
NMR and DFT Studies on Solvation Phenomena in Bioorganic Molecules, Natural Products and Model Compounds: Current and Future Perspectives for Atomic-Level Structures and Mechanistic Catalytic Reactions



Abstract
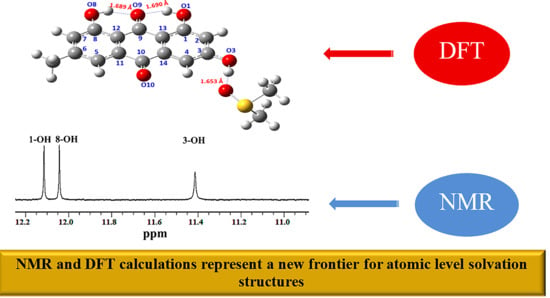
1. Introduction
- (i)
- Implicit solvation models, which treat the solvent as a continuous dielectric medium surrounding the solute molecule. Typical examples include the polarizable continuum model (PCM) [47], the integral equation formalism PCM (IEF = PCM) [48] model, the conductor-like polarizable continuum model (CPCM) [49], the conductor-like screening model (COSMO), and the conductor-like screening model for real solvents (COSMO-RS) [50]. The solvent is treated as a conductor (ε = ), and a scaling factor is used for a given solvent.
- (ii)
- Solvation models, which include explicit solvent molecules in the calculation. This can be done using molecular dynamics simulations or manually placing solvent molecules in the vicinity of potential polar or charged sites of the solute.
- (iii)
- Hybrid models, which combine implicit and explicit solvation. This can be done by including a limited number of explicit solvent molecules around, e.g., polar or charged groups, and by using an implicit model for the bulk solvent (Figure 1).

- (i)
- A systematic investigation of the factors that determine the accuracy of the chemical shift computations of 1H NMR of labile hydrogens, 14,15N, 17O, and 31P NMR.
- (ii)
- The role of explicit inclusion of solvent molecules in the calculation of chemical shifts, including the effects of increasing the number of explicit solvent molecules and the cooperative hydrogen bonding effect, using reliable and fast methods.
- (iii)
- The effects of the choice of the density functional, the size of the basis set, the effect of zero-point vibrational correction and the importance of geometry optimization in DFT calculations.
- (iv)
- The dynamic nature of molecules in solutions results in chemical shifts that are weighted averages of all accessible low-energy solvation structures, which can significantly add to the computational cost.
- (v)
- A systematic investigation of the role of solvents in various atomistic reaction mechanisms with the combined use of calculated and experimentally determined activation free energies.
2. 1H NMR and DFT Studies of Solvation Phenomena in Labile Hydrogens
2.1. Phenols
2.2. Alcohols
2.3. Amides and Amines
2.4. Pyrimidine Basis
2.5. Carboxylic Groups
2.6. Enol–Enol Tautomerism
2.7. Mixed Solvents
2.8. Ionic Compounds
2.9. Hydroperoxides (R–O–O–H)
3. Nitrogen NMR and DFT Studies on Solvation Phenomena
3.1. Nitrogen Heterocycles
3.2. Amides and Amines
3.3. Effects of Protonation
4. Oxygen–17 NMR and DFT Studies on Solvation Phenomena
4.1. H2O and Alcohols
4.2. Carbonyls
4.3. Amides
4.4. Nucleobases
4.5. Organic Acids and Peracids
4.6. Amino Acids
5. Phosphorous–31 NMR and DFT Studies on Solvation Phenomena
6. Computational vs. Experimental Activation Energies, ΔG‡, as a Tool for the Role of Solvents in Atomistic Reaction Mechanisms
7. Software
8. Conclusions and Prospects for Future Research
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Reichardt, C. Solvents and solvent effects: An introduction. Org. Process Res. Dev. 2007, 11, 105–113. [Google Scholar] [CrossRef]
- Cramer, C.J.; Truhlar, D.G. A universal approach to solvation modeling. Acc. Chem. Res. 2008, 41, 760–768. [Google Scholar] [CrossRef]
- El Seoud, O.A. Understanding solvation. Pure Appl. Chem. 2009, 81, 697–707. [Google Scholar] [CrossRef]
- Reichardt, C.; Welton, T. Solvents and Solvent Effects in Organic Chemistry, 4th ed.; John Wiley & Sons: Hoboken, NJ, USA, 2011. [Google Scholar]
- Mammino, L.; Kabanda, M.M. Considering the Medium when Studying Biologically Active Molecules: Motivation, Options and Challenges. In Frontiers in Computational Chemistry; Ul-Haq, Z., Madura, J.D., Eds.; Bentham Science Publishers: Sharjah, United Arab Emirates, 2014; pp. 197–256. [Google Scholar]
- Widmer, D.R.; Schwartz, B.J. Solvents can control solute molecular identity. Nat. Chem. 2018, 10, 910–916. [Google Scholar] [CrossRef]
- Yu, M.; Chen, C.; Wu, J.; Deng, J.; Li, B.; Li, Z. Solvation: Basic conception, visual observation and potential applications. Int. J. Hydrogen Energy 2025, 136, 413–426. [Google Scholar] [CrossRef]
- Chang, T.-M.; Dang, L.X. Recent advances in molecular simulations of ion solvation at liquid interfaces. Chem. Rev. 2006, 106, 1305–1322. [Google Scholar] [CrossRef] [PubMed]
- Persson, I. Structures of hydrated metal ions in solid state and aqueous solution. Liguids 2022, 2, 210–242. [Google Scholar] [CrossRef]
- Persson, I. Hydrated metal ions in aqueous solution: How regular are their structures? Pure Appl. Chem. 2010, 82, 1901–1917. [Google Scholar] [CrossRef]
- Dopfer, O.; Fujii, M. Probing solvation dynamics around and biological molecules at a single-molecular level. Chem. Rev. 2016, 116, 5432–5463. [Google Scholar] [CrossRef]
- Burrows, C.J.; Harper, J.B.; Sander, W.; Tantillo, D.J. Solvation effects in organic chemistry. J. Org. Chem. 2022, 87, 1599–1601. [Google Scholar] [CrossRef]
- Reichardt, C. Solvation effects in organic chemistry: A short historical overview. J. Org. Chem. 2022, 87, 1616–1629. [Google Scholar] [CrossRef]
- Das, M.; Gogoi, A.R.; Sunoj, R.B. Molecular insights on solvent effects in organic reactions as obtained through computational chemistry tools. J. Org. Chem. 2022, 87, 1630–1640. [Google Scholar] [CrossRef]
- Katzberger, P.; Riniker, S. A general graph neural network based implicit solvation model for organic molecules in water. Chem. Sci. 2024, 15, 10794–10802. [Google Scholar] [CrossRef] [PubMed]
- Görbitz, C.H.; Hersleth, H.-P. On the inclusion of solvent molecules in the crystal structures of organic compounds. Acta Crystallogr. Sect. B 2000, 56, 526–534. [Google Scholar] [CrossRef]
- Oxtoby, N.S.; Blake, A.J.; Champness, N.R.; Wilson, C. Water superstructures within organic arrays; Hydrogen-bonded water sheets, chains and clusters. Chem. Eur. J. 2005, 11, 4643–4654. [Google Scholar] [CrossRef] [PubMed]
- Mascal, M.; Infantes, L.; Chisholm, J. Water oligomers in crystal hydrates—What’s news and what isn’t? Angew. Chem. Int. Ed. 2006, 45, 32–36. [Google Scholar]
- Fucke, K.; Steed, J.W. X-ray and neutron diffraction in the study of organic crystalline hydrates. Water 2010, 2, 333–350. [Google Scholar] [CrossRef]
- Becker, M. Dynamic solvation fields: A paradigm shift in solvent effects on chemical reactivity. Phys. Chem. Chem. Phys. 2025, 27, 24146–24158. [Google Scholar] [CrossRef]
- Waghorne, W.E. Using computational chemistry to explore experimental solvent parameters–solvent basicity, acidity and polarity/polarizability. Pure Appl. Chem. 2020, 92, 1539–1551. [Google Scholar] [CrossRef]
- Lucht, K.; Loose, D.; Ruschmeier, M.; Strotkötter, V.; Dyker, G.; Morgenstern, K. Hydrophilicity and microsolvation of an organic molecule resolved on the sub-molecular level by scanning tunneling microscopy. Angew. Chem. Int. Ed. 2018, 57, 1266–1270. [Google Scholar] [CrossRef]
- Choi, E.H.; Lee, Y.; Heo, J.; Ihee, H. Reactions dynamics studied via femtosecond X-ray liquidography at X-ray free-electron lasers. Chem. Sci. 2022, 13, 8457–8490. [Google Scholar] [CrossRef]
- Bhagat, S.; Chaubey, B.; Pal, S. Solvent-based NMR approaches for the assessment of molecular interactions: A review of current practices. Magn. Reson. Chem. 2026, 6, 114–139. [Google Scholar] [CrossRef]
- Otting, G. NMR studies of water bound to biological molecules. Prog. Nucl. Magn. Reason. Spectrosc. 1997, 31, 259–285. [Google Scholar] [CrossRef]
- Jorge, C.; Marques, B.S.; Valentine, K.G.; Joshua Wand, A. Chapter Three—Characterizing protein hydration dynamics using solution NMR Spectroscopy. Methods Enzymol. 2019, 615, 77–101. [Google Scholar]
- Bagno, A.; Scorrano, G. Selectivity in proton transfer, hydrogen bonding and solvation. Acc. Chem. Res. 2000, 33, 609–616. [Google Scholar] [CrossRef]
- Diaz, M.D.; Berger, S. Preferential solvation of a tetrapeptide by trifluoroethanol as studied by intermolecular NOE. Magn. Reson. Chem. 2001, 39, 369–373. [Google Scholar] [CrossRef]
- Gerig, J.T. Investigation of ethanol-peptide and water-peptide interactions through intermolecular nuclear Overhauser effects and molecular dynamics simulations. J. Phys. Chem. B 2013, 117, 4880–4892. [Google Scholar] [CrossRef] [PubMed]
- Gerig, J.T. Examination of solvent interactions with Trp-cage in 1,1,1,3,3,3-hexafluoro-2-propanol-water at 298 K through MD simulations and intermolecular nuclear Overhauser effects. J. Phys. Chem. B 2023, 127, 5062–5071. [Google Scholar] [CrossRef]
- Bifulco, G.; Dambruoso, P.; Gomez-Paloma, L.; Riccio, R. Determination of relative configuration in organic compounds by NMR spectroscopy and computational methods. Chem. Rev. 2007, 107, 3744–3779. [Google Scholar] [CrossRef]
- Barone, V.; Improta, R.; Rega, N. Quantum mechanical computations and spectroscopy: From small rigid molecules in the gas phase to large flexible molecules in solution. Acc. Chem. Res. 2008, 41, 605–616. [Google Scholar] [CrossRef] [PubMed]
- Mulder, F.A.; Filatov, M. NMR chemical shift data and ab initio shielding calculations: Emerging tools for protein structure determination. Chem. Soc. Rev. 2010, 39, 578–590. [Google Scholar] [CrossRef]
- Lodewyk, M.W.; Siebert, M.R.; Tantillo, D.J. Computational prediction of 1H and 13C chemical shifts: A useful tool for natural product, mechanistic, and synthetic organic chemistry. Chem. Rev. 2012, 112, 1839–1862. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Vasquez, A. State of the art and perspectives in the application of quantum chemical prediction of 1H and 13C chemical shifts and scalar couplings for structural elucidation of organic compounds. Magn. Reson. Chem. 2017, 55, 29–32. [Google Scholar] [CrossRef] [PubMed]
- Marcarino, M.O.; Zanardi, M.M.; Cicetti, S.; Sarotti, A.M. NMR calculations with quantum methods: Development of new tools for structure elucidation and beyond. Acc. Chem. Res. 2020, 53, 1922–1932. [Google Scholar] [CrossRef]
- Howarth, A.; Ermanis, K.; Goodman, J.M. DP4-AI automated NMR data analysis: Straight from spectrometer to structure. Chem. Sci. 2020, 11, 4351–4359. [Google Scholar] [CrossRef]
- Casabianca, L.B. Calculating nuclear magnetic resonance chemical shifts in solvated systems. Magn. Reson. Chem. 2020, 58, 611–624. [Google Scholar] [CrossRef] [PubMed]
- Cartes, I.; Cuadrado, C.; Garanas, A.H.; Sarotti, A.M. Machine learning in computational NMR-Aided structure elucidation. Front. Nat. Prod. 2023, 2, 1122426. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Kupka, T.; Ruscic, B.; Botto, R.E. Toward Hartree-Fock and density functional complete basis-set-predicted NMR parameters. J. Phys. Chem. A 2002, 106, 10396–10407. [Google Scholar] [CrossRef]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange-correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Kupka, T.; Stachow, M.; Mieradka, M.; Kaminsky, J.; Pluta, T. Convergence of nuclear magnetic shieldings in the Kohn-Sham limit for several small molecules. J. Chem. Theory Comput. 2010, 6, 1580–1589. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Leniak, A.; Pietrus, W.; Swiderska, A.; Kurczab, R. From NMR to AI: Do we need 1H NMR experimental spectra to obtain high quality log D prediction models? J. Chem. Inf. Model. 2025, 65, 2924–2939. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef]
- Mennucci, B.; Cancès, E.; Tomasi, J. Evaluation of solvent effects in isotropic and anisotropic dielectrics and in ionic solutions with a unified integral equation method: Theoretical bases, computational implementation, and numerical applications. J. Phys. Chem. B 1997, 101, 10506–10517. [Google Scholar] [CrossRef]
- Takano, Y.; Houk, K.N. Benchmarking the conductor-like polarizable continuum model (CPCM) for aqueous solvation free energies of neutral and ionic organic molecules. J. Chem. Theory Comput. 2005, 1, 70–77. [Google Scholar] [CrossRef]
- Klamt, A. The COSMO and COSMO-RS solvation models. WIREs Comput. Mol. Sci. 2011, 1, 699–709. [Google Scholar] [CrossRef]
- Kaplanskiy, M.V.; Shitov, D.A.; Tolstoy, P.M.; Tupikina, E.Y. Deconstructing 1H NMR chemical shifts in strong hydrogen bonds: A computational investigation of solvation, dynamics and nuclear delocalization effects. J. Chem. Inf. Model. 2025, 65, 5019–5034. [Google Scholar] [CrossRef]
- Charisiadis, P.; Exarchou, V.; Troganis, A.N.; Gerothanassis, I.P. Exploring the “forgotten”–OH NMR spectral region in natural products. Chem. Commun. 2010, 46, 3589–3591. [Google Scholar] [CrossRef]
- Charisiadis, P.; Primikyri, A.; Exarchou, V.; Tzakos, A.G.; Gerothanassis, I.P. Unprecedented ultra-high-resolution hydroxy group 1H NMR spectroscopic analysis of plant extracts. J. Nat. Prod. 2011, 74, 2462–2466. [Google Scholar] [CrossRef]
- Kontogianni, V.G.; Charisiadis, P.; Primikyri, A.; Pappas, C.G.; Exarchou, V.; Tzakos, A.G.; Gerothanassis, I.P. Hydrogen bonding probes of phenol–OH groups. Org. Biomol. Chem. 2013, 11, 1013–1025. [Google Scholar] [CrossRef]
- Charisiadis, P.; Kontogianni, V.; Tsiafoulis, C.; Tzakos, A.; Siskos, M.; Gerothanassis, I.P. 1H-NMR as a structural and analytical tool of intra-and intermolecular hydrogen bonds of phenol-containing natural products and model compounds. Molecules 2014, 19, 13643–13682. [Google Scholar] [CrossRef]
- Abraham, M.H.; Abraham, R.J.; Acree, W.E.; Aliev, A.E.; Leo, A.J.; Whaley, W.L. An NMR method for the quantitative assessment of intramolecular hydrogen bonding; application to physicochemical, environmental, and biochemical properties. J. Org. Chem. 2014, 79, 11075–11083. [Google Scholar] [CrossRef]
- Charisiadis, P.; Kontogianni, V.G.; Tsiafoulis, C.G.; Tzakos, A.G.; Gerothanassis, I.P. Determination of polyphenolic phytochemicals using highly deshielded -OH 1H NMR signals. Phytochem. Anal. 2017, 28, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Charisiadis, P.; Venianakis, T.; Papaemmanouil, C.D.; Primikyri, A.; Tzakos, A.G.; Siskos, M.G.; Gerothanassis, I.P. On the use of strong proton donors as a tool for overcoming line broadening in NMR: A comment. Magn. Reson. Chem. 2025, 63, 170–179. [Google Scholar] [CrossRef]
- Siskos, M.; Kontogianni, V.G.; Tsiafoulis, C.; Tzakos, A.; Gerothanassis, I.P. Investigation of solute-solvent interactions in phenol compounds: Accurate ab initio solvents effects on 1H NMR chemical shifts. Org. Biomol. Chem. 2013, 11, 7400–7411. [Google Scholar] [CrossRef] [PubMed]
- Siskos, M.G.; Tzakos, A.G.; Gerothanassis, I.P. Accurate ab initio calculations of O–H···O and O–H···−O proton chemical shifts: Towards elucidation of the nature of the hydrogen bond and prediction of hydrogen bond distances. Org. Biomol. Chem. 2015, 13, 8852–8868. [Google Scholar] [CrossRef]
- Siskos, M.G.; Choudhary, M.I.; Tzakos, A.G.; Gerothanassis, I.P. 1H ΝΜR chemical shift assignment, structure and conformational elucidation of hypericin with the use of DFT calculations–The challenge of accurate positions of labile hydrogens. Tetrahedron 2016, 72, 8287–8293. [Google Scholar] [CrossRef]
- Siskos, M.G.; Choudhary, M.I.; Gerothanassis, I.P. Hydrogen atomic positions of O–H···O hydrogen bonds in solution and in the solid state: The synergy of quantum chemical calculations with 1H-NMR chemical shifts and X-ray diffraction methods. Molecules 2017, 22, 415. [Google Scholar] [CrossRef]
- Siskos, M.G.; Choudhary, M.I.; Gerothanassis, I.P. Refinement of labile hydrogen positions based on DFT calculations of 1H NMR chemical shifts: Comparison with X-ray and neutron diffraction methods. Org. Biomol. Chem. 2017, 15, 4655–4666. [Google Scholar] [CrossRef]
- Siskos, M.G.; Choudhary, M.I.; Gerothanassis, I.P. DFT-calculated structures based on 1H NMR chemical shifts in solution vs. structures solved by single-crystal X-ray and crystalline-sponge methods: Assessing specific sources of discrepancies. Tetrahedron 2018, 74, 4728–4737. [Google Scholar] [CrossRef]
- Picric Acid Hazards. Available online: http://oag.ca.gov/sites/all/files/agweb/pdfs/cci/safety/picric.pdf (accessed on 5 January 2026).
- Whitty, A. Cooperativity and biological complexity. Nat. Chem. Biol. 2008, 4, 435–439. [Google Scholar] [CrossRef]
- Hunter, C.A.; Anderson, H.L. What is cooperativity? Angew. Chem. Int. Ed. 2009, 48, 7488–7499. [Google Scholar] [CrossRef]
- Mahadevi, S.A.; Sastry, N.G. Cooperativity in noncovalent interactions. Chem. Rev. 2016, 116, 2775–2825. [Google Scholar] [CrossRef] [PubMed]
- Trevisan, L.; Bond, A.D.; Hunter, C.A. Substituent effects on cooperativity in three-component H-bond networks involving phenol-phenol interactions. J. Am. Chem. Soc. 2025, 147, 1319–1326. [Google Scholar] [CrossRef] [PubMed]
- Lomas, J.S.; Rosenberg, R.E. Cooperativity and intermolecular hydrogen bonding in donor-acceptor complexes of phenol and polyhydroxybenzenes. J. Phys. Org. Chem. 2023, 36, e4506. [Google Scholar] [CrossRef]
- Rosenberg, R.E.; Lomas, J.S. Cooperativity and topological hydrogen bonding in aromatic diol complexes. J. Phys. Org. Chem. 2024, 37, e4578. [Google Scholar] [CrossRef]
- Mari, S.H.; Varras, P.C.; Wahab, A.T.; Choudhary, I.M.; Siskos, M.G.; Gerothanassis, I.P. Solvents-dependent structures of natural products based on the combined use of DFT calculations and 1H-NMR chemical shifts. Molecules 2019, 24, 2290. [Google Scholar] [CrossRef]
- Lee, M.S.; Cha, E.Y.; Sul, J.Y.; Song, I.S.; Kim, J.Y. Chrysophanic acid blocks proliferation of colon cancer cells by inhibiting EGFR/mTOR pathway. Phytother. Res. 2011, 25, 833–837. [Google Scholar] [CrossRef]
- Hsu, S.C.; Chung, J.G. Anticancer potential of emodin. BioMedicine 2012, 2, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Wijesekara, I.; Zhang, C.; Van Ta, Q.; Vo, T.S.; Li, Y.X.; Kim, S.K. Physcion from marine-derived fungus Microsporum sp. induces apoptosis in human cervical carcinoma HeLa cells. Microbiol. Res. 2014, 169, 255–261. [Google Scholar] [CrossRef]
- Argay, G.; Kalman, A.; Kovacevic, N.; Grubisic, D.; Ribar, B. Crystal structure of 1,8-dihydroxy-3-methyl-9, 10-anthracendion, C15H10O4. Z. Für Krist.-Cryst. Mater. 1996, 211, 723–724. [Google Scholar] [CrossRef]
- Zhu, J.C.; Liang, Y.; Wang, H.S.; Pan, Y.M.; Zhang, Y. 1,3,8-trihydroxy-6-methylanthraquinone monohydrate. Acta Crystallogr. Sect. E 2007, 63, o233–o235. [Google Scholar] [CrossRef]
- Hopf, H.; Jones, P.G.; Goclik, E.; Aust, P.; Rödiger, J. A new polymorph of physcion. Cryst. Struct. Commun. 2012, 68, o317–o319. [Google Scholar] [CrossRef]
- Deringer, V.L.; Hoepfner, V.; Dronskowski, R. Accurate hydrogen positions in organic crystals: Assessing a quantum-chemical aide. Cryst. Growth Des. 2012, 12, 1014–1021. [Google Scholar] [CrossRef]
- Rybarczyk-Pirek, A.J.; Malecka, M.; Palusiak, M. Use of quantum theory of atoms in molecules in the search of appropriate hydrogen atom locations in X-ray diffraction based studies. Cryst. Growth Des. 2016, 16, 6841–6848. [Google Scholar] [CrossRef]
- Falk, H. From the photosensitizer hypericin to the photoreceptor stentorin—The chemistry of phenanthroperylene quinones. Angew. Chem. Int. Ed. 1999, 38, 3116–3136. [Google Scholar] [CrossRef]
- Tatsis, E.; Exarchou, V.; Troganis, A.; Gerothanaassis, I.P. 1H-NMR determination of hypericin and pseudohypericin in complex natural mixtures by the use of strongly deshielded OH groups. Anal. Chim. Acta 2008, 607, 219–226. [Google Scholar] [CrossRef]
- Skalkos, D.; Tatsis, E.; Gerothanassis, I.P.; Troganis, A. Towards a consensus structure of hypericin in solution: Direct evidence for a single tautomer and different ionization states in protic and nonprotic solvents by the use of variable temperature gradient 1H NMR. Tetrahedron 2002, 58, 4925–4929. [Google Scholar] [CrossRef]
- Smirnov, A.; Fulton, D.B.; Andreotti, A.; Petrich, J.W. Exploring ground-state heterogeneity of hypericin and hypocrellin A and B: Dynamic and 2D NOESY NMR study. J. Am. Chem. Soc. 1999, 121, 7979–7988. [Google Scholar] [CrossRef]
- Freeman, D.; Frolow, F.; Kapinus, E.; Lavie, D.; Lavie, G.; Meruelo, D.; Mazur, Y. Acidic properties of hypericin and its octahydroxy analogue in the ground and excited states. J. Chem. Soc. Chem. Commun. 1994, 891–892. [Google Scholar] [CrossRef]
- Kawahata, M.; Komogawa, S.; Ohara, K.; Fujita, M.; Yamaguchi, K. High-resolution X-ray structure of methyl salicylate, a time-honored oily medicinal drug, solved by crystalline sponge method. Tetrahedron Lett. 2016, 57, 4633–4636. [Google Scholar] [CrossRef]
- Cardenal, A.D.; Ramadhar, T.R. Application of crystalline matrices for the structural determination of organic molecules. ACS Cent. Sci. 2021, 7, 406–414. [Google Scholar] [CrossRef] [PubMed]
- Lomas, J.S. 1H NMR spectra of butane-1, 4-diol and other 1, 4-diols: DFT calculation of shifts and coupling constants. Magn. Reson. Chem. 2014, 52, 87–97. [Google Scholar] [CrossRef]
- Lomas, J.S. 1H NMR spectra of alcohols in hydrogen bonding solvents: DFT/GIAO calculations of chemical shifts. Magn. Reson. Chem. 2016, 54, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Lomas, J.S. Cooperativity in alkane-1,2- and 1,3-polyols: NMR, QTAIM, and IQA study of O–H···OH and C–H···OH bonding. Magn. Reson. Chem. 2020, 58, 666–684. [Google Scholar] [CrossRef]
- Lomas, J.S. Intermolecular O–H···O and C–H···O hydrogen bond cooperativity in D-glucopyranose and D-galactopyranose—A DFT/GIAO, QTAIM/IQA and NCI approach. Magn. Reson. Chem. 2018, 56, 748–766. [Google Scholar] [CrossRef]
- De Almeida, M.V.; Couri, M.R.C.; De Assis, J.V.; Anconi, C.P.A.; Dos Santos, H.F.; De Almeida, W.B. 1H NMR Analysis of O-methyl-inositol isomers: A joint experimental and theoretical study. Magn. Reson. Chem. 2012, 50, 608−614. [Google Scholar] [CrossRef]
- De Souza, L.A.; Da Silva, H.C.; De Almeida, W.B. Structural determination of antioxidant and anticancer flavonoid rutin in solution through DFT calculations of 1H NMR chemical shifts. Chem. Open 2018, 7, 902–913. [Google Scholar] [CrossRef]
- Napolitano, J.G.; Lankin, D.C.; Chena, S.-N.; Paulia, G.F. Complete 1H NMR spectral analysis of ten chemical markers of Ginko biloba. Magn. Reson. Chem. 2012, 50, 569–575. [Google Scholar] [CrossRef]
- Da Silva, H.C.; Hernandes, I.S.; De Almeida, W.B. Modeling solvent effects in quantum chemical calculation of relative energies and NMR chemical shifts for azithromycin. J. Phys. Chem. A 2025, 129, 2200–2216. [Google Scholar] [CrossRef]
- Hernandes, I.S.; Da Silva, H.C.; Dos Santos, H.F.; Ávila, E.P.; De Almeida, M.V.; De Almeida, W.B. Quantum chemical investigation of predominant conformation of the antibiotic azithromycin in water and DMSO solutions: Thermodynamic and NMR analysis. R. Soc. Open Sci. 2023, 10, 230409. [Google Scholar] [CrossRef]
- Neglur, R.; Hosten, E.; Aucamp, M.; Liebenberg, W.; Grooff, D. Water and the relationship to the crystal structure stability of azithromycin. Thermal investigations of solvatomorphism, amorphism and polymorphism. J. Therm. Anal. Calorim. 2018, 132, 373–384. [Google Scholar] [CrossRef]
- Hernandes, I.S.; Da Silva, H.C.; Dos Santos, H.F.; Ávila, E.P.; De Almeida, M.V.; Gomes, M.G.R.; Paschoal, D.F.S.; De Almeida, W.B. An investigation of the predominant structure of antibiotic azithromycin in chloroform solution through NMR and thermodynamic analysis. Phys. Chem. Chem. Phys. 2022, 24, 22845−22858. [Google Scholar] [CrossRef]
- Barber, J. Assignments of the 13C and 1H NMR spectra of azithromycin in CDCl3. Magn. Reson. Chem. 1991, 29, 740−743. [Google Scholar] [CrossRef]
- Brennan, R.J.; Barber, J. Full assignments of the 13C and 1H NMR spectra of azithromycin in buffered D2O and DMSO-d6. Magn. Reson. Chem. 1992, 30, 327−333. [Google Scholar] [CrossRef]
- Mohammad, S.; Alam, M.F.; Islam, M.M.; Parvin, N.; Islam, M.N.; Mamun, M.I.R. Assessment of quality of azithromycin, a macrolide antibiotic by NMR spectroscopy. Bangladesh Pharm. J. 2021, 24, 37−44. [Google Scholar] [CrossRef]
- Bagno, A.; Rastrelli, F.; Saielli, G. Toward the complete prediction of the 1H and 13C NMR spectra of complex organic molecules by DFT methods: Application to natural substances. Chem.-Eur. J. 2006, 12, 5514−5525. [Google Scholar] [CrossRef]
- Krivdin, L.B. Computational NMR of carbohydrates: Theoretical background, applications and perspectives. Molecules 2021, 26, 2450. [Google Scholar] [CrossRef]
- Hafsa, N.E.; Berjanskii, M.V.; Arndt, D.; Wishart, D.S. Rapid and reliable protein structure determination via chemical shift threading. J. Biomol. NMR 2018, 70, 33–51. [Google Scholar] [CrossRef]
- Kjaergaard, M.; Brander, S.; Poulsen, F.M. Random coil chemical shift of intrinsically disordered proteins: Effects of temperature and pH. J. Biomol. NMR 2011, 49, 139–149. [Google Scholar] [CrossRef]
- Tzakos, A.G.; Fuchs, P.; van Nuland, N.A.J.; Troganis, A.; Tselios, T.; Deraos, J.; Matsoukas, J.; Gerothanassis, I.P.; Bonvin, A.M.J.J. NMR and molecular dynamics studies of an autoimmune myelin basic protein peptide and its antagonist: Structural implications for the MHC II (1-Au)-peptide complex from docking calculations. Eur. J. Biochem. 2004, 271, 3399–3413. [Google Scholar] [CrossRef] [PubMed]
- Molchanov, S.; Gryff-Keller, A. Solvation of amides in DMSO and CDCl3: An attempt at quantitative DFT-based interpretation of 1H and 13C NMR chemical shifts. J. Phys. Chem. A 2017, 121, 9645–9653. [Google Scholar] [CrossRef]
- Da Silva, H.C.; De Almeida, W.B. Theoretical calculations of 1H NMR chemical shifts for nitrogenated compounds in chloroform solution. Chem. Phys. 2020, 528, 110479−1104892. [Google Scholar] [CrossRef]
- Kubica, D.; Molchanov, S.; Gryff-Keller, A. Solvation of uracil and its derivatives by DMSO: A DFT-supported 1H NMR and 13C NMR study. J. Phys. Chem. A 2017, 121, 1841–1848. [Google Scholar] [CrossRef]
- Venianakis, T.; Primikyri, A.; Alexandri, E.; Papamokos, G.; Gerothanassis, I.P. Molecular models of three ω-3 fatty acids based on NMR and DFT calculations of 1H NMR chemical shifts. J. Mol. Liq. 2021, 342, 117460. [Google Scholar] [CrossRef]
- Venianakis, T.; Siskos, M.; Papamokos, G.; Gerothanassis, I.P. NMR and DFT studies of monounsaturated and ω-3 polyunsaturated free fatty acids in the liquid state reveal a novel atomistic structural model of DHA. J. Mol. Liq. 2023, 376, 121459. [Google Scholar] [CrossRef]
- Takahashi, O.; Kohno, Y.; Nishio, M. Relevance of weak hydrogen bonds in the conformational of organic compounds and bioconjugates: Evidence from recent experimental data and high-level ab initio MO calculations. Chem. Rev. 2010, 110, 6049–6076. [Google Scholar] [CrossRef] [PubMed]
- Venianakis, T.; Siskos, M.G.; Papamokos, G.; Gerothanassis, I.P. Structural studies of monounsaturated and ω-3 polyunsaturated free fatty acids in solution with the combined use of NMR and DFT Calculations—Comparison with the liquid state. Molecules 2023, 28, 6049–6144. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, H.C.; Hernandes, I.S.; De Almeida, W.B. Quantum chemical NMR spectroscopic structural analysis in solution: The investigation of 3-indoleacetic acid dimer formation in chloroform and DMSO solution. Magn. Reson. Chem. 2025, 63, 292–313. [Google Scholar] [CrossRef]
- Tjahjono, M.; Cheng, S.; Li, C.; Garland, M. Self-association of acetic acid in dilute deuterated chloroform. Wide-range spectral reconstructions and analysis using FTIR spectroscopy, BTEM, and DFT. J. Phys. Chem. A 2010, 114, 12168–12175. [Google Scholar] [CrossRef]
- Nagy, P.I. Competing intramolecular vs. intermolecular hydrogen bonds in solution. Int. J. Mol. Sci. 2014, 15, 19562–19633. [Google Scholar] [CrossRef] [PubMed]
- Claramunt, R.M.; López, C.; Santa Maria, M.D.; Sanz, D.; Elguero, J. The use of NMR spectroscopy to study tautomerism. Prog. Nucl. Magn. Reson. 2006, 49, 169–206. [Google Scholar] [CrossRef]
- Antonov, L. (Ed.) Tautomerism: Methods and Theories; Wiley-VCH Verlag: Weinhein, Germany, 2014. [Google Scholar]
- Hansen, P.E. Methods to distinguish tautomeric cases from static ones. In Tautomerism: Ideas, Compounds, Applications; Antonov, L., Ed.; Wiley-VCH: Weinhein, Germany, 2014. [Google Scholar]
- Siskos, M.G.; Varras, P.C.; Gerothanassis, I.P. DFT Calculations of O-H····O 1H NMR chemical shifts in investigating enol-enol tautomeric equilibria: Probing the impacts of intramolecular hydrogen bonding vs stereoelectronic interactions. Tetrahedron 2020, 76, 130979. [Google Scholar] [CrossRef]
- Bekiroglou, S.; Kenne, L.; Sandstrom, C. 1H NMR studies of maltose, maltoheptaose, α-, β- and γ-cyclodextrins, and complexes in aqueous solutions with hydroxy protons as structural probes. J. Org. Chem. 2003, 68, 1671–1678. [Google Scholar] [CrossRef]
- Bagchi, B. Water in Biological and Chemical Processes: From Structure and Dynamics to Function; Cambridge University Press: Cambridge, UK, 2013. [Google Scholar]
- Fatima, S.; Varras, P.C.; Wahab-tul, A.; Choudhary, M.I.; Siskos, M.G.; Gerothanassis, I.P. On the molecular basis of H2O/DMSO eutectic mixtures by using natural products as molecular sensors: A combined NMR and DFT Study. Phys. Chem. Chem. Phys. 2021, 23, 15645–15658. [Google Scholar] [CrossRef]
- Trindle, C.; Shillady, D. Electronic Structure Modeling: Connections Between Theory and Software; CRC Press: Boca Raton, FL, USA, 2008. [Google Scholar]
- Fortes, A.D.; Ponsonby, J.; Kirichek, O.; Garcia-Sakai, V. On the crystal structures and phase transitions of hydrates in the binary dimethyl sulfoxide-water system. Acta Crystalogr. Sect. B 2020, 76, 733–748. [Google Scholar] [CrossRef]
- Perrin, C.L. Are short, low-barrier hydrogen bonds unusually strong? Acc. Chem. Res. 2010, 43, 1550–1557. [Google Scholar] [CrossRef]
- Harris, T.K.; Zhao, Q.; Mildvan, A.S. NMR studies of strong hydrogen bonds in enzymes and in a model compound. J. Mol. Struct. 2000, 552, 97–109. [Google Scholar] [CrossRef]
- Tolstoy, P.M.; Koeppe, B.; Denisov, G.S.; Limbach, H.-H. Combined NMR and UV-Vis spectroscopy in the solution state: Study of the geometries of strong OHO hydrogen bonds of phenols with carboxylic acids. Angew. Chem. Int. Ed. 2009, 48, 5745–5747. [Google Scholar] [CrossRef]
- Koeppe, B.; Tolstoy, P.M.; Limbach, H.-H. Reactions pathways of proton transfer in hydrogen-bonded phenol-carboxylate complexes explored by combined UV-Vis and NMR spectroscopy. J. Am. Chem. Soc. 2011, 133, 7897–7908. [Google Scholar] [CrossRef] [PubMed]
- Koeppe, B.; Guo, J.; Tolstoy, P.M.; Denisov, G.S.; Limbach, H.-H. Solvent and H/D isotope effects on the proton transfer pathways in heteroconjucated hydrogen-bonded phenol-carboxylic acid anions observed by the combined UV-Vis and NMR spectroscopy. J. Am. Chem. Soc. 2013, 135, 7553–7566. [Google Scholar] [CrossRef]
- Limbach, H.-H.; Tolstoy, P.M.; Perez-Hernandez, N.; Guo, J.; Shenderovich, I.G.; Denisov, G.S. OHO hydrogen bond geometries and NMR chemical shifts: From equilibrium structures to geometric H/D isotope effects, with applications for water, protonated water, and compressed ice. Isr. J. Chem. 2009, 49, 199–216. [Google Scholar] [CrossRef]
- Pylaeva, S.; Allolio, C.; Koeppe, B.; Denisov, G.S.; Limbach, H.-H.; Sebastiani, D.; Tolstoy, P.M. Proton transfer pathways in a short hydrogen bond caused be solvation shell fluctuation: An ab initio MD and NMR/UV study of N(OHO)− bonded system. Phys. Chem. Chem. Phys. 2015, 17, 4634–4644. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.-C.; Sim, E.; Burke, K. Avoiding unbound anions in density functional calculations. J. Phys. Chem. 2011, 134, 171103. [Google Scholar] [CrossRef]
- Nattino, F.; Dupont, C.; Marzani, N.; Andreussi, O. Functional extrapolations to tame unbound anions in density-functional theory calculations. J. Chem. Theory Comput. 2019, 15, 6313–6322. [Google Scholar] [CrossRef]
- Lacerda, E.G., Jr.; Kamounah, F.S.; Coutinho, K.; Sauer, S.P.A.; Hansen, P.E.; Hammerich, O. Computational prediction of 1H and 13C NMR chemical shifts of protonated alkylpyrroles: Electron correlation and not solvation is the salvation. ChemPhysChem 2019, 20, 78–91. [Google Scholar] [CrossRef]
- Cadet, J.; Di Mascio, P. Peroxides in biological systems. In PATAI’s Chemistry of Functional Groups; Wiley: Chichester, UK, 2009; pp. 1–85. [Google Scholar]
- Shahidi, F.; Zhong, Y. Lipid oxidation and improving the oxidative stability. Chem. Soc. Rev. 2010, 39, 4067–4079. [Google Scholar] [CrossRef]
- Frankel, E.N. Photooxidation of unsaturated fats. In Lipid Oxidation, 2nd ed.; Oily Press Lipid Library Series: Davis, CA, USA; University of California, 2005; Chapter 3; pp. 51–66. [Google Scholar]
- Pratt, D.A.; Tallmon, K.A.; Porter, N.A. Free radical oxidation of polyunsaturated lipids: New mechanistic insights and the development of peroxyl radical clocks. Acc. Chem. Res. 2011, 44, 458–467. [Google Scholar] [CrossRef]
- Kontogianni, V.G.; Gerothanassis, I.P. Analytical and structural tools of lipid hydroperoxides: Present state and future perspectives. Molecules 2022, 27, 2139. [Google Scholar] [CrossRef]
- Ahmed, R.; Varras, P.C.; Siskos, M.G.; Siddiqui, H.; Choudhary, M.I.; Gerothanassis, I.P. NMR and computational studies as analytical and high-resolution structural tool for complex hydroperoxides and endo-hydroperoxides of fatty acids in solution. Molecules 2020, 25, 4902. [Google Scholar] [CrossRef]
- Chung, L.W.; Sameera, W.M.C.; Ramozzi, R.; Page, A.J.; Hatanaka, M.; Petrova, G.P.; Harris, T.V.; Li, X.; Ke, Z.; Liu, F.; et al. The ONIOM method and its applications. Chem. Rev. 2015, 115, 5678–5796. [Google Scholar] [CrossRef]
- Krivdin, L.B. Calculation of 15N NMR chemical shifts: Recent advances and perspectives. Prog. Nucl. Magn. Reson. Spectrosc. 2017, 102–103, 98–119. [Google Scholar] [CrossRef] [PubMed]
- Semenov, V.A.; Samul’tsev, D.O.; Krivdin, L.B. Quantum-chemical calculations of NMR chemical shifts of organic molecules: XIII. Accuracy of the calculation of 15N NMR chemical shifts of azines with account taken of solvation effects. Russ. J. Org. Chem. 2014, 50, 381–388. [Google Scholar] [CrossRef]
- Chernyshev, K.A.; Semenov, V.A.; Krivdin, L.B. Quantum-chemical calculations of NMR chemical shifts of organic molecules: VIII. Solvation effects on 15N NMR chemical shifts of nitrogen-containing heterocycles. Russ. J. Org. Chem. 2013, 49, 379–383. [Google Scholar] [CrossRef]
- Semenov, V.A.; Samul’tsev, D.O.; Krivdin, L.B. Solvent effects in the GIAO-DFT calculations of the 15N NMR chemical shifts of azoles and azines. Magn. Reson. Chem. 2014, 52, 686–693. [Google Scholar] [CrossRef]
- Samultsev, D.O.; Semenov, V.A.; Krivdin, L.B. On the accuracy factors and computational cost of the GIAO-DFT calculation of 15N NMR chemical shifts of amides. Magn. Reson. Chem. 2017, 55, 1015–1021. [Google Scholar] [CrossRef] [PubMed]
- Samultsev, D.O.; Semenov, V.A.; Krivdin, L.B. Calculation of 15N NMR chemical shifts of amines in the framework of the density functional theory. Russ. Chem. Bul. 2017, 66, 2248–2252. [Google Scholar] [CrossRef]
- Beltrame, P.; Cadoni, E.; Floris, C.; Gelli, G.; Lai, A. 15N and 13C NMR study of protonated monoaminopyridines in CDCl3–DMSO. Spectrochim. Acta A 2002, 58, 2693–2697. [Google Scholar] [CrossRef]
- Sanz, D.; Perova, A.; Claramunt, R.M.; Pinilla, E.; Torres, M.R.; Elguero, J. Protonation effects on the chemical shifts of Schiff bases derived from 3-hydroxypyridin-4-carboxaldehyde. Arch. Org. Chem. 2010, 2010, 102–113. [Google Scholar] [CrossRef]
- Semenov, V.A.; Samultsev, D.O.; Krivdin, L.B. Theoretical and experimental study of 15N NMR protonation shifts. Magn. Reson. Chem. 2015, 53, 433–441. [Google Scholar] [CrossRef]
- Gerothanassis, I.P. Oxygen-17 NMR spectroscopy: Basic principles and applications (Part I). Prog. NMR Spectrosc. 2010, 56, 95–197. [Google Scholar] [CrossRef]
- Gerothanassis, I.P. Oxygen-17 NMR spectroscopy: Basic principles and applications (Part II). Prog. Nucl. Magn. Reson. Spectrosc. 2010, 57, 1–110. [Google Scholar] [CrossRef] [PubMed]
- Castiglione, F.; Mele, A.; Raos, G. 17O NMR: A “rare and sensitive” probe of molecular interactions and dynamics. Annu. Rep. NMR Spectrosc. 2015, 85, 143–193. [Google Scholar]
- Krivdin, L.B. 17O nuclear magnetic resonance: Recent advances and applications. Magn. Reson. Chem. 2023, 61, 507–529. [Google Scholar] [CrossRef]
- Wong, W.; Poli, F. Solid state 17O NMR studies of biomolecules. Annu. Rep. NMR Spectrosc. 2014, 83, 145–220. [Google Scholar]
- Wu, G. 17O NMR studies of organic and biological molecules in aqueous solution and in the solid state. Prog. Nucl. Magn. Reson. Spectrosc. 2019, 114, 135–191. [Google Scholar] [CrossRef]
- Wu, G.; Palmer, J. Recent developments in 17O NMR studies of organic and biological molecules in the solid state. Annu. Rep. NMR Spectrosc. 2021, 103, 1–46. [Google Scholar]
- Malkin, V.G.; Malkina, O.L.; Steinebrunner, G.; Huber, H. Solvent effect on the NMR chemical shieldings in water calculated by a combination of molecular dynamics and density functional theory. Chem. Eur. J. 1996, 2, 452–457. [Google Scholar] [CrossRef]
- Pfrommer, B.G.; Mauri, F.; Louie, G.C. NMR chemical shifts of ice and liquid water: The effects of condensation. J. Am. Chem. Soc. 2000, 122, 123–129. [Google Scholar] [CrossRef]
- Sebastiani, D.; Parrinello, M. Ab-initio study of NMR chemical shifts of water under normal and supercritical conditions. Chem. Phys. Chem. 2002, 3, 675–679. [Google Scholar] [CrossRef] [PubMed]
- Pennanen, T.S.; Vaara, J.; Lantto, P.; Sillanpää, A.J.; Laasonen, K.; Jokisaari, J. Nuclear magnetic shielding and quadrupole coupling tensors in liquid water: A combined molecular dynamics simulation and quantum chemical study. J. Am. Chem. Soc. 2004, 126, 11093–11102. [Google Scholar] [CrossRef] [PubMed]
- Karadakov, P.B. Electron correlation and basis set effects on the 17O and 1H nuclear magnetic shieldings in water clusters (H2O)n (n = 2–5). J. Mol. Struct. 2002, 602–603, 293–301. [Google Scholar] [CrossRef]
- Bilalbegovic, G. Nuclear magnetic resonance parameters of water hexamers. J. Phys. Chem. A 2010, 114, 715–720. [Google Scholar] [CrossRef]
- Klein, R.A.; Mennucci, B.; Tomasi, J. Ab initio calculations of 17O NMR-chemical shifts for water. The limits of PCM theory and the role of hydrogen-bond geometry and cooperativity. J. Phys. Chem. A 2004, 108, 5851–5863. [Google Scholar] [CrossRef]
- De Dios, A.C. 17O chemical shifts in water. J. Phys. Chem. A 2025, 129, 11203–11212. [Google Scholar] [CrossRef]
- Kupka, T. Prediction of water’s isotropic nuclear shieldings and indirect nuclear spin–spin coupling constants (SSCCs) using correlation-consistent and polarization-consistent basis sets in the Kohn–Sham basis set limit. Magn. Reson. Chem. 2009, 47, 210–221. [Google Scholar] [CrossRef]
- Auer, A.A. High-level ab-initio calculation of gas-phase NMR chemical shifts and secondary isotope effects of methanol. Chem. Phys. Lett. 2009, 467, 230–232. [Google Scholar] [CrossRef]
- Kupka, T. Complete basis set prediction of methanol isotropic nuclear magnetic shieldings and indirect nuclear spin–spin coupling constants (SSCC) using polarization-consistent and XZP basis sets and B3LYP and BHandH density functionals. Magn. Reson. Chem. 2009, 47, 674–683. [Google Scholar] [CrossRef] [PubMed]
- Makulski, W. 1H, 13C, and 17O nuclear magnetic shielding of methanol and its deuterated isotopomers from gas phase measurements. J. Mol. Struct. 2008, 872, 81–86. [Google Scholar] [CrossRef]
- Cossi, M.; Crescenzi, O. Different models for the calculation of solvent effects on 17O nuclear magnetic shielding. J. Chem. Phys. 2003, 118, 8863–8872. [Google Scholar] [CrossRef]
- Kozminski, W.; Jackowski, K. Application of adiabatic inversion pulses for elimination of baseline distortions in Fourier transform NMR. A natural abundance 17O NMR spectrum for gaseous acetone. Magn. Reson. Chem. 2000, 38, 459–462. [Google Scholar] [CrossRef]
- Pavone, M.; Crescenzi, O.; Morelli, G.; Rega, N.; Barone, V. Solvent effects on the UV (η → π*) and NMR (17O) spectra of acetone in aqueous solution: Development and validation of a modified AMBER force field for an integrated MD/DFT/PCM approach. Theor. Chem. Acc. 2006, 116, 456–461. [Google Scholar] [CrossRef][Green Version]
- Gerothanassis, I.P. Multinuclear and multidimensional NMR methodology for studying individual water molecules bound to peptides and proteins in solution: Principles and applications. Prog. Nucl. Magn. Reson. 1994, 26, 171–237. [Google Scholar] [CrossRef]
- Gerothanassis, I.P.; Vakka, C. 17O NMR chemical shifts as a tool to study specific hydration sites of amides and peptides—Correlation with the IR amide I stretching vibration. J. Org. Chem. 1994, 59, 2341–2348. [Google Scholar] [CrossRef]
- Gerothanassis, I.P.; Vakka, C.; Troganis, A. 17O NMR studies of the solvation state of cis/trans isomers of amides and model protected peptides. J. Magn. Reson. B 1996, 111, 220–229. [Google Scholar] [CrossRef]
- Diez, E.; San Fabian, J.; Gerothanassis, I.P.; Esteban, A.L.; Abboud, J.-L.M.; Contreras, R.H.; De Kowalewski, D.G. Solvent effects on oxygen-17 chemical shifts in amides. Quantitative linear solvation shift relationships. J. Magn. Reson. 1997, 124, 8–19. [Google Scholar] [CrossRef]
- De Kowalewski, D.G.; Kowalewski, V.J.; Contreras, R.H.; Diez, E.; Casanueva, J.; San Fabian, J.; Esteban, A.L.; Galache, M.P. Solvent effects on oxygen-17 chemical shifts of methyl formate: Linear solvation shift relationships. J. Magn. Reson. 2001, 148, 1–10. [Google Scholar] [CrossRef]
- Kamlet, M.J.; Abboud, J.-L.M.; Abraham, M.H.; Taft, R.W. Linear solvation energy relationships. 23. A comprehensive collection of the solvatochromic parameters,.pi.*,.alpha., and.beta., and some methods for simplifying the generalized solvatochromic equation. J. Org. Chem. 1983, 48, 2877–2887. [Google Scholar] [CrossRef]
- Cosi, M.; Crescenzi, O. Solvent effects on 17O nuclear magnetic shielding: N-methyl formamide in polar and apolar solutions. Theor. Chem. Acc. 2004, 111, 162–167. [Google Scholar] [CrossRef]
- Mennucci, B.; Martinez, J.M. How to model solvation of peptides? Insights from a quantum mechanical and molecular dynamics study of N-methylacetamide. 2. 15N and 17O nuclear shielding in water and in acetone. J. Phys. Chem. B 2005, 109, 9830–9838. [Google Scholar] [CrossRef] [PubMed]
- Bednarek, E.; Dobrowolski, J.C.; Dobroz-Teperek, K.; Kozerski, L.; Lewandowski, W.; Mazurek, A.P. Theoretical and experimental 1H, 13C, 15N, and 17O NMR chemical shifts for 5-halogenouracils. J. Mol. Struct. 2000, 554, 233–243. [Google Scholar] [CrossRef]
- Gester, R.M.; Bistafa, C.; Georg, H.C.; Coutinho, K.; Canuto, S. Theoretically describing the 17O magnetic shielding constant of biomolecular systems: Uracil and 5-fluorouracil in water environment. Theor. Chem. Acc. 2014, 133, 1424. [Google Scholar] [CrossRef]
- Chandrasekaran, S.; Wilson, W.D.; Boykin, D.W. Oxygen-17 NMR studies on 5-substituted uracils. J. Org. Chem. 1985, 50, 829–831. [Google Scholar] [CrossRef]
- Rivelino, R.; Cabral, C.B.J.; Coutinho, K.; Canuto, S. Electronic polarization in liquid acetonitrile: A sequential Monte Carlo/quantum mechanics investigation. Chem. Phys. Lett. 2005, 407, 13–17. [Google Scholar] [CrossRef]
- Castiglione, F.; Baggioli, A.; Citterio, A.; Mele, A.; Raos, G. Organic peracids: A structural puzzle for 17O NMR and ab initio chemical shift calculations. J. Phys. Chem. A 2012, 116, 1814–1819. [Google Scholar] [CrossRef]
- Baggioli, A.; Crescenzi, O.; Field, M.J.; Castiglione, F.; Raos, G. Computational 17O-NMR spectroscopy of organic acids and peracids: Comparison of solvation models. Phys. Chem. Chem. Phys. 2013, 15, 1130–1140. [Google Scholar] [CrossRef]
- Wasylishen, R.E.; Bryce, D.L. A revised experimental absolute magnetic shielding scale for oxygen. J. Chem. Phys. 2002, 117, 10061–10066. [Google Scholar] [CrossRef]
- Aikens, C.M.; Gordon, M.S. Incremental solvation of nonionized and zwitterionic glycine. J. Am. Chem. Soc. 2006, 128, 12835–12850. [Google Scholar] [CrossRef] [PubMed]
- Caputo, M.C.; Provasi, P.F.; Sauer, S.P.A. The role of explicit solvent molecules in the calculation of NMR chemical shifts of glycine in water. Theor. Chem. Acc. 2018, 137, 88. [Google Scholar] [CrossRef]
- Rusakova, I.L.; Rusakov, Y.Y. Modern quantum chemistry methodology for predicting 31P nuclear magnetic resonance chemical shifts. Int. J. Mol. Sci. 2026, 27, 704–779. [Google Scholar] [CrossRef] [PubMed]
- Rusakov, Y.Y.; Rusakova, I.L.; Semenov, V.A.; Samultsev, D.O.; Fedorov, S.V.; Krivdin, L.B. Calculation of 15N and 31P NMR chemical shifts of azoles, phospholes, and phosphazoles: A gateway to higher accuracy at less computational cost. J. Phys. Chem. A 2018, 122, 6746–6759. [Google Scholar] [CrossRef]
- Maryasin, B.; Zipse, H. Theoretical studies of 31P NMR spectral properties of phosphanes and related compounds in solution. Phys. Chem. Chem. Phys. 2011, 13, 5150−5158. [Google Scholar] [CrossRef]
- Calcagno, F.; Maryasin, B.; Garavelli, M.; Avagliano, D.; Rivalta, I. Modeling solvent effects and convergence of 31P NMR shielding calculations with COBRAMM. J. Comput. Chem. 2024, 45, 1562–1575. [Google Scholar] [CrossRef] [PubMed]
- Přecechtělová, J.; Munzarová, M.L.; Vaara, J.; Novotný, J.; Dračínský, M.; Sklenář, V. Toward reproducing sequence trends in phosphorus chemical shifts for nucleic acids by MD/DFT calculations. J. Chem. Theory Comput. 2013, 9, 1641–1656. [Google Scholar] [CrossRef]
- Fukal, J.; Budešínský, M.; Páv, O.; Jurecka, P.; Zgarbová, M.; Šebera, J.; Sychrovský, V. The Ad-MD method to calculate NMR shift including effects due to conformational dynamics: The 31P NMR shift in DNA. J. Comput. Chem. 2022, 43, 132–143. [Google Scholar] [CrossRef]
- Fukal, J.; Páv, O.; Buděšínský, M.; Šebera, J.; Sychrovský, V. The benchmark of 31P NMR parameters in phosphate: A case study on structurally constrained and flexible phosphate. Phys. Chem. Chem. Phys. 2017, 19, 31830–31841. [Google Scholar] [CrossRef]
- Ruiz-Lopez, M.F.; Francisco, J.S.; Martins-Costa, M.T.C.; Anglada, J.M. Molecular reactions at aqueous interfaces. Nat. Rev. Chem. 2020, 4, 459–475. [Google Scholar] [CrossRef]
- Cortes-Clerget, M.; Yu, J.; Kincaid, J.R.A.; Walde, P.; Gallou, F.; Lipshutz, B.H. Water as the reaction medium in organic chemistry: From our worst enemy to our best friend. Chem. Sci. 2021, 12, 4237–4266. [Google Scholar] [CrossRef] [PubMed]
- Gröger, H.; Gallou, F.; Lipshutz, B.H. Where chemocatalysis meets biocatalysis: In water. Chem. Rev. 2023, 123, 5262–5296. [Google Scholar] [CrossRef]
- Mehr, H.S.M.; Fukuyama, K.; Bishop, S.; Lelj, F.; MacLachlan, M.J. Deuteration of aromatic rings under very mild conditions through keto-enamine tautomeric amplification. J. Org. Chem. 2015, 80, 5144–5150. [Google Scholar] [CrossRef]
- Banaldo, F.; Mattivi, F.; Catorci, D.; Arapitsas, P.; Guella, G. H/D exchange processes in flavonoids: Kinetics and mechanistic investigations. Molecules 2021, 26, 3544. [Google Scholar] [CrossRef] [PubMed]
- Fayaz, A.; Siskos, M.G.; Varras, P.C.; Choudhary, M.I.; Wahab, A.T.; Gerothanassis, I.P. The unique catalytic role of water in aromatic C–H activation at neutral pH: A combined NMR and DFT study of polyphenolic compounds. Phys. Chem. Chem. Phys. 2020, 22, 17401–17411. [Google Scholar] [CrossRef]
- Chalkidou, P.; Venianakis, T.; Papamokos, G.; Siskos, M.; Gerothanassis, I.P. Can we predict specific numbers of catalytically important molecules of water in H/D exchange in aromatic systems? A combined NMR and DFT study. New J. Chem. 2025, 49, 3877–3889. [Google Scholar] [CrossRef]
- Jordheim, M.; Fossen, T.; Songstad, J.; Anderson, Ø.M. Reactivity of anthocyanins and pyranoanthocyanins. Studies on aromatic hydrogen–deuterium exchange reactions in methanol. J Agric. Food. Chem. 2007, 55, 8261–8268. [Google Scholar] [CrossRef]
- Zuccon, G.; Longo, E.; Tugarinov, V.; Boselli, E.; Ceccon, A. Probing self-association of (+)-catechin coupled with hydrogen-deuterium exchange by solution NMR spectroscopy. Chem. Phys. Chem. 2025, 26, e202500325. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.J.; Morfin, C.; Paul, T.; Kyne, H.; Zhang, X.; Thomas-Tran, R.; Venkataramani, C.; Ghaffari, H.; Subramanian, M.; Marray, B.; et al. Hydrogen/deuterium exchange for chiral stability assessment in acidic methine-containing compounds. Anal. Chem. 2025, 97, 26107. [Google Scholar] [CrossRef]
- Dračínský, Μ. NMR spectroscopy studies of hydrogen bonding. Coord. Chem. Rev. 2026, 549, 217284. [Google Scholar] [CrossRef]


























































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | δDMSO-d6 | Δδ/ΔΤ e | δAcetone-d6 | Δδ/ΔΤ e | Δδ(δDMSO-d6 -δAcetone-d6) | δCD3CN | Δδ/ΔΤ e | Δδ(δDMSO-d6 -δCD3CN) | δCDCl3 | Δδ/ΔΤ e | Δδ(δDMSO-d6 -δCDCl3) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| (1) C-1 OH | 9.36 | −5.4 | 8.29 | −9.0 | 1.07 | 6.90 | −6.7 | 2.46 | 4.65 | −5.8 | 4.71 |
| (2) C-1 OH | 8.57 | −6.9 | (8.26) f 7.58 | −8.9 | 0.99 | 6.42 | −5.7 | 2.15 | 4.88 | −5.9 | 3.69 |
| 9C-2 OH | 8.71 | −7.1 | 7.65 | −9.2 | 1.06 | 6.52 | −5.8 | 2.19 | 5.02 | −5.7 | 3.69 |
| (3) C-5 OH | 12.95 | −2.1 | 13.03 | −1.4 | −0.08 | 12.95 | −0.8 | 0.00 | 12.93 | c | 0.02 |
| C-4΄OH | 10.56 | −8.9 | 9.26 | −8.9 | 1.30 | 7.70 | −8.5 | 2.86 | 5.36 | c | 5.20 |
| 1:1 PhOH + Solvent Complex | B3LYP 6-31+G(d) Geometry Optimization, Gas Phase | B3LYP 6-31+G(d) Geometry Optimization, CPCM | B3LYP 6-311++G(d,p) Geometry Optimization, Gas Phase | B3LYP 6-311++G(d,p) Geometry Optimization, CPCM | Experimental Values |
|---|---|---|---|---|---|
| PhOH + CHCl3 | 3.96 | 4.61 | 3.85 | 4.49 | 4.65 |
| PhOH + MeCN | 6.43 | 6.80 | 6.42 | 6.79 | 6.90 |
| PhOH + acetone | 8.60 | 8.95 | 8.48 | 8.83, 8.71 a | 8.29 |
| PhOH + DMSO | 9.02 | 9.31 | 9.08 | 9.37 | 9.36 |
| Compound | Group | δCDCl3 | δacetone-d6 | Δδ (δacetone-d6-δCDCl3) | δDMSO-d6 | Δδ (δDMSO-d6-δCDCl3) |
|---|---|---|---|---|---|---|
| Chrysophanol (1) | C(1)–OH | 12.11 | 12.03 | −0.08 | 11.96 | −0.15 |
| C(8)–OH | 12.00 | 11.95 | −0.05 | 11.87 | −0.13 | |
| C(4)–H | 7.64 | 7.62 | −0.02 | 7.56 | −0.08 | |
| C(5)–H | 7.81 | 7.70 | −0.11 | 7.71 | −0.10 | |
| C(3)–H | 7.65 | 7.82 | 0.17 | 7.80 | 0.15 | |
| C(7)–H | 7.27 | 7.35 | 0.08 | 7.38 | 0.11 | |
| C(2)–H | 7.09 | 7.19 | 0.10 | 7.22 | 0.13 | |
| C(6)–CH3 | 2.45 | 2.45 | 0.00 | 2.44 | −0.01 | |
| Emodin (2) | C(1)–OH | 12.26 | 12.21 | −0.05 | 12.11 | −0.15 |
| C(8)–OH | 12.08 | 12.09 | 0.01 | 12.04 | −0.04 | |
| C(5)–H | 7.61 | 7.58 | −0.03 | 7.50 | −0.11 | |
| C(4)–H | 7.26 | 7.27 | 0.01 | 7.18 | −0.08 | |
| C(7)–H | 7.07 | 7.15 | 0.08 | 7.11 | 0.04 | |
| C(2)–H | 6.65 | 6.67 | 0.02 | 6.57 | −0.08 | |
| C(3)–OH | 6.18 | 10.21 | 4.03 | 11.41 | 5.23 | |
| C(6)–CH3 | 2.41 | 2.47 | 0.06 | 2.41 | 0.00 | |
| Physcion (3) | C(1)–OH | 12.30 | 12.24 | −0.06 | 12.17 | −0.13 |
| C(8)–OH | 12.10 | 12.05 | −0.05 | 11.97 | −0.13 | |
| C(2)–H | 6.67 | 6.80 | 0.13 | 6.68 | 0.01 | |
| C(4)–H | 7.35 | 7.28 | −0.07 | 7.20 | −0.15 | |
| C(5)–H | 7.61 | 7.59 | −0.02 | 7.53 | −0.08 | |
| C(7)–H | 7.06 | 7.16 | 0.10 | 7.20 | 0.14 | |
| C(3)–OCH3 | 3.92 | 4.00 | 0.08 | 3.92 | 0.00 | |
| C(6)–CH3 | 2.43 | 2.47 | 0.04 | 2.42 | −0.01 |
| Hypericin Complex | Method | Correlation Coefficient (R2) | Mean Square Error | Slope |
|---|---|---|---|---|
| HyH + acetone 1:1 | B3LYP/6-31+G(d) (gas phase) | 0.9903 (0.9208) b | 0.2707 (2.1175) b | 1.0924 (1.0304) b |
| B3LYP/6-31+G(d) (CPCM) | 0.9917 (0.8926) b | 0.2320 (2.8740) b | 1.070 (0.9928) b | |
| TPSSh/TZVP (gas phase) | 0.9966 (0.9167) b | 0.0942 (2.2277) b | 0.9888 (0.9276) b | |
| TPSSh/TZVP (CPCM) | 0.9946 (0.871) b | 0.1506 (3.4516) b | 0.9589 (0.8777) b | |
| CAM-B3LYP (CPCM) | 0.9901 (0.8771) b | 0.2781 (3.2863) b | 1.0701 (0.9852) b | |
| HyH + acetone 1:2 | B3LYP/6-31+G(d) (gas phase) | 0.9943 (0.9146) b | 0.1597 (2.284) b | 1.0978 (1.0299) b |
| B3LYP/6-31+G(d) (CPCM) | 0.9982 (0.8673) b | 0.3288 (3.5511) b | 1.0788 (0.9885) b | |
| TPSSh/TZVP (gas phase) | 0.9973 (0.9238) b | 0.0763 (2.0393) b | 1.0049 (0.946) b | |
| TPSSh/TZVP (CPCM) | 0.9937 (0.877) | 0.1753 (3.6063) b | 0.9755 (0.8904) b | |
| CAM-B3LYP (CPCM) | 0.9858 (0.8521) b | 0.3977 (3.9557) b | 1.0729 (0.9757) b | |
| Hy- | B3LYP/6-31+G(d) (gas phase) | 0.9926 | 0.3324 | 0.9605 |
| B3LYP/6-31+G(d) (IEF-PCM in DMSO) | 0.9981 | 0.0848 | 1.0561 | |
| TPSSh/TZVP (gas phase) | 0.998 | 0.0912 | 0.9079 | |
| TPSSh/TZVP (IEF-PCM in DMSO) | 0.9994 | 0.0271 | 0.9537 | |
| CAM-B3LYP (IEF-PCM in DMSO) | 0.9981 | 0.0833 | 1.0603 | |
| X-ray crystal structure | 0.9678 | 1.4428 | 0.7472 |
| DFT | Energy a | ΔG b | H6 | H5 | H4 | H3 | CH3 | OH | |
|---|---|---|---|---|---|---|---|---|---|
| ωB97XD/631G+d | A | −1088.153109 | 0.0 (77.61%) | 8.35 | 7.08 | 7.77 | 7.22 | 4.01 | 10.35 |
| ωB97XD/631G+d | Β | −1088.151165 | 1.22 (9.90%) | 8.26 | 7.14 | 7.84 | 7.35 | 3.89 | 10.91 |
| ωB97XD/631G+d | C | −1088.150705 | 1.59 (5.30%) | 8.15 | 7.09 | 7.73 | 7.22 | 3.96 | 9.72 |
| ωB97XD/631G+d | D | −1088.150860 | 1.41 (7.18%) | 8.00 | 7.14 | 7.70 | 7.15 | 3.89 | 10.04 |
| Experimental | 7.79 | 6.95 | 7.52 | 6.99 | 3.90 | 10.49 |
| Diol | Proton | C6D6 (Experimental) | C6H6 (Calculated) | CDCl3 a | DMSO b,c |
|---|---|---|---|---|---|
| 1 | OH | 1.07 | 1.20 | — | 4.35 |
| CH2 | 1.33 | 1.58 | 1.69 | 1.43 | |
| CH2OH | 3.29 | 3.69 | 3.71 | 3.39 | |
| 2 | OH1 | 1.25 | 1.14 | — | 4.35 |
| OH8 | 1.24 | 1.13 | — | 4.33 | |
| CH | 1.37 | 1.38 | 1.68 | 1.41 | |
| CH | 1.39 | 1.60 | 1.70 | 1.48 | |
| CH | 1.23 | 1.33 | 1.52 | 1.32 | |
| CH | 1.27 | 1.62 | 1.62 | 1.33 | |
| CHOH | 3.32 | 3.67 | 3.68 | 3.37 | |
| CHOH | 3.34 | 3.70 | 3.71 | 3.38 | |
| CHOH | 3.49 | 3.79 | 3.87 | 3.58 | |
| Me | 0.95 | 1.11 | 1.22 | 1.03 | |
| 3 | OH | 1.36 | 1.35 | — | 4.33 |
| CH | 1.30 | 1.51 | 1.61 | 1.40 | |
| CH | 1.32 | 1.40 | 1.55 | 1.29 | |
| CHOH | 3.51 | 3.79 | 3.85 | 3.55 | |
| Me | 0.97 | 1.10 | 1.22 | 1.03 | |
| 4 | OH | 1.04 | 0.50 | — | 4.06 |
| CH2 | 1.40 | 1.50 | 1.58 | 1.37 | |
| Me | 1.05 | 1.11 | 1.24 | 1.06 | |
| 5 | OH | 0.58 | 0.24 | — | 4.28 |
| CH-trans | 1.28 | 1.56 | 1.66 | 1.41 | |
| CH-cis | 1.52 | 1.65 | 1.74 | 1.55 | |
| CHOH | 3.36 | 3.69 | 3.80 | 3.52 | |
| 6 | OH | 0.59 | 0.24 | — | 4.42 |
| CH-ax | 1.06 | 1.21 | 1.36 | 1.15 | |
| CH-eq | 1.65 | 1.89 | 1.98 | 1.74 | |
| CHOH | 3.25 | 3.56 | 3.68 | 3.36 |
| Alcohol | Proton | Acetonitrile | Acetone | ||||
|---|---|---|---|---|---|---|---|
| δexpt | δcomp a | δcomp b | δexpt | δcomp a | δcomp b | ||
| Methanol | OH | 2.13 | 1.62 | 2.69 | 3.08 | 3.61 | 4.61 |
| CH3 | 3.27 | 3.26 | 3.29 | 3.31 | 3.49 | 3.50 | |
| Ethanol | OH | 2.43 | 2.28 | 3.27 | 3.34 | 3.99 | 4.85 |
| CH2 | 3.54 | 3.54 | 3.58 | 3.57 | 3.70 | 3.69 | |
| CH3 | 1.12 | 0.99 | 0.99 | 1.12 | 1.13 | 1.20 | |
| Tert-butyl-alcohol | OH | 2.36 | 2.09 | 3.00 | 3.18 | 3.95 | 4.80 |
| CH3 | 1.17 | 1.05 | 1.09 | 1.19 | 1.18 | 1.21 | |
| Strs. | Regression Slope a | Regression Intercept a | Adjusted R2 (adj-R2) b |
|---|---|---|---|
| 27 | 0.952 (±0.041) | 0.515 (±0.198) | 0.973 (±1.342) |
| 28 | 1.033 (±0.058) | −0.096 (±0.281) | 0.954 (±2.698) |
| 30 | 0.959 (±0.040) | 0.408 (±0.195) | 0.974 (±1.295) |
| 32 | 0.960 (±0.025) | 0.427 (±0.122) | 0.990 (±0.506) |
| 34 | 0.948 (±0.044) | 0.499 (±0.214) | 0.968 (±1.565) |
| Solute | Solvation Mode b | RMSD | HCO | N−H c | RMSD | C=O | CH3 | |
|---|---|---|---|---|---|---|---|---|
| E-NMF | 0S | 0.175 | 0.13 | 0.14 | 0.78 | 1.37 | 4.62 | 1.61 |
| E-NMF | 2S | 0.094 | 0.18 | 0.11 | 0.33 | 1.14 | 3.05 | 1.01 |
| E-NMF | 0.90(2S) + 0.10(D) | 0.051 | 0.09 | 0.11 | −0.03 | 1.08 | 2.49 | 0.99 |
| Z-NMF | 0S | 0.198 | 0.08 | 0.13 | 0.91 | 1.24 | 3.91 | 0.83 |
| Z-NMF | 2S | 0.124 | 0.11 | 0.10 | 0.53 | 0.99 | 1.64 | −0.06 |
| Z-NMF | 0.74(2S) + 0.26(D′) | 0.058 | 0.15 | 0.13 | 0.00 | 0.98 | 1.59 | 0.06 |
| F | 0S | 0.321 | 0.02 | 0.79 E | 1.26 Z | 1.26 | 3.97 | |
| F | 2S | 0.210 | 0.08 | 0.48 E | 0.82 Z | 1.01 | 1.75 | |
| F | D | 0.564 | −0.42 | 0.67 E | −2.37 Z | 1.35 | −4.57 | |
| F | D′ | 0.313 | 0.23 | −1.04 E | 1.08 Z | 1.02 | 1.82 | |
| F | 0.41(2S) + 0.25(D) + 0.34(D′) | 0.044 | −0.08 | 0.00 E | 0.03 Z | 0.94 | 0.18 |
| Solute | Solvation Mode b | RMSD | HCO | N−H d | RMSD c | C=O | CH3 | |
|---|---|---|---|---|---|---|---|---|
| E-NMF | 0S | 0.500 | −0.16 | −0.14 | 2.38 | 1.22 | 2.48 | 0.66 |
| E-NMF | NH·S | 0.053 | −0.02 | 0.01 | −0.09 | 1.21 | 2.44 | 0.65 |
| E-NMF | 0.04(0S) + 0.96(NH·S) | 0.050 | −0.03 | 0.01 | 0.00 | 1.22 | 2.44 | 0.88 |
| Z-NMF | 0S | 0.523 | −0.26 | −0.14 | 2.49 | 1.13 | 1.34 | 0.00 |
| Z-NMF | NH·S | 0.082 | −0.09 | 0.06 | −0.27 | 1.13 | 1.24 | 0.44 |
| Z-NMF | 0.11(0S) + 0.89(NH·S) | 0.057 | −0.12 | 0.05 | 0.00 | 1.13 | 1.25 | 0.39 |
| F | 0S | 0.623 | −0.37 | 1.81 E | 2.28 Z | 1.26 | 2.78 | |
| F | N(H·S)2 | 0.213 | −0.04 | −0.19 E | −0.90 Z | 1.16 | 1.41 | |
| F | 0.25(0S) + 0.75(N(H·S)2) | 0.090 | −0.18 | 0.26 E | −0.17 Z | 1.18 | 1.75 | |
| F | 0.31(NH E·S) + 0.19(NH Z·S) + 0.50(N(H·S)2) | 0.062 | −0.17 | 0.00 E | 0.00 Z | 1.17 | 1.58 |
| Solvatomer | 1-NH···O | 3-NH···O |
|---|---|---|
| U·2DMSO | 1.765 | 1.835 |
| DHU·2DMSO | 1.879 | 1.890 |
| T·2DMSO | 1.777 | 1.845 |
| ax-DHT·2DMSO | 1.874 | 1.883 |
| eq-DHT·2DMSO | 1.879 | 1.892 |
| HMU·3DMSO a | 1.832 | 1.801 |
| Solute | Po | P1 | P3 | P13 | Ps1 | Ps3 |
|---|---|---|---|---|---|---|
| U | 0.06 ± 0.01 | 0.17 ± 0.02 | 0.20 | 0.57 | 0.74 | 0.77 |
| T | 0.06 ± 0.01 | 0.22 ± 0.02 | 0.15 | 0.57 | 0.79 | 0.72 |
| HMU | 0.06 ± 0.01 | 0.21 ± 0.02 | 0.07 | 0.70 | 0.91 | 0.77 |
| DHU | 0.06 ± 0.01 | 0.13 ± 0.02 | 0.26 | 0.55 | 0.68 | 0.81 |
| DHT | 0.06 ± 0.01 | 0.13 ± 0.02 | 0.26 | 0.54 | 0.67 | 0.80 |
| FFA | Intermolecular Interaction | δ(COOH) (ppm) | Complexation Energy (kcal/Mole–Gas Phase) |
|---|---|---|---|
| CA | COO-H···DMSO | 13.4 (11.94) | −18.0 a |
| CA dimer parallel | COO-H···DMSO | 14.4, 13.9 b | −15.7 c,d |
| CA dimer antiparallel | COO-H··· DMSO | 14.1, 14.2 b | 15.9 c,d |
| OA | COO-H···DMSO | 13.4 (11.94) | |
| ALA | COO-H··· DMSO | 13.4 (11.95) | |
| EPA | COO-H···DMSO | 13.1 (12.01) | |
| DHA | COO-H···DMSO | 13.5 (12.08) |
| 1 (FHF)− | 2 (H5O2)+ | 3 (PyHPy)+ | |
|---|---|---|---|
| Experimental values | |||
| 16.6 | 21.3 | 21.73 | |
| Computed values I. Stationary QC I.1. Solvation effects | |||
| Vacuum | 18.4 | 21.3 | 21.6 |
| Implicit solvent | 18.3 | 21.2 | 20.0 |
| Explicit solvent | 18.3 | 19.7 | 20.8 |
| Cluster–continuum | 18.2 | 21.0 | 20.1 |
| I.2. Nuclear delocalization 1D SE | |||
| Vacuum | 17.2 | 19.7 | 23.0 |
| Implicit solvent | 16.9 | 19.4 | 21.9 |
| 2D SE | |||
| Vacuum | 17.6 | 20.2 | 23.4 |
| Implicit solvent | 17.3 | 19.9 | 22.1 |
| 3D SE | |||
| Vacuum | 17.6 | 20.1 | 22.7 |
| Implicit solvent | 17.3 | 19.9 | 20.8 |
| I.3. Relativistic effects | |||
| Levy−Leblond | 17.9 | 21.0 | 21.7 |
| Four-component (4c) | 17.7 | 21.0 | 21.7 |
Vacuum | 18.4 | II. Thermal motion 20.9 | 24.2 |
| Explicit solvent | 17.7 | 20.2 | 22.0 |
| 15N NMR Chemical Shift (δ, ppm) b | |||||
|---|---|---|---|---|---|
| Compound | Solvent | Gas Phase | IEF-PCM | Supermolecule (1:1) | Experiment |
 | C6H6 | −56.1 (5.0) | −61.6 (0.5) | −64.6 (3.5) | −61.1 |
| CHCl3 | −56.1 (12.6) | −65.5 (3.2) | −75.0 (6.3) | −68.7 | |
| CH2Cl2 | −56.1 (9.2) | −67.4 (2.1) | −65.2 (0.1) | −65.3 | |
| (CH3)2CO | −56.1 (5.7) | −68.8 (7.0) | −68.3 (6.5) | −61.8 | |
| CH3OH | −56.1 (25.0) | −69.3 (11.8) | −87.7 (6.6) | −81.1 | |
| C2H5OH | −56.1 (24.2) | −69.0 (11.3) | −82.1 (1.8) | −80.3 | |
| H2O | −56.1 (28.2) | −69.7 (14.6) | −82.2 (2.1) | −84.3 | |
 | c-C6H12 | 45.6 (10.3) | 36.8 (1.5) | 38.8 (3.5) | 35.3 |
| CCl4 | 45.6 (16.2) | 35.8 (6.4) | 35.3 (5.9) | 29.4 | |
| C6H6 | 45.6 (17.8) | 35.7 (7.9) | 29.5 (1.7) | 27.8 | |
| CHCl3 | 45.6 (26.6) | 29.0 (10.0) | 21.1 (2.1) | 19.0 | |
| CH2Cl2 | 45.6 (25.4) | 25.8 (5.6) | 24.8 (4.6) | 20.2 | |
| (CH3)2CO | 45.6 (19.7) | 23.3 (2.6) | 23.5 (2.4) | 25.9 | |
| CH3OH | 45.6 (39.3) | 22.5 (16.2) | 6.3 (0.0) | 6.3 | |
| C2H5OH | 45.6 (35.1) | 22.9 (12.4) | 10.2 (0.3) | 10.5 | |
| H2O | 45.6 (51.8) | 21.7 (27.9) | 9.3 (15.5) | −6.2 | |
| Compound | Name | Protonation Shift | ||||
|---|---|---|---|---|---|---|
| Gas Phase | Polarizable Continuum Model | Supermolecule in Polarizable Continuum | Counter Ion in Polarizable Continuum | Experiment | ||
| 1 | Pyridine | −139.3 | −119.4 | −99.9 | −87.7 | −107.4 |
| 2 | N-methylimidazole | −114.9 | −92.3 | −84.5 | −64.1 | −85.6 |
| 3 | Acetone oxime | −153.9 | −134.1 | −129.3 | −108.8 | −117.5 |
| 4 | Triethylamine | +23.7 | +18.0 | +9.8 | +4.3 | +9.9 |
| No. of Solvent Molecules | Without PCM | With PCM |
|---|---|---|
| 0 | 0.0 (0.0) | −50.2 (−64.6) |
| 2 | −59.2 (−54.2) | −92.6 (−94.2) |
| 5 | −75.3 (−67.7) | −96.0 (−92.5) |
| 10 | −81.4 | |
| 20 | −87.2 | |
| 40 | −97.3 |
| Compound | Gas | PCM a | ASEC(PCM) b | ASEC(Iter) c | Exp. d | |
|---|---|---|---|---|---|---|
| Uracil | ||||||
| O2 | σ | −7.8 (−11.6) | 35.5 (33.0) | 50.6 (47.3) | 57.2 (54.1) | 55.5 |
| O4 | σ | −113.5 (−117.1) | −41.1 (−43.1) | −17.2 (−20.4) | −13.5 (−16.4) | −13.5 |
| 5-Flurouracil | ||||||
| O2 | σ | 3.6 (−0.2) | 43.7 (41.0) | 49.4 (45.9) | 56.5 (53.3) | 57.5 |
| O4 | σ | −93.5 (−96.4) | −25.5 (−27.0) | −13.6 (−16.4) | −12.6 (−15.1) | −6.5 |
| Compound | Buffer Concentration | pD | H-8 | H-6 | ||||
|---|---|---|---|---|---|---|---|---|
| (kcal mol−1) | (kcal mol−1) | (kcal mol−1) | (kcal mol−1) | (kcal mol−1) | (kcal mol−1) | |||
| Taxifolin | 25 mM | 6.0 | 20.31 ± 1.57 | 4.70 ± 0.79 | 25.01 | 18.91 ± 1.18 | 5.62 ± 0.59 | 24.53 |
| 25 mM | 7.6 | 18.79 ± 1.59 | 5.80 ± 0.80 | 24.59 | 14.01 ± 1.47 | 9.17 ± 0.82 | 23.18 | |
| 50 mM | 7.6 | 19.34 ± 1.18 | 5.05 ± 0.65 | 24.39 | 20.63 ± 1.26 | 2.24 ± 0.49 | 22.87 | |
| 25 mM | 9.6 | 18.43 ± 1.79 | 6.24 ± 0.72 | 24.67 | 16.67 ± 0.55 | 6.15 ± 0.55 | 22.81 | |
| 50 mM | 9.6 | 19.62 ± 0.77 | 4.07 ± 0.65 | 23.69 | 16.96 ± 0.54 | 5.51 ± 0.58 | 22.46 | |
| 1 M | 9.6 | 15.96 ± 1.22 | 7.98 ± 0.78 | 23.94 | 11.76 ± 0.75 | 10.38 ± 0.98 | 22.14 | |
| Phloroglucinol | H-2,4,6 | |||||||
| 25 mM | 6.9 | 17.46 ± 0.30 | 3.50 ± 0.09 | 20.96 | ||||
| 25 mM | 7.9 | 16.05 ± 0.78 | 3.69 ± 0.26 | 19.74 | ||||
| 25 mM | 8.9 | 16.55 ± 1.15 | 2.86 ± 0.29 | 19.41 | ||||
| Neutral Phloroglucinol | +2H2O | +3H2O | Exp. | |||
|---|---|---|---|---|---|---|
| “in-out” | “in-in” “in-in A” | 20.96 | ||||
| APFD/6-31+G(d) | 20.59 | 26.28 (10.94%) | 20.13 (58.52%) 19.75 (30.51%) | |||
| B3LYP/6-31+G(d)/GD3BJ | 23.15 | 27.52 (14.19%) | 21.66 (46.80%) 21.54 (38.19%) | |||
| PBE0/6-31+G(d)/GD3BJ | 23.16 | 27.76 (9.65%) | 21.65 (51.47%) 21.48 (38.75%) | |||
| ωB97XD/6-31+G(d) | 26.19 | 30.86 (17.12%) | 25.08 (52.04%) 24.76 (30.36%) | |||
| CAM-B3LYP/6-31+G(d)/GD3BJ | 24.68 | 29.71 (16.10%) | 23.99 (46.39%) 23.62 (37.37%) | |||
| Neutral taxifolin | +2H2O | +3H2O | Exp. | |||
| C-6 | C-8 | C-6 | C-8 | C-6 | C-8 | |
| APFD/6-31+G(d) | 20.79 | 20.77 | 22.98 | 23.55 | 24.53 | 25.01 |
| B3LYP/6-31+G(d)/GD3BJ | 20.81 | 21.93 | 25.63 | 24.72 | ||
| PBE0/6-31+G(d)/GD3BJ | 20.80 | 21.28 | 25.18 | 24.67 | ||
| ωB97XD/6-31+G(d) | 25.08 | 24.19 | 27.38 | 27.37 | ||
| CAM-B3LYP/6-31+G(d) | 24.78 | 25.67 | 29.12 | 28.81 | ||
| CAM-B3LYP/6-31+G(d)/GD3BJ | 22.69 | 23.59 | 27.26/27.05 | 27.66 | ||
| Ionic phloroglucinol | +2H2O | +3H2O | Exp. | |||
| on OH | C=O (A) | C=O (B) | 19.74 | |||
| APFD/6-31+G(d) | 11.83 | 19.15 | a | 11.26 | ||
| B3LYP/6-31+G(d)/GD3BJ | 13.01 (0.26%) | 19.91 (33.85%) | 19.86 (65.89%) | 11.63 | ||
| PBE0/6-31+G(d)/GD3BJ | 13.05 | 19.71 | a | 11.48 | ||
| ωB97XD/6-31+G(d) | 15.21 (0.37%) | 22.16 (41.71%) | 21.37 (57.92%) | 14.07 | ||
| CAM-B3LYP/6-31+G(d) | 14.39 (0.32%) | 21.69 (60.95%) | 20.58 (38.73%) | 13.36 | ||
| CAM-B3LYP/6-31+G(d)/GD3BJ | 14.08 (0.30%) | 21,32 (39.49%) | 20.47 (60.20%) | 12.95 | ||
| Ionic taxifolin | +2H2O | +3H2O | Exp. | |||
| a | C-6 | C-8 | C-6 | C-8 | ||
| B3LYP/6-31+G(d)/GD3BJ | a | 23.83 | 24.27 | 23.18 | 24.59 | |
| PBE0/6-31+G(d)/GD3BJ | a | 24.76 | 26.00 | |||
| ωB97XD/6-31+G(d) | a | 24.51 | 26.18 | |||
| CAM-B3LYP-D/6-31+G(d) | a | 23.44 | 22.80 | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.
Share and Cite
Siskos, M.G.; Gerothanassis, I.P. NMR and DFT Studies on Solvation Phenomena in Bioorganic Molecules, Natural Products and Model Compounds: Current and Future Perspectives for Atomic-Level Structures and Mechanistic Catalytic Reactions. Molecules 2026, 31, 703. https://doi.org/10.3390/molecules31040703
Siskos MG, Gerothanassis IP. NMR and DFT Studies on Solvation Phenomena in Bioorganic Molecules, Natural Products and Model Compounds: Current and Future Perspectives for Atomic-Level Structures and Mechanistic Catalytic Reactions. Molecules. 2026; 31(4):703. https://doi.org/10.3390/molecules31040703
Chicago/Turabian StyleSiskos, Michael G., and Ioannis P. Gerothanassis. 2026. "NMR and DFT Studies on Solvation Phenomena in Bioorganic Molecules, Natural Products and Model Compounds: Current and Future Perspectives for Atomic-Level Structures and Mechanistic Catalytic Reactions" Molecules 31, no. 4: 703. https://doi.org/10.3390/molecules31040703
APA StyleSiskos, M. G., & Gerothanassis, I. P. (2026). NMR and DFT Studies on Solvation Phenomena in Bioorganic Molecules, Natural Products and Model Compounds: Current and Future Perspectives for Atomic-Level Structures and Mechanistic Catalytic Reactions. Molecules, 31(4), 703. https://doi.org/10.3390/molecules31040703