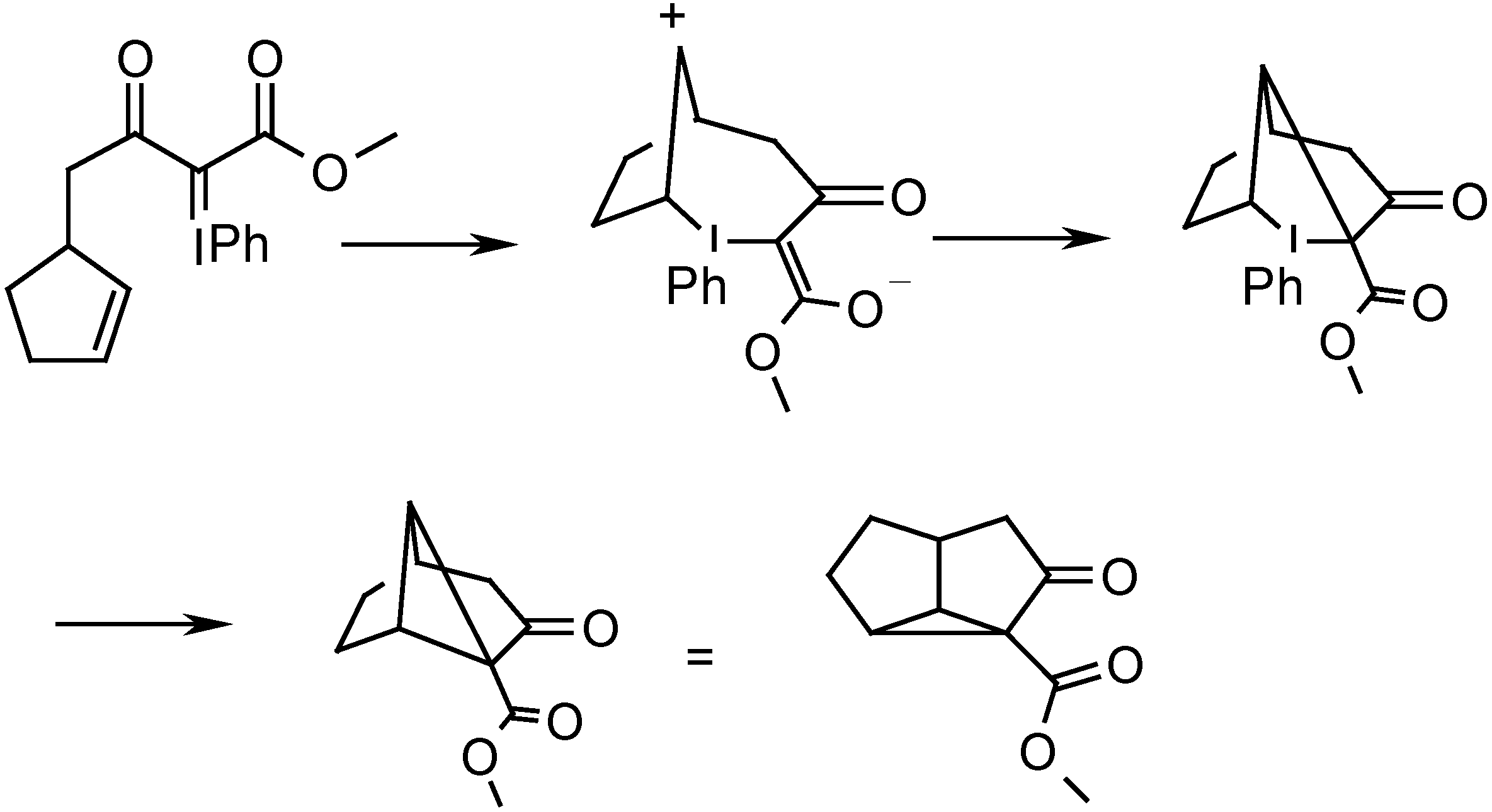
Results and Discussion

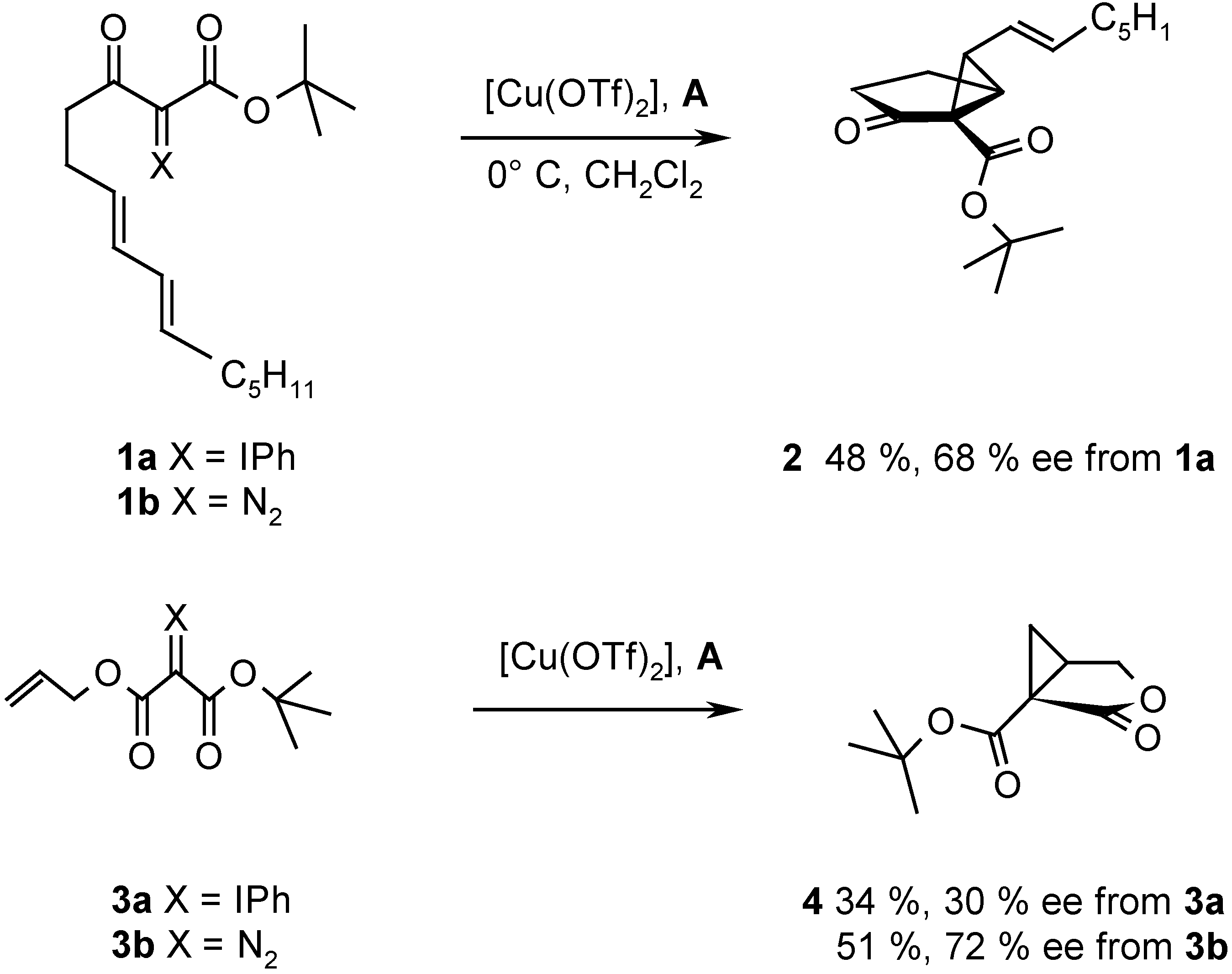
{kind=link}
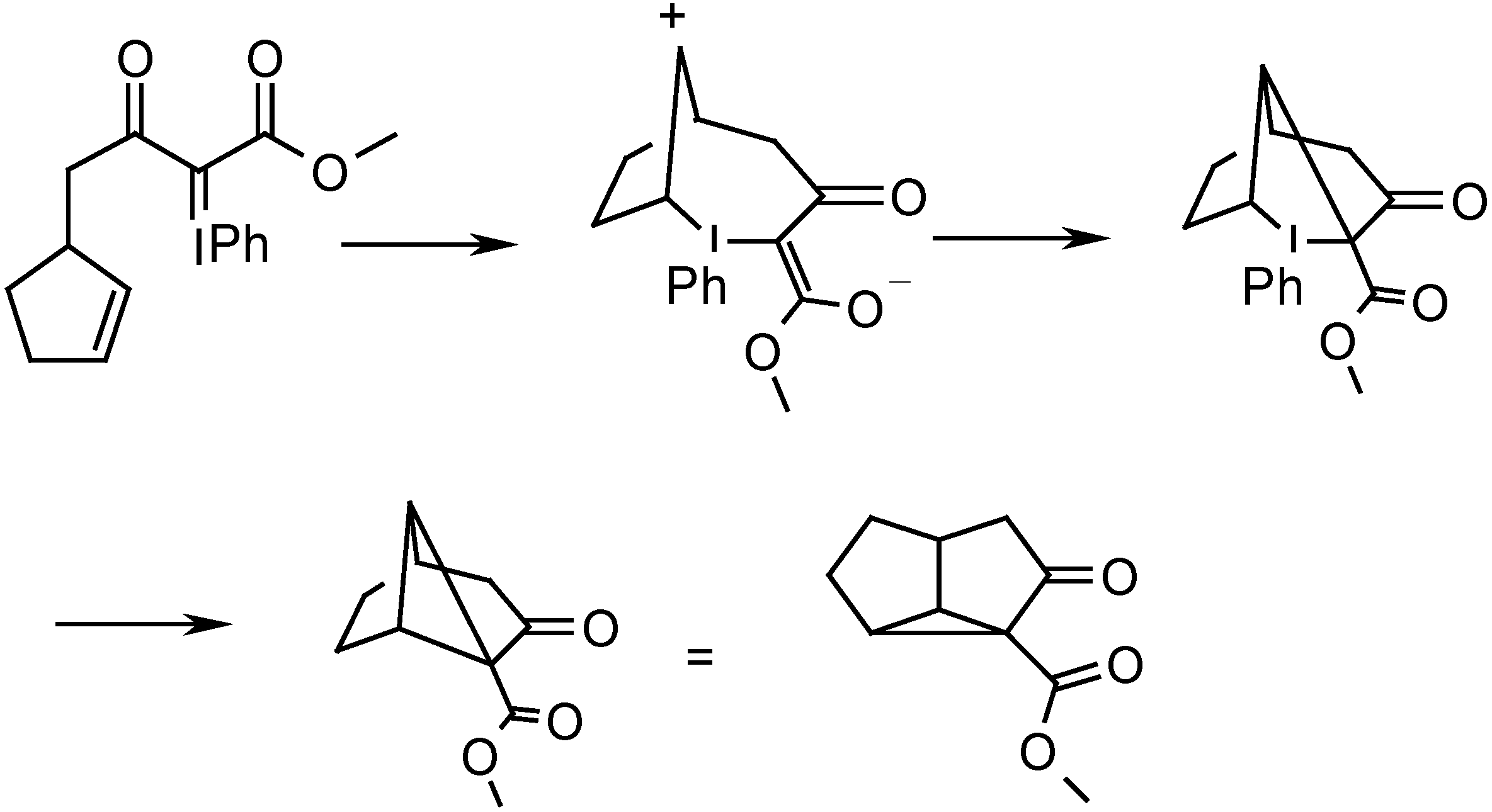{kind=link}
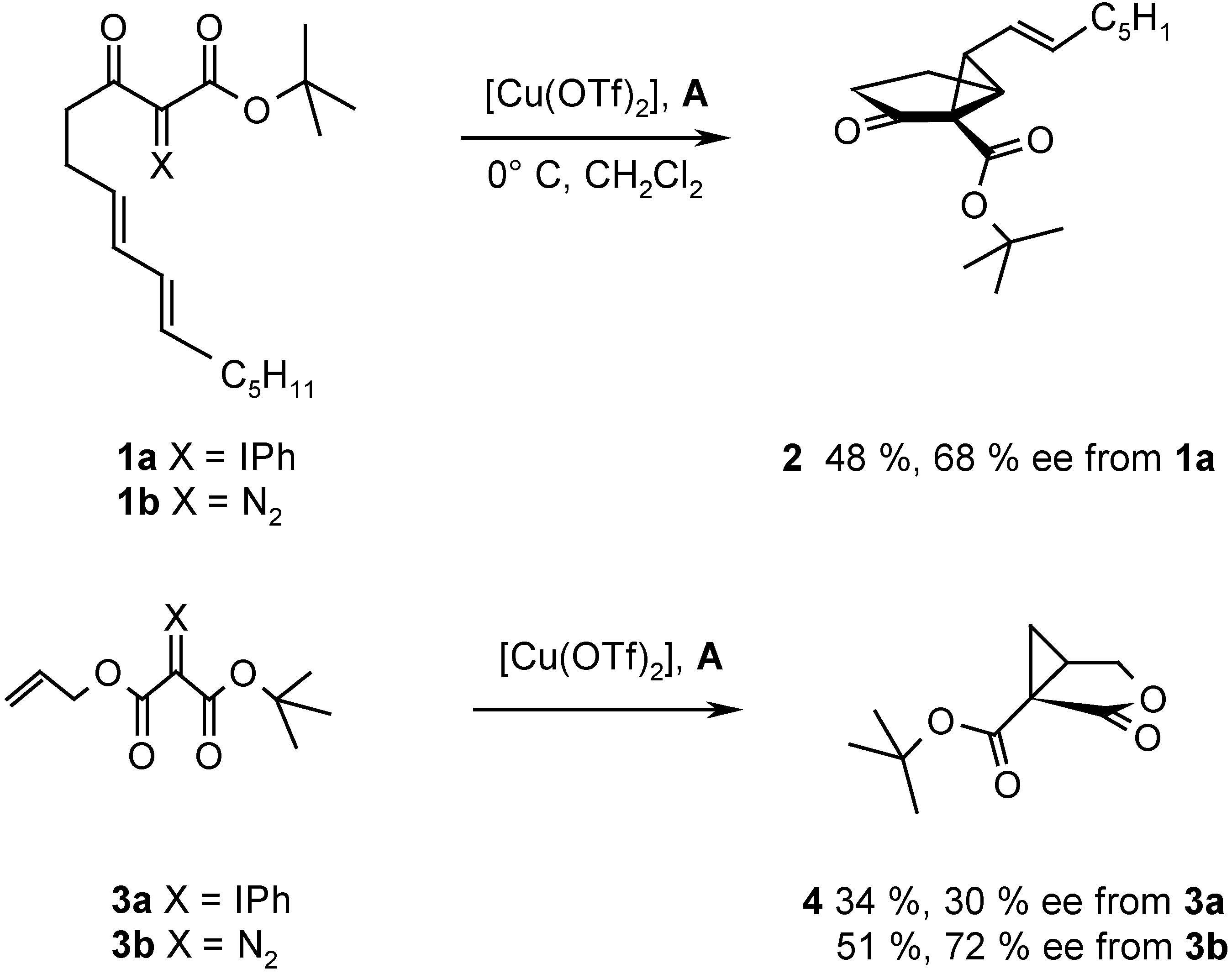{kind=link}
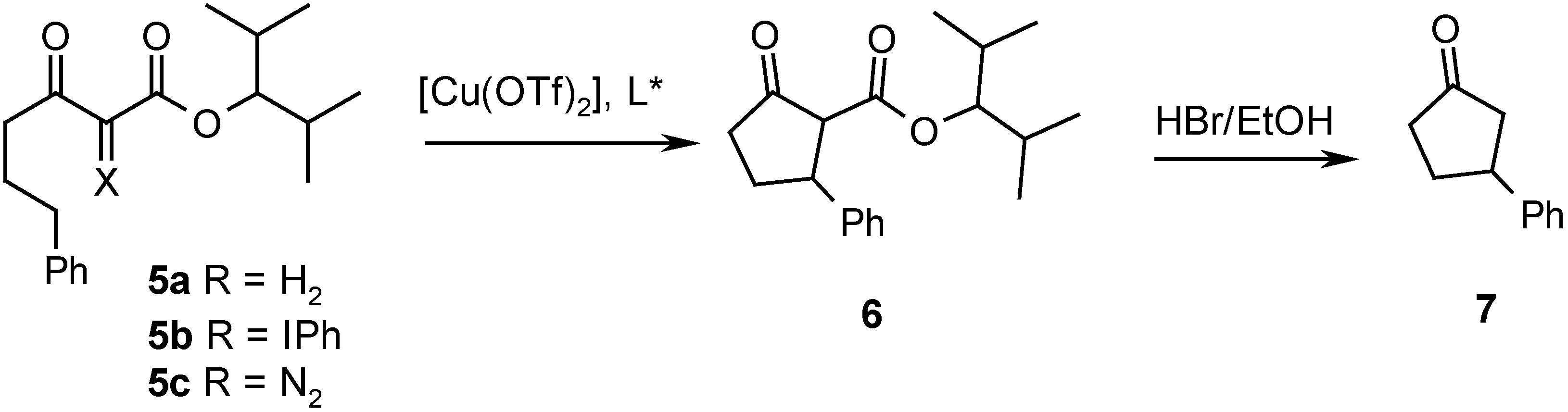{kind=link}
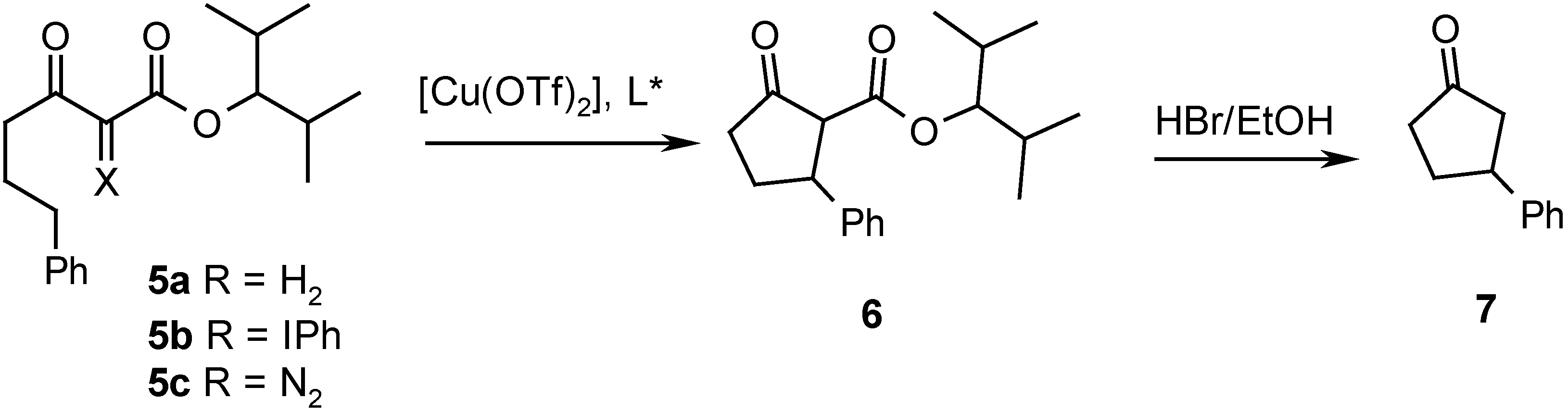
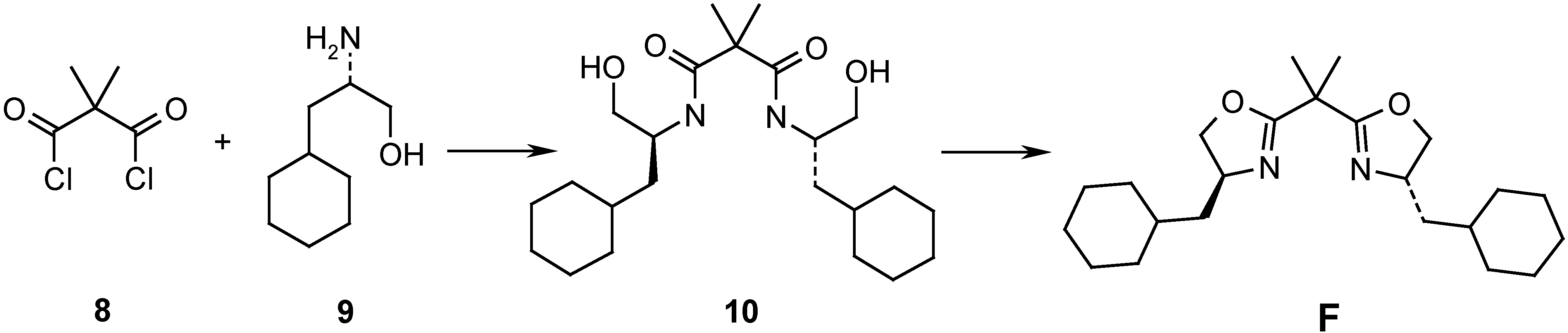{kind=link}
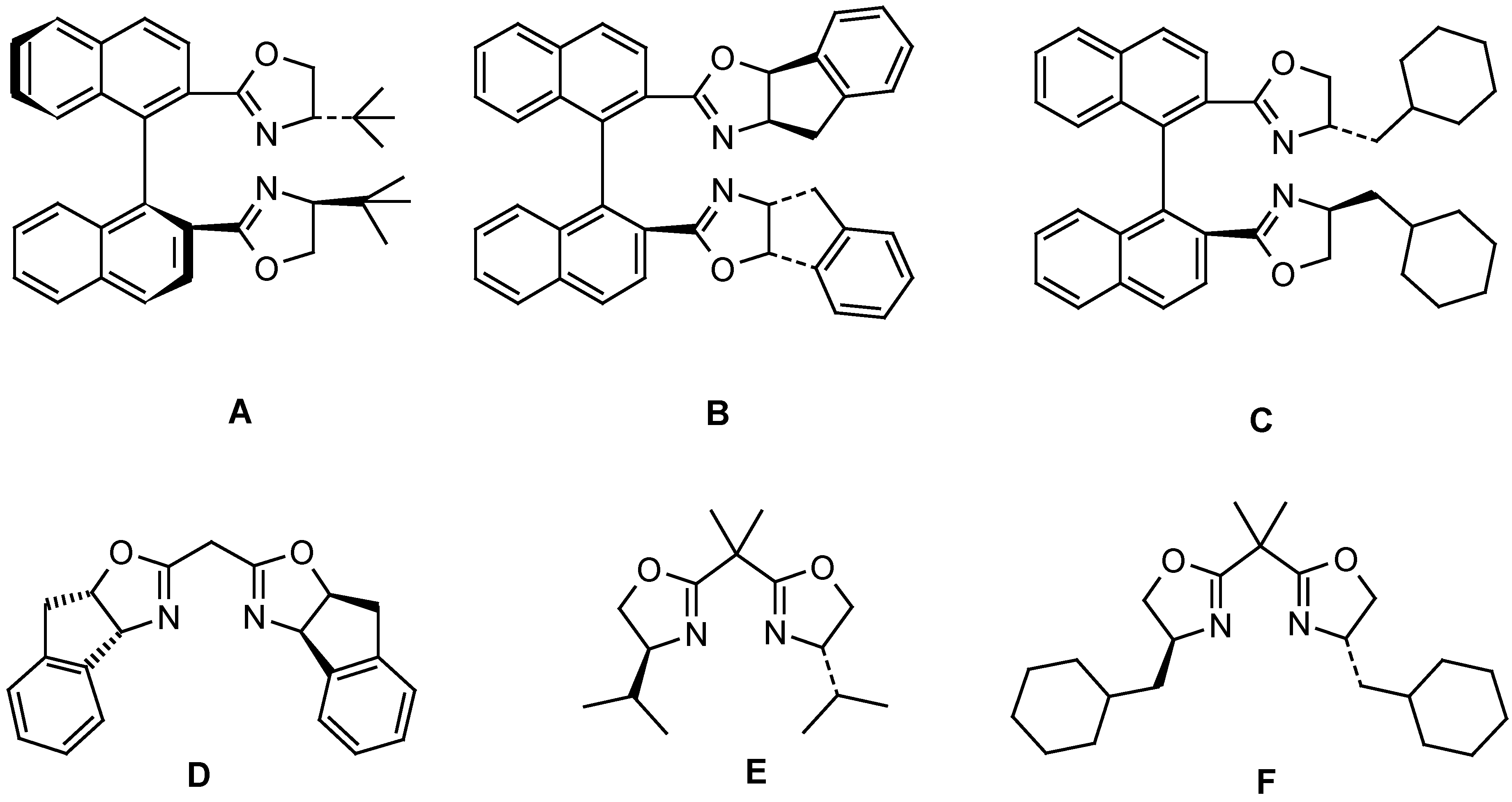
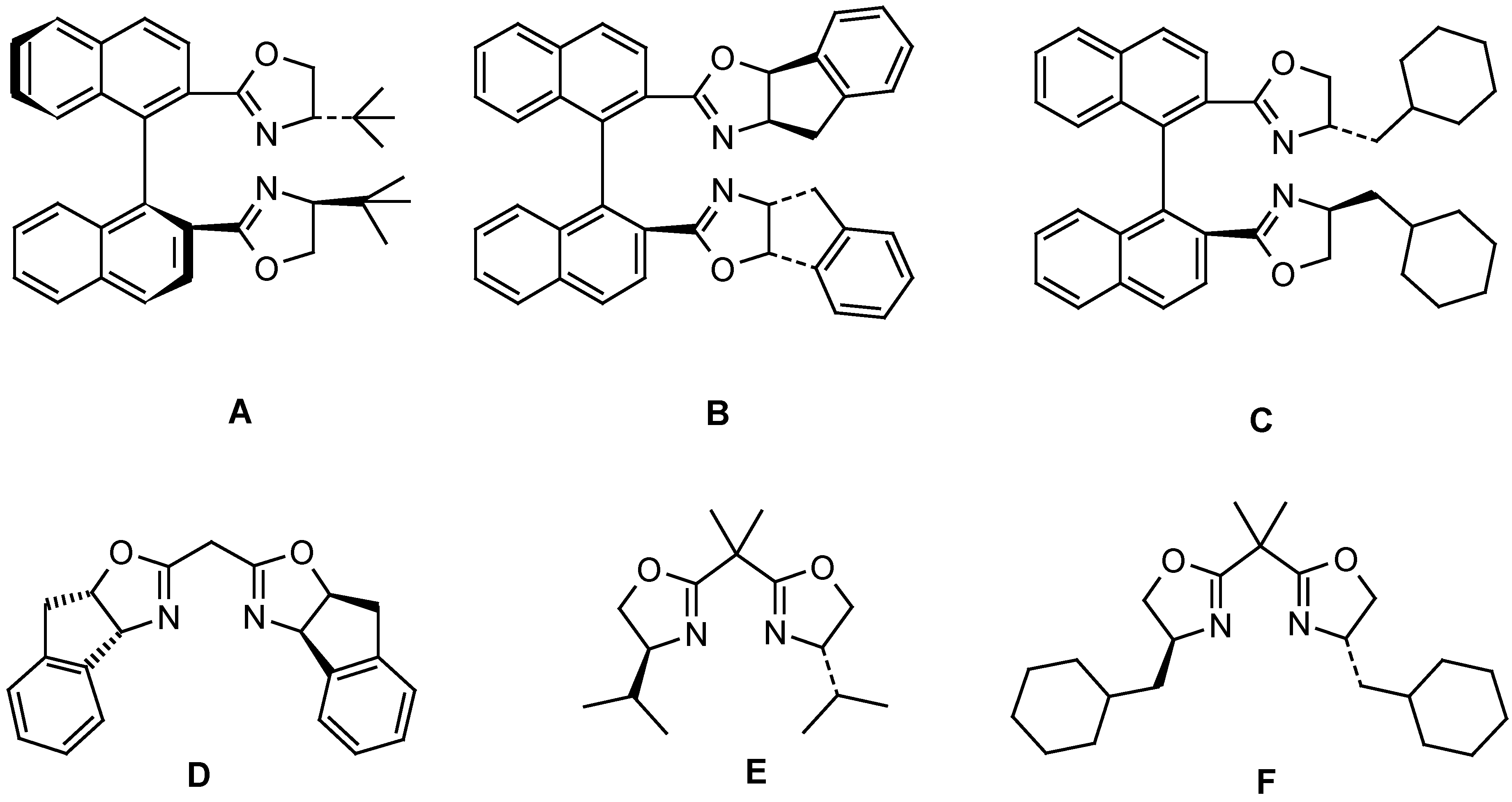
| Entry | Ligand | Yield from 5b (%)a) | ee from 5b (%) | Yield from 5c (%)b) | ee from 5c (%) |
| 1 | A | 55 | 22 | -- | -- |
| 2 | B | 49 | 70 | 17 | 51 |
| 3 | C | 46 | 59 | 35 | 60 |
| 4 | D | 52 | 72 | 14 | 31 |
| 5 | E | 51 | 42 | 38 | 15 |
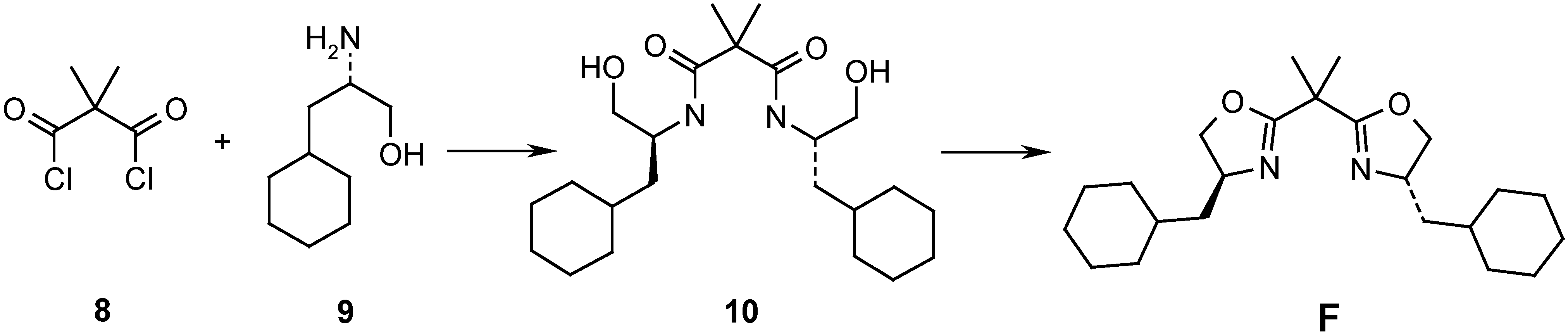
| 6 | F | 49 | 38 | 32 | 18 |
Conclusions
Experimental
General
Acknowledgments
References and Notes
- Camacho, M. B.; Clark, A. E.; Liebrecht, T. A.; DeLuca, J. P. A phenyliodonium ylide as a precursor for dicarboethoxycarbenes: Demonstration of a strategy for carbene generation. J. Am. Chem. Soc. 2000, 122, 5210–5211. [Google Scholar] [CrossRef]
- Hayasi, Y.; Okada, T.; Kawanisi, M. Cyclic diacylcarbenes generated from iodonium ylides and diazodiketones. Bull. Chem. Soc. Jpn. 1970, 43, 2506–2511. [Google Scholar] [CrossRef]
- Hadjiarapoglou, L.; Varvoglis, A.; Alcock, N. W.; Pike, G. A. Reactivity of Phenyliodonium bis(arylsulphonyl)methylides towards alkenes and alkynes: Crystal structure of 9-phenylsulfonyl-1,2,3,4,4a,9a-hexahydro-1,4-methanofluorene. J. Chem. Soc. Perkin Trans. 1 1988, 2839–2846. [Google Scholar] [CrossRef] Hadjiarapoglou, L.; Spyroudis, S.; Varvoglis, A. Phenyliodonium bis(phenylsulfonyl)methylide: A new hypervalent iodonium ylide. J. Am. Chem. Soc. 1985, 107, 7178–7179. [Google Scholar] [CrossRef] Hadjiarapoglou, L. The chemistry of photolytically and thermally generated α-ketocarbenes from iodonium ylides of β−diketones. Tetrahedron Lett. 1987, 28, 4449–4450. [Google Scholar] [CrossRef]
- Hood, J. N. C.; Lloyd, D.; McDonald, W. A.; Shepherd, T. M. The effects of different copper (and some other) catalysts on the conversion of triphenyl- and tetraphenyl-diazocyclopentadienes and of some phenyliodonium α,α’-dicarbonylylides into arsonium and other ylides. Tetrahedron 1982, 38, 3355–3358. [Google Scholar] Saito, T.; Kikuchi, K.; Kondo, A. A new and simple synthesis of heterocycle-fused [c]thiophenes: Reaction of heteroaromatic thioketones with bis(arylsulfonyl)diazomethanes and phenyliodonium bis(phenylsulfonyl)-methylide. Synthesis 1995, 87–91. [Google Scholar] [CrossRef] Saito, T.; Gon, S.; Kikuchi, H.; Motoki, S. 2, 223–235. Fairfax, D. J.; Austin, D. J.; Xu, S. L.; Padwa, A. Alternatives to α-diazo ketones for tandem cyclization-cycloaddition and carbenoid-alkyne metathesis strategies. Novel cyclic enol ether formation via carbonyl ylide rearrangement reactions. J. Chem. Soc. Perkin Trans. 1 1992, 2837–2844. [Google Scholar] [CrossRef]
- Müller, P.; Fernandez, D. Carbenoid reactions in rhodium(II)-catalyzed decomposition of iodonium ylides. Helv. Chim. Acta 1995, 78, 947–958. [Google Scholar] [CrossRef] Müller, P.; Fernandez, D.; Nury, P.; Rossier, J.-C. Metal-catalyzed carbenoid reactions with iodonium and sulfonium ylides. J. Phys. Org. Chem. 1998, 11, 321–333. [Google Scholar] [CrossRef]
- Doyle, M. P.; McKervey, M. A.; Ye, T. Modern Catalytic Methods for Organic Synthesis with Diazo Compounds: From Cyclopropanes to Ylides; Wiley: New York, 1998; Chapter 2. [Google Scholar] Doyle, M. P.; Forbes, D. C. Recent advances in asymmetric catalytic metal carbene transformations. Chem. Rev. 1998, 98, 911–935. [Google Scholar] [CrossRef] [PubMed]
- Moriarty, R. M.; Prakash, O.; Vaid, R. K.; Zhao, L. A novel intramolecular cyclopropanation using iodonium ylides. J. Am. Chem. Soc. 1989, 111, 6443–6444. [Google Scholar] [CrossRef] Moriarty, R. M.; Kim, J.; Guo, L. Intramolecular cyclopropanation using iodonium ylides: The 3,5-cyclovitamin D ring A synthon. Tetrahedron Lett. 1993, 34, 4129–4132. [Google Scholar] [CrossRef] Moriarty, R. M.; May, E. J.; Prakash, O. Intramolecular cyclization of aryl substituted iodonium ylides with copper(I) chloride. Tetrahedron Lett. 1997, 38, 4333–4336. [Google Scholar] [CrossRef] Gallos, J. K.; Koftis, T. V.; Koumbis, A. E. Synthesis of enantiomerically pure bicyclo[3.1.0]hexanes from D-ribose by intramolecular cyclopropanation. J. Chem. Soc. Perkin Trans. 1 1994, 611–612. [Google Scholar] [CrossRef]
- Müller, P.; Boléa, C. Asymmetric induction in Cu-catalyzed intramolecular cyclopropanations of phenyliodonium ylides. Synlett 2000, 826–828. [Google Scholar]
- Preliminary communication: Müller, P.; Boléa, C. Enantioselective intramolecular CH-insertions upon Cu-catalyzed decomposition of phenyliodonium ylides 4th electronic Conference on Synthetic Organic Chemistry, ecsoc.-4:. http://www.mdpi.org/ecsoc-4a0015/0015.htm.
- Hashimoto, S.; Watanabe, N.; Sato, T.; Shiro, M.; Ikegami, S. Enhancement of enantioselectivity in intramolecular CH-insertion reactions of α-diazo β-keto esters catalyzed by chiral dirhodium(II) carboxylates. Tetrahedron Lett. 1993, 32, 5109–5112. [Google Scholar] [CrossRef] Hashimoto, S.; Watanabe, N.; Ikegami, S. Enantioselective intramolecular CH insertion reactions of α-diazo β-keto esters catalyzed by dirhodium(II) tetrakis[N-phthaloyl-(S)-phenylalaninate]: The effect of the substituent at the insertion site on enantioselectivity. Synlett 1994, 353–355. [Google Scholar] [CrossRef] Hashimoto, S.; Watanabe, N.; Kawano, K.; Ikegami, S. Double asymmetric induction in intramolecular CH insertion reactions of α-diazo β-keto esters. Synth. Commun. 1994, 24, 3277–3287. [Google Scholar] [CrossRef]
- Schank, K.; Lick, C. Ozonolytic fragmentation of phenyliodonium β-diketonates, a convenient synthesis of unsolvated vic-triketones. Synthesis 1983, 392–395. [Google Scholar] [CrossRef]
- Müller, P.; Maîtrejean, E. Rhodium(II)- and copper(I)-catalyzed intramolecular carbon-hydrogen bond insertions with metal carbenoids derived from diazo ketones. Collect. Czech. Chem. Commun. 1999, 64, 1807–1825. [Google Scholar] [CrossRef]
- Doyle, M. P.; Davies, S. B.; Hu, W. Dirhodium(II) tetrakis[methyl 2-oxaazetidine-4-carboxylate]: A chiral dirhodium(II) carboxamidate of exceptional reactivity and selectivity. Org. Lett. 2000, 2, 1145–1147. [Google Scholar] [CrossRef] [PubMed]
- Doyle, M. P.; Winchester, W. R.; Protopopova, M. N.; Müller, P.; Bernardinelli, G.; Ene, D.; Motallebi, S. Tetrakis[(4S)-4-phenyloxazolidin-2-one]dirhodium(II) and its catalytic applications for metal carbene transformations. Helv. Chim. Acta 1993, 76, 2227–2235. [Google Scholar] [CrossRef] Müller, P.; Nury, P. Desymmetrization of meso-N-sulfonated aziridines with chiral nonracemic nucleophiles and bases. Helv. Chim. Acta 2000. submitted. [Google Scholar]
- Andrus, M. B.; Asgari, D.; Sclafani, J. A. Efficient synthesis of 1,1'-binaphthyl and 2,2'-bi-o-tolyl-2,2'-bis(oxazoline)s and preliminary use for the catalytic asymmetric allylic oxidation of cyclohexene. J. Org. Chem. 1997, 62, 9365–9368. [Google Scholar] [CrossRef]
- Ghosh, A. K.; Mathivanan, P.; Cappiello, J. Conformationally constrained bis(oxazoline) derived chiral catalysts: A highly effective enantioselective Diels-Alder Reaction. Tetrahedron Lett. 1996, 37, 3815–1818. [Google Scholar] [CrossRef]
- Evans, D. A.; Woerpel, K. A.; Hinman, M. M.; Faul, M. M. Bis(oxazolines) as chiral ligands in metal-catalyzed asymmetric reactions. Catalytic asymmetric cyclopropanation of olefins. J. Am. Chem. Soc. 1991, 113, 726–728. [Google Scholar] [CrossRef] Evans, D. A.; Peterson, G. S.; Johnson, J. S.; Barnes, D. M.; Campos, K. R.; Woerpel, K. A. An improved procedure for the preparation of 2,2-bis[2-[4(S)-tert-butyl-1,3-oxazolinyl]]propane [(S,S)-tert-butyl bis(oxazoline)] and derived copper(II) complexes. J. Org. Chem. 1998, 63, 4541–4544. [Google Scholar] [CrossRef]
- Müller, P.; Boléa, C. Carbenoid pathways in Cu-catalyzed intramolecular cyclopropanations of phenyliodonium ylides. Helv. Chim. Acta 2001. in preparation. [Google Scholar]
- Sample Availability:. Samples not available.
© 2001 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes
Share and Cite
Müller, P.; Boléa, C. Enantioselective Intramolecular CH-Insertions upon Cu-Catalyzed Decomposition of Phenyliodonium Ylides. Molecules 2001, 6, 258-266. https://doi.org/10.3390/60300258
Müller P, Boléa C. Enantioselective Intramolecular CH-Insertions upon Cu-Catalyzed Decomposition of Phenyliodonium Ylides. Molecules. 2001; 6(3):258-266. https://doi.org/10.3390/60300258
Chicago/Turabian StyleMüller, Paul, and Christelle Boléa. 2001. "Enantioselective Intramolecular CH-Insertions upon Cu-Catalyzed Decomposition of Phenyliodonium Ylides" Molecules 6, no. 3: 258-266. https://doi.org/10.3390/60300258
APA StyleMüller, P., & Boléa, C. (2001). Enantioselective Intramolecular CH-Insertions upon Cu-Catalyzed Decomposition of Phenyliodonium Ylides. Molecules, 6(3), 258-266. https://doi.org/10.3390/60300258