
Lipid Peroxidation in Cancer Therapy: Molecular Mechanisms Involving Oxidative Stress, Cell Death, and Therapeutic Response

 , , , and
, , , and 
Abstract
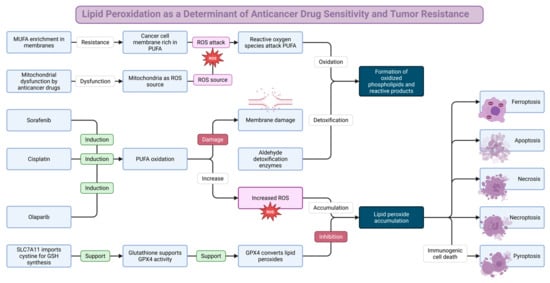
1. Introduction
1.1. Search Strategy and Study Selection
1.2. Initiation
1.3. Propagation
1.4. Termination
2. Lipid Peroxidation Products
3. Effects of Lipid Peroxidation on Anticancer Drug Efficacy
4. Lipid Peroxidation and Chemoresistance
4.1. An Integrated Model of Lipid-Peroxidation Resistance
4.2. Tumor Adaptation
4.3. NRF2
4.4. GPX4 and Drug-Tolerant Persister Cells
4.5. FSP1 and CoQ10
4.6. SLC7A11
4.7. ALDH
4.8. Paraoxonase-2 (PON2)
4.9. Tumour-Type Specificity
5. Lipid Remodeling Resistance
5.1. PUFAs
5.2. MUFA via SCD1
5.3. ACSL and LPCAT
5.4. DHODH and Additional GPX4-Independent Defenses
6. Cell Death Dependent on Lipid Peroxidation
6.1. Ferroptosis
6.2. Apoptosis
6.3. Necrosis
6.4. Necroptosis
6.5. Pyroptosis
6.6. Cuproptosis
6.7. Immunogenic Cell Death
6.8. Lipid-Induced Cell Death
6.8.1. Organelle Stress and Metabolic Dysfunction
6.8.2. Membrane Destabilization
6.8.3. Terminal Cellular Failure
6.8.4. Stages of Lipid-Induced Cellular Damage
6.8.5. Mitochondrial Response to Lipid Stress
6.8.6. Endoplasmic Reticulum Stress Pathway
7. Dual Pro- and Anti-Tumor Roles of Lipid Peroxidation
8. Translational Challenges and Opportunities
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Xiao, L.; Xian, M.; Zhang, C.; Guo, Q.; Yi, Q. Lipid Peroxidation of Immune Cells in Cancer. Front. Immunol. 2024, 14, 1322746. [Google Scholar] [CrossRef]
- Zheng, Y.; Sun, J.; Luo, Z.; Li, Y.; Huang, Y. Emerging Mechanisms of Lipid Peroxidation in Regulated Cell Death and Its Physiological Implications. Cell Death Dis. 2024, 15, 859. [Google Scholar] [CrossRef]
- Ayala, A.; Muñoz, M.F.; Argüelles, S. Lipid Peroxidation: Production, Metabolism, and Signaling Mechanisms of Malondialdehyde and 4-Hydroxy-2-Nonenal. Oxid. Med. Cell. Longev. 2014, 2014, 360438. [Google Scholar] [CrossRef]
- Harayama, T.; Riezman, H. Understanding the Diversity of Membrane Lipid Composition. Nat. Rev. Mol. Cell Biol. 2018, 19, 281–296. [Google Scholar] [CrossRef]
- Su, L.J.; Zhang, J.H.; Gomez, H.; Murugan, R.; Hong, X.; Xu, D.; Jiang, F.; Peng, Z.Y. Reactive Oxygen Species-Induced Lipid Peroxidation in Apoptosis, Autophagy, and Ferroptosis. Oxid. Med. Cell. Longev. 2019, 2019, 5080843. [Google Scholar] [CrossRef]
- Yin, H.; Xu, L.; Porter, N.A. Free Radical Lipid Peroxidation: Mechanisms and Analysis. Chem. Rev. 2011, 111, 5944–5972. [Google Scholar] [CrossRef] [PubMed]
- Pratt, D.A.; Tallman, K.A.; Porter, N.A. Free Radical Oxidation of Polyunsaturated Lipids: New Mechanistic Insights and the Development of Peroxyl Radical Clocks. Acc. Chem. Res. 2011, 44, 458. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, M.S.; Ruiz, J.; Watts, J.L. Polyunsaturated Fatty Acids Drive Lipid Peroxidation during Ferroptosis. Cells 2023, 12, 804. [Google Scholar] [CrossRef] [PubMed]
- Gęgotek, A.; Skrzydlewska, E. Biological Effect of Protein Modifications by Lipid Peroxidation Products. Chem. Phys. Lipids 2019, 221, 46–52. [Google Scholar] [CrossRef]
- Sakai, K.; Kino, S.; Masuda, A.; Takeuchi, M.; Ochi, T.; Osredkar, J.; Rejc, B.; Gersak, K.; Ramarathnam, N.; Kato, Y. Determination of HEL (Hexanoyl-Lysine Adduct): A Novel Biomarker for Omega-6 PUFA Oxidation. Subcell. Biochem. 2014, 77, 61–72. [Google Scholar] [CrossRef]
- Zarkovic, N. 4-Hydroxynonenal as a Bioactive Marker of Pathophysiological Processes. Mol. Asp. Med. 2003, 24, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Jaganjac, M.; Milkovic, L.; Gegotek, A.; Cindric, M.; Zarkovic, K.; Skrzydlewska, E.; Zarkovic, N. The Relevance of Pathophysiological Alterations in Redox Signaling of 4-Hydroxynonenal for Pharmacological Therapies of Major Stress-Associated Diseases. Free Radic. Biol. Med. 2020, 157, 128–153. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Li, W.; Kong, A.-N.T. Anti-Oxidative Stress Regulator NF-E2-Related Factor 2 Mediates the Adaptive Induction of Antioxidant and Detoxifying Enzymes by Lipid Peroxidation Metabolite 4-Hydroxynonenal. Cell Biosci. 2012, 2, 40. [Google Scholar] [CrossRef]
- Esterbauer, H.; Schaur, R.J.; Zollner, H. Chemistry and Biochemistry of 4-Hydroxynonenal, Malonaldehyde and Related Aldehydes. Free Radic. Biol. Med. 1991, 11, 81–128. [Google Scholar] [CrossRef]
- Niki, E. Biomarkers of Lipid Peroxidation in Clinical Material. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2014, 1840, 809–817. [Google Scholar] [CrossRef] [PubMed]
- Weismann, D.; Binder, C.J. The Innate Immune Response to Products of Phospholipid Peroxidation. Biochim. Biophys. Acta (BBA)-Biomembr. 2012, 1818, 2465–2475. [Google Scholar] [CrossRef]
- Que, X.; Hung, M.Y.; Yeang, C.; Gonen, A.; Prohaska, T.A.; Sun, X.; Diehl, C.; Määttä, A.; Gaddis, D.E.; Bowden, K.; et al. Oxidized Phospholipids Are Proinflammatory and Proatherogenic in Hypercholesterolemic Mice. Nature 2018, 558, 301. [Google Scholar] [CrossRef]
- Bochkov, V.N.; Oskolkova, O.V.; Birukov, K.G.; Levonen, A.L.; Binder, C.J.; Stöckl, J. Generation and Biological Activities of Oxidized Phospholipids. Antioxid. Redox Signal. 2010, 12, 1009–1059. [Google Scholar] [CrossRef]
- Friedli, O.; Freigang, S. Cyclopentenone-Containing Oxidized Phospholipids and Their Isoprostanes as pro-Resolving Mediators of Inflammation. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2017, 1862, 382–392. [Google Scholar] [CrossRef]
- Tyurin, V.A.; Tyurina, Y.Y.; Kochanek, P.M.; Hamilton, R.; DeKosky, S.T.; Greenberger, J.S.; Bayir, H.; Kagan, V.E. Chapter Nineteen Oxidative Lipidomics of Programmed Cell Death. Methods Enzymol. 2008, 442, 375–393. [Google Scholar]
- Yang, K.; Han, X. Lipidomics: Techniques, Applications, and Outcomes Related to Biomedical Sciences. Trends Biochem. Sci. 2016, 41, 954. [Google Scholar] [CrossRef]
- Clemente, S.M.; Martínez-Costa, O.H.; Monsalve, M.; Samhan-Arias, A.K. Targeting Lipid Peroxidation for Cancer Treatment. Molecules 2020, 25, 5144. [Google Scholar] [CrossRef]
- Ma, C.; Hu, H.; Liu, H.; Zhong, C.; Wu, B.; Lv, C.; Tian, Y. Lipotoxicity, Lipid Peroxidation and Ferroptosis: A Dilemma in Cancer Therapy. Cell Biol. Toxicol. 2025, 41, 75. [Google Scholar] [CrossRef]
- Borović Šunjić, S.; Jaganjac, M.; Vlainić, J.; Halasz, M.; Žarković, N. Lipid Peroxidation-Related Redox Signaling in Osteosarcoma. Int. J. Mol. Sci. 2024, 25, 4559. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.E.; Zhang, L.; Ma, K.; Riegman, M.; Chen, F.; Ingold, I.; Conrad, M.; Turker, M.Z.; Gao, M.; Jiang, X.; et al. Ultrasmall Nanoparticles Induce Ferroptosis in Nutrient-Deprived Cancer Cells and Suppress Tumour Growth. Nat. Nanotechnol. 2016, 11, 977–985. [Google Scholar] [CrossRef] [PubMed]
- Kong, F.-H.; Ye, Q.-F.; Miao, X.-Y.; Liu, X.; Huang, S.-Q.; Xiong, L.; Wen, Y.; Zhang, Z.-J. Current Status of Sorafenib Nanoparticle Delivery Systems in the Treatment of Hepatocellular Carcinoma. Theranostics 2021, 11, 5464–5490. [Google Scholar] [CrossRef]
- Yuan, S.; Wei, C.; Liu, G.; Zhang, L.; Li, J.; Li, L.; Cai, S.; Fang, L. Sorafenib Attenuates Liver Fibrosis by Triggering Hepatic Stellate Cell Ferroptosis via HIF-1α/SLC7A11 Pathway. Cell Prolif. 2022, 55, e13158. [Google Scholar] [CrossRef] [PubMed]
- Lachaier, E.; Louandre, C.; Godin, C.; Saidak, Z.; Baert, M.; Diouf, M.; Chauffert, B.; Galmiche, A. Sorafenib Induces Ferroptosis in Human Cancer Cell Lines Originating from Different Solid Tumors. Anticancer Res. 2014, 34, 6417–6422. [Google Scholar]
- Liu, M.; Zhu, W.; Pei, D. System Xc−: A Key Regulatory Target of Ferroptosis in Cancer. Investig. New Drugs 2021, 39, 1123–1131. [Google Scholar] [CrossRef]
- Song, X.; Zhu, S.; Chen, P.; Hou, W.; Wen, Q.; Liu, J.; Xie, Y.; Liu, J.; Klionsky, D.J.; Kroemer, G.; et al. AMPK-Mediated BECN1 Phosphorylation Promotes Ferroptosis by Directly Blocking System Xc– Activity. Curr. Biol. 2018, 28, 2388–2399.e5. [Google Scholar] [CrossRef]
- Liu, X.; Estaras, M.; Cosialls, E.; Peng, L.; Santofimia-Castaño, P.; Iovanna, J. Synergistic Antitumor Activity of Sorafenib and the NUPR1 Inhibitor LZX-2-73 in Multiple Cancer Models. Cell Death Dis. 2025, 16, 839. [Google Scholar] [CrossRef]
- Praharaj, P.P.; Singh, A.; Patra, S.; Bhutia, S.K. Co-Targeting Autophagy and NRF2 Signaling Triggers Mitochondrial Superoxide to Sensitize Oral Cancer Stem Cells for Cisplatin-Induced Apoptosis. Free Radic. Biol. Med. 2023, 207, 72–88. [Google Scholar] [CrossRef]
- Roh, J.-L.; Kim, E.H.; Jang, H.J.; Park, J.Y.; Shin, D. Induction of Ferroptotic Cell Death for Overcoming Cisplatin Resistance of Head and Neck Cancer. Cancer Lett. 2016, 381, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Zając-Grabiec, A.; Bartusek, K.; Sroczyńska, K.; Librowski, T.; Gdula-Argasińska, J. Effect of Eicosapentaenoic Acid Supplementation on Murine Preadipocytes 3T3-L1 Cells Activated with Lipopolysaccharide and/or Tumor Necrosis Factor-α. Life 2021, 11, 977. [Google Scholar] [CrossRef] [PubMed]
- Yates, C.M.; Calder, P.C.; Ed Rainger, G. Pharmacology and Therapeutics of Omega-3 Polyunsaturated Fatty Acids in Chronic Inflammatory Disease. Pharmacol. Ther. 2014, 141, 272–282. [Google Scholar] [CrossRef]
- Zhang, Q.; Deng, T.; Zhang, H.; Zuo, D.; Zhu, Q.; Bai, M.; Liu, R.; Ning, T.; Zhang, L.; Yu, Z.; et al. Adipocyte-Derived Exosomal MTTP Suppresses Ferroptosis and Promotes Chemoresistance in Colorectal Cancer. Adv. Sci. 2022, 9, 2203357. [Google Scholar] [CrossRef]
- Hangauer, M.J.; Viswanathan, V.S.; Ryan, M.J.; Bole, D.; Eaton, J.K.; Matov, A.; Galeas, J.; Dhruv, H.D.; Berens, M.E.; Schreiber, S.L.; et al. Drug-Tolerant Persister Cancer Cells Are Vulnerable to GPX4 Inhibition. Nature 2017, 551, 247–250. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Tomita, Y.; Kanazawa, T.; Shinohara, H.; Sakano, M.; Ishibashi, S.; Ikeda, M.; Kinoshita, M.; Minami, J.; Yamamoto, K.; et al. Lipid Peroxidation Regulators GPX4 and FSP1 as Prognostic Markers and Therapeutic Targets in Advanced Gastric Cancer. Int. J. Mol. Sci. 2024, 25, 9203. [Google Scholar] [CrossRef]
- Bersuker, K.; Hendricks, J.M.; Li, Z.; Magtanong, L.; Ford, B.; Tang, P.H.; Roberts, M.A.; Tong, B.; Maimone, T.J.; Zoncu, R.; et al. The CoQ Oxidoreductase FSP1 Acts Parallel to GPX4 to Inhibit Ferroptosis. Nature 2019, 575, 688–692. [Google Scholar] [CrossRef]
- Majumder, S.; Crabtree, J.S.; Golde, T.E.; Minter, L.M.; Osborne, B.A.; Miele, L. Targeting Notch in Oncology: The Path Forward. Nat. Rev. Drug Discov. 2021, 20, 125–144. [Google Scholar] [CrossRef]
- Mai, T.T.; Hamaï, A.; Hienzsch, A.; Cañeque, T.; Müller, S.; Wicinski, J.; Cabaud, O.; Leroy, C.; David, A.; Acevedo, V.; et al. Salinomycin Kills Cancer Stem Cells by Sequestering Iron in Lysosomes. Nat. Chem. 2017, 9, 1025–1033. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chen, Y.; Wang, X.; Tian, H.; Wang, Y.; Jin, J.; Shan, Z.; Liu, Y.; Cai, Z.; Tong, X.; et al. Stem Cell Factor SOX2 Confers Ferroptosis Resistance in Lung Cancer via Upregulation of SLC7A11. Cancer Res. 2021, 81, 5217–5229. [Google Scholar] [CrossRef] [PubMed]
- Fumarola, S.; Cecati, M.; Sartini, D.; Ferretti, G.; Milanese, G.; Galosi, A.B.; Pozzi, V.; Campagna, R.; Morresi, C.; Emanuelli, M.; et al. Bladder Cancer Chemosensitivity Is Affected by Paraoxonase-2 Expression. Antioxidants 2020, 9, 175. [Google Scholar] [CrossRef]
- Schiavoni, V.; Emanuelli, M.; Campagna, R.; Cecati, M.; Sartini, D.; Milanese, G.; Galosi, A.B.; Pozzi, V.; Salvolini, E. Paraoxonase-2 ShRNA-Mediated Gene Silencing Suppresses Proliferation and Migration, While Promotes Chemosensitivity in Clear Cell Renal Cell Carcinoma Cell Lines. J. Cell. Biochem. 2024, 125, e30572. [Google Scholar] [CrossRef]
- Turska-Skrodzka, W.; Ostrowska, A.; Krętowski, A.J.; Niemira, M. The Functional Role of Ferroptosis in the Development and Progression of Common Cancers. Exp. Mol. Pathol. 2026, 146, 105050. [Google Scholar] [CrossRef]
- Xiao, Q.; Xia, M.; Tang, W.; Zhao, H.; Chen, Y.; Zhong, J. The Lipid Metabolism Remodeling: A Hurdle in Breast Cancer Therapy. Cancer Lett. 2024, 582, 216512. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Lai, Y.; Lu, F.; Wang, W. Targeting ACSLs to Modulate Ferroptosis and Cancer Immunity. Trends Endocrinol. Metab. 2025, 36, 677–690. [Google Scholar] [CrossRef]
- Glibetic, N.; Weichhaus, M. Metabolic Regulation of Ferroptosis in Breast Cancer. Int. J. Mol. Sci. 2025, 26, 9686. [Google Scholar] [CrossRef]
- Gulhati, P.; Zaytseva, Y.Y.; Valentino, J.D.; Stevens, P.D.; Kim, J.T.; Sasazuki, T.; Shirasawa, S.; Lee, E.Y.; Weiss, H.L.; Dong, J.; et al. Sorafenib Enhances the Therapeutic Efficacy of Rapamycin in Colorectal Cancers Harboring Oncogenic KRAS and PIK3CA. Carcinogenesis 2012, 33, 1782. [Google Scholar] [CrossRef]
- Lu, Y.; Liang, H.; Li, X.; Chen, H.; Yang, C. Pan-Cancer Analysis Identifies LPCATs Family as a Prognostic Biomarker and Validation of LPCAT4/WNT/β-Catenin/c-JUN/ACSL3 in Hepatocellular Carcinoma. Aging 2023, 15, 4699. [Google Scholar] [CrossRef]
- Tang, S.; Chen, L. The Recent Advancements of Ferroptosis of Gynecological Cancer. Cancer Cell Int. 2024, 24, 351. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Meng, Y.; Li, D.; Yao, L.; Le, J.; Liu, Y.; Sun, Y.; Zeng, F.; Chen, X.; Deng, G. Ferroptosis in Cancer: From Molecular Mechanisms to Therapeutic Strategies. Signal Transduct. Target. Ther. 2024, 9, 55. [Google Scholar] [CrossRef]
- Zhang, J.; Tan, B.; Wu, H.; Han, T.; Fang, D.; Cai, H.; Hu, B.; Kang, A. Scutellaria Baicalensis Extracts Restrict Intestinal Epithelial Cell Ferroptosis by Regulating Lipid Peroxidation and GPX4/ACSL4 in Colitis. Phytomedicine 2025, 141, 156708. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Jiang, X.; Jing, D.; Lin, Y.; Gao, R.; Zhao, Q.; Da, H.; Ren, Y.; Cao, Q.; Liu, N.; et al. ACSL4 Knockdown Inhibits Colorectal Cancer Progression through Stimulating Anti-Tumor Immunity. Neoplasia 2025, 67, 101194. [Google Scholar] [CrossRef]
- Castillo, A.F.; Orlando, U.D.; Maloberti, P.M.; Prada, J.G.; Dattilo, M.A.; Solano, A.R.; Bigi, M.M.; Ríos Medrano, M.A.; Torres, M.T.; Indo, S.; et al. New Inhibitor Targeting Acyl-CoA Synthetase 4 Reduces Breast and Prostate Tumor Growth, Therapeutic Resistance and Steroidogenesis. Cell. Mol. Life Sci. 2021, 78, 2893–2910. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Liu, Y.; Liao, Y.; Zhu, C.; Zou, Z. GPX4, Ferroptosis, and Diseases. Biomed. Pharmacother. 2024, 174, 116512. [Google Scholar] [CrossRef]
- Liu, W.; Wu, G.; Wang, J.; Wu, S.; Chen, Z. Co-Treatment with Triptolide and RSL3 Induces Hepatocellular Carcinoma Cell Apoptosis and Ferroptosis. Mol. Med. Rep. 2025, 32, 202. [Google Scholar] [CrossRef]
- Chaudhry, G.-e.-S.; Akim, A.M.; Sung, Y.Y.; Muhammad, T.S.T. Cancer and Apoptosis. Methods Mol. Biol. 2022, 2543, 191–210. [Google Scholar] [CrossRef]
- Kiraz, Y.; Adan, A.; Kartal Yandim, M.; Baran, Y. Major Apoptotic Mechanisms and Genes Involved in Apoptosis. Tumor Biol. 2016, 37, 8471–8486. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A Review of Programmed Cell Death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Romero, A.; Novoa, B.; Figueras, A. The Complexity of Apoptotic Cell Death in Mollusks: An Update. Fish Shellfish Immunol. 2015, 46, 79–87. [Google Scholar] [CrossRef]
- Ashkenazi, A.; Dixit, V.M. Death Receptors: Signaling and Modulation. Science 1998, 281, 1305–1308. [Google Scholar] [CrossRef] [PubMed]
- Metkar, S.S.; Wang, B.; Ebbs, M.L.; Kim, J.H.; Lee, Y.J.; Raja, S.M.; Froelich, C.J. Granzyme B Activates Procaspase-3 Which Signals a Mitochondrial Amplification Loop for Maximal Apoptosis. J. Cell Biol. 2003, 160, 875–885. [Google Scholar] [CrossRef]
- Grodzovski, I.; Lichtenstein, M.; Galski, H.; Lorberboum-Galski, H. IL-2-Granzyme A Chimeric Protein Overcomes Multidrug Resistance (MDR) through a Caspase 3-Independent Apoptotic Pathway. Int. J. Cancer 2011, 128, 1966–1980. [Google Scholar] [CrossRef]
- Wu, C.C.; Bratton, S.B. Regulation of the Intrinsic Apoptosis Pathway by Reactive Oxygen Species. Antioxid. Redox Signal. 2013, 19, 546. [Google Scholar] [CrossRef]
- Bratton, S.B.; Salvesen, G.S. Regulation of the Apaf-1–Caspase-9 Apoptosome. J. Cell Sci. 2010, 123, 3209–3214. [Google Scholar] [CrossRef]
- Zhong, H.; Xiao, M.; Zarkovic, K.; Zhu, M.; Sa, R.; Lu, J.; Tao, Y.; Chen, Q.; Xia, L.; Cheng, S.; et al. Mitochondrial Control of Apoptosis through Modulation of Cardiolipin Oxidation in Hepatocellular Carcinoma: A Novel Link between Oxidative Stress and Cancer. Free Radic. Biol. Med. 2017, 102, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Petrosillo, G.; Ruggiero, F.M.; Pistolese, M.; Paradies, G. Reactive Oxygen Species Generated from the Mitochondrial Electron Transport Chain Induce Cytochrome c Dissociation from Beef-Heart Submitochondrial Particles via Cardiolipin Peroxidation. Possible Role in the Apoptosis. FEBS Lett. 2001, 509, 435–438. [Google Scholar] [CrossRef] [PubMed]
- Kagan, V.E.; Tyurin, V.A.; Jiang, J.; Tyurina, Y.Y.; Ritov, V.B.; Amoscato, A.A.; Osipov, A.N.; Belikova, N.A.; Kapralov, A.A.; Kini, V.; et al. Cytochrome C Acts As A Cardiolipin Oxygenase Required for Release of Proapoptotic Factors. Nat. Chem. Biol. 2005, 1, 223–232. [Google Scholar] [CrossRef]
- McMillin, J.B.; Dowhan, W. Cardiolipin and Apoptosis. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2002, 1585, 97–107. [Google Scholar] [CrossRef]
- Das, U.N. Essential Fatty Acids, Lipid Peroxidation and Apoptosis. Prostaglandins Leukot. Essent. Fat. Acids 1999, 61, 157–163. [Google Scholar] [CrossRef]
- Sadati, S.; Khalaji, A.; Bonyad, A.; Khoshdooz, S.; Sadat, K.; Kolbadi, H.; Bahrami, A.; Saeid Moeinfar, M.; Morshedi, M.; Ghamsaraian, A.; et al. NF-ΚB and Apoptosis: Colorectal Cancer Progression and Novel Strategies for Treatment. Eur. J. Med. Res. 2025, 30, 616. [Google Scholar] [CrossRef]
- Page, S.; Fischer, C.; Baumgartner, B.; Haas, M.; Kreusel, U.; Loidl, G.; Hayn, M.; Ziegler-Heitbrock, H.W.L.; Neumeier, D.; Brand, K. 4-Hydroxynonenal Prevents NF-ΚB Activation and Tumor Necrosis Factor Expression by Inhibiting IκB Phosphorylation and Subsequent Proteolysis. J. Biol. Chem. 1999, 274, 11611–11618. [Google Scholar] [CrossRef] [PubMed]
- Khalid, N.; Azimpouran, M. Necrosis Pathology. In The Brain, the Nervous System, and Their Diseases: Volumes 1–3; Jaypee Digital: New Delhi, India, 2023; Volume 2, pp. 701–702. [Google Scholar] [CrossRef]
- Zong, W.X.; Thompson, C.B. Necrotic Death as a Cell Fate. Genes Dev. 2006, 20, 1–15. [Google Scholar] [CrossRef]
- Chen, D.; Song, M.; Mohamad, O.; Yu, S.P. Inhibition of Na+/K+-ATPase Induces Hybrid Cell Death and Enhanced Sensitivity to Chemotherapy in Human Glioblastoma Cells. BMC Cancer 2014, 14, 716. [Google Scholar] [CrossRef]
- Noh, M.R.; Padanilam, B.J. Cell Death Induced by Acute Renal Injury: A Perspective on the Contributions of Accidental and Programmed Cell Death. Am. J. Physiol.-Ren. Physiol. 2024, 327, F4–F20. [Google Scholar] [CrossRef]
- Herson, P.S.; Lee, K.; Pinnock, R.D.; Hughes, J.; Ashford, M.L.J. Hydrogen Peroxide Induces Intracellular Calcium Overload by Activation of a Non-Selective Cation Channel in an Insulin-Secreting Cell Line. J. Biol. Chem. 1999, 274, 833–841. [Google Scholar] [CrossRef]
- Zhivotovsky, B.; Orrenius, S. Calcium and Cell Death Mechanisms: A Perspective from the Cell Death Community. Cell Calcium 2011, 50, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Festjens, N.; Vanden Berghe, T.; Vandenabeele, P. Necrosis, a Well-Orchestrated Form of Cell Demise: Signalling Cascades, Important Mediators and Concomitant Immune Response. Biochim. Biophys. Acta (BBA)-Bioenerg. 2006, 1757, 1371–1387. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular Mechanisms of Cell Death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Ros, U.; Pedrera, L.; Garcia-Saez, A.J. Partners in Crime: The Interplay of Proteins and Membranes in Regulated Necrosis. Int. J. Mol. Sci. 2020, 21, 2412. [Google Scholar] [CrossRef] [PubMed]
- Petrie, E.J.; Hildebrand, J.M.; Murphy, J.M. Insane in the Membrane: A Structural Perspective of MLKL Function in Necroptosis. Immunol. Cell Biol. 2017, 95, 152–159. [Google Scholar] [CrossRef]
- Dondelinger, Y.; Declercq, W.; Montessuit, S.; Roelandt, R.; Goncalves, A.; Bruggeman, I.; Hulpiau, P.; Weber, K.; Sehon, C.A.; Marquis, R.W.; et al. MLKL Compromises Plasma Membrane Integrity by Binding to Phosphatidylinositol Phosphates. Cell Rep. 2014, 7, 971–981. [Google Scholar] [CrossRef] [PubMed]
- Canli, Ö.; Alankus, Y.B.; Grootjans, S.; Vegi, N.; Hültner, L.; Hoppe, P.S.; Schroeder, T.; Vandenabeele, P.; Bornkamm, G.W.; Greten, F.R. Glutathione Peroxidase 4 Prevents Necroptosis in Mouse Erythroid Precursors. Blood 2016, 127, 139–148. [Google Scholar] [CrossRef]
- Tong, Y.; Wu, Y.; Ma, J.; Ikeda, M.; Ide, T.; Griffin, C.T.; Ding, X.Q.; Wang, S. Comparative Mechanistic Study of RPE Cell Death Induced by Different Oxidative Stresses. Redox Biol. 2023, 65, 102840. [Google Scholar] [CrossRef]
- Wang, H.; Sun, L.; Su, L.; Rizo, J.; Liu, L.; Wang, L.F.; Wang, F.S.; Wang, X. Mixed Lineage Kinase Domain-like Protein MLKL Causes Necrotic Membrane Disruption upon Phosphorylation by RIP3. Mol. Cell 2014, 54, 133–146. [Google Scholar] [CrossRef]
- Ouyang, X.; Zhou, J.; Lin, L.; Zhang, Z.; Luo, S.; Hu, D. Pyroptosis, Inflammasome, and Gasdermins in Tumor Immunity. Innate Immun. 2023, 29, 3–13. [Google Scholar] [CrossRef]
- Fang, Y.; Tian, S.; Pan, Y.; Li, W.; Wang, Q.; Tang, Y.; Yu, T.; Wu, X.; Shi, Y.; Ma, P.; et al. Pyroptosis: A New Frontier in Cancer. Biomed. Pharmacother. 2020, 121, 109595. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, Z.; Ruan, J.; Pan, Y.; Magupalli, V.G.; Wu, H.; Lieberman, J. Inflammasome-Activated Gasdermin D Causes Pyroptosis by Forming Membrane Pores. Nature 2016, 535, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Kolb, R.; Liu, G.H.; Janowski, A.M.; Sutterwala, F.S.; Zhang, W. Inflammasomes in Cancer: A Double-Edged Sword. Protein Cell 2014, 5, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.; Zhang, X.; Liu, N.; Tang, L.; Peng, C.; Chen, X. Pyroptosis: Mechanisms and Diseases. Signal Transduct. Target. Ther. 2021, 6, 128. [Google Scholar] [CrossRef] [PubMed]
- Tong, X.; Tang, R.; Xiao, M.; Xu, J.; Wang, W.; Zhang, B.; Liu, J.; Yu, X.; Shi, S. Targeting Cell Death Pathways for Cancer Therapy: Recent Developments in Necroptosis, Pyroptosis, Ferroptosis, and Cuproptosis Research. J. Hematol. Oncol. 2022, 15, 174. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Bettadapura, S.N.; Smeltzer, M.S.; Zhu, H.; Wang, S. Pyroptosis and Pyroptosis-Inducing Cancer Drugs. Acta Pharmacol. Sin. 2022, 43, 2462. [Google Scholar] [CrossRef]
- Gao, W.; Wang, X.; Zhou, Y.; Wang, X.; Yu, Y. Autophagy, Ferroptosis, Pyroptosis, and Necroptosis in Tumor Immunotherapy. Signal Transduct. Target. Ther. 2022, 7, 196. [Google Scholar] [CrossRef]
- Aachoui, Y.; Sagulenko, V.; Miao, E.A.; Stacey, K.J. Inflammasome-Mediated Pyroptotic and Apoptotic Cell Death, and Defense against Infection. Curr. Opin. Microbiol. 2013, 16, 319–326. [Google Scholar] [CrossRef]
- de Gassart, A.; Martinon, F. Pyroptosis: Caspase-11 Unlocks the Gates of Death. Immunity 2015, 43, 835–837. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Li, J.; Wu, R.; Li, Y.; Zhang, C. Targeting Pyroptosis for Cancer Immunotherapy: Mechanistic Insights and Clinical Perspectives. Mol. Cancer 2025, 24, 131. [Google Scholar] [CrossRef]
- Kim, M.S.; Lebron, C.; Nagpal, J.K.; Chae, Y.K.; Chang, X.; Huang, Y.; Chuang, T.; Yamashita, K.; Trink, B.; Ratovitski, E.A.; et al. Methylation of the DFNA5 Increases Risk of Lymph Node Metastasis in Human Breast Cancer. Biochem. Biophys. Res. Commun. 2008, 370, 38–43. [Google Scholar] [CrossRef]
- Qiao, L.; Wu, X.; Zhang, J.; Liu, L.; Sui, X.; Zhang, R.; Liu, W.; Shen, F.; Sun, Y.; Xi, X. A-NETA Induces Pyroptosis of Epithelial Ovarian Cancer Cells through the GSDMD/Caspase-4 Pathway. FASEB J. 2019, 33, 12760–12767. [Google Scholar] [CrossRef]
- Wang, Y.; Yin, B.; Li, D.; Wang, G.; Han, X.; Sun, X. GSDME Mediates Caspase-3-Dependent Pyroptosis in Gastric Cancer. Biochem. Biophys. Res. Commun. 2018, 495, 1418–1425. [Google Scholar] [CrossRef]
- Lin, W.; Chen, Y.; Wu, B.; Chen, Y.; Li, Z. Identification of the Pyroptosis-related Prognostic Gene Signature and the Associated Regulation Axis in Lung Adenocarcinoma. Cell Death Discov. 2021, 7, 161. [Google Scholar] [CrossRef]
- Zhang, Q.; Tan, Y.; Zhang, J.; Shi, Y.; Qi, J.; Zou, D.; Ci, W. Pyroptosis-Related Signature Predicts Prognosis and Immunotherapy Efficacy in Muscle-Invasive Bladder Cancer. Front. Immunol. 2022, 13, 782982. [Google Scholar] [CrossRef]
- Li, S.R.; Bu, L.L.; Cai, L. Cuproptosis: Lipoylated TCA Cycle Proteins-Mediated Novel Cell Death Pathway. Signal Transduct. Target. Ther. 2022, 7, 158. [Google Scholar] [CrossRef]
- Tsvetkov, P.; Coy, S.; Petrova, B.; Dreishpoon, M.; Verma, A.; Abdusamad, M.; Rossen, J.; Joesch-Cohen, L.; Humeidi, R.; Spangler, R.D.; et al. Copper Induces Cell Death by Targeting Lipoylated TCA Cycle Proteins. Science 2022, 375, 1254. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Min, J.; Wang, F. Copper Homeostasis and Cuproptosis in Health and Disease. Signal Transduct. Target. Ther. 2022, 7, 378. [Google Scholar] [CrossRef]
- MacDonald, G.; Nalvarte, I.; Smirnova, T.; Vecchi, M.; Aceto, N.; Doelemeyer, A.; Frei, A.; Lienhard, S.; Wyckoff, J.; Hess, D.; et al. Memo Is a Copper-Dependent Redox Protein with an Essential Role in Migration and Metastasis. Sci. Signal. 2014, 7, ra56. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zeng, X.; Wu, Y.; Liu, Y.; Zhang, X.; Song, Z. Cuproptosis-Related Risk Score Predicts Prognosis and Characterizes the Tumor Microenvironment in Hepatocellular Carcinoma. Front. Immunol. 2022, 13, 925618. [Google Scholar] [CrossRef] [PubMed]
- Xing, T.; Li, L.; Chen, Y.; Ju, G.; Li, G.; Zhu, X.; Ren, Y.; Zhao, J.; Cheng, Z.; Li, Y.; et al. Targeting the TCA Cycle through Cuproptosis Confers Synthetic Lethality on ARID1A-Deficient Hepatocellular Carcinoma. Cell Rep. Med. 2023, 4, 101264. [Google Scholar] [CrossRef]
- Li, J.; Chen, S.; Liao, Y.; Wang, H.; Zhou, D.; Zhang, B. Arecoline Is Associated With Inhibition of Cuproptosis and Proliferation of Cancer-Associated Fibroblasts in Oral Squamous Cell Carcinoma: A Potential Mechanism for Tumor Metastasis. Front. Oncol. 2022, 12, 925743. [Google Scholar] [CrossRef]
- Li, Y.; Liu, J.; Chen, Y.; Weichselbaum, R.R.; Lin, W. Nanoparticles Synergize Ferroptosis and Cuproptosis to Potentiate Cancer Immunotherapy. Adv. Sci. 2024, 11, 2310309. [Google Scholar] [CrossRef]
- Choi, M.; Shin, J.; Lee, C.E.; Chung, J.Y.; Kim, M.; Yan, X.; Yang, W.H.; Cha, J.H. Immunogenic Cell Death in Cancer Immunotherapy. BMB Rep. 2023, 56, 275. [Google Scholar] [CrossRef]
- Jiang, J.; Yan, Y.; Yang, C.; Cai, H. Immunogenic Cell Death and Metabolic Reprogramming in Cancer: Mechanisms, Synergies, and Innovative Therapeutic Strategies. Biomedicines 2025, 13, 950. [Google Scholar] [CrossRef]
- Ahmed, A.; Tait, S.W.G. Targeting Immunogenic Cell Death in Cancer. Mol. Oncol. 2020, 14, 2994. [Google Scholar] [CrossRef] [PubMed]
- Palanivelu, L.; Liu, C.H.; Lin, L.T. Immunogenic Cell Death: The Cornerstone of Oncolytic Viro-Immunotherapy. Front. Immunol. 2023, 13, 1038226. [Google Scholar] [CrossRef]
- Liu, Z.; Xu, X.; Liu, K.; Zhang, J.; Ding, D.; Fu, R. Immunogenic Cell Death in Hematological Malignancy Therapy. Adv. Sci. 2023, 10, 2207475. [Google Scholar] [CrossRef]
- Choi, J.H.; Kagan, J.C. Oxidized Phospholipid Damage Signals as Modulators of Immunity. Open Biol. 2025, 15, 240391. [Google Scholar] [CrossRef]
- Dou, L.; Fang, Y.; Yang, H.; Ai, G.; Shen, N. Immunogenic Cell Death: A New Strategy to Enhancing Cancer Immunotherapy. Hum. Vaccin. Immunother. 2024, 20, 2437918. [Google Scholar] [CrossRef]
- Chiaravalli, M.; Spring, A.; Agostini, A.; Piro, G.; Carbone, C.; Tortora, G. Immunogenic Cell Death: An Emerging Target in Gastrointestinal Cancers. Cells 2022, 11, 3033. [Google Scholar] [CrossRef]
- Delcheva, G.; Stefanova, K.; Stankova, T. Ceramides—Emerging Biomarkers of Lipotoxicity in Obesity, Diabetes, Cardiovascular Diseases, and Inflammation. Diseases 2024, 12, 195. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M. Lipotoxicity as a Therapeutic Target in Obesity and Diabetic Cardiomyopathy. J. Pharm. Pharm. Sci. 2024, 27, 12568. [Google Scholar] [CrossRef] [PubMed]
- Ceja-Galicia, Z.A.; Cespedes-Acuña, C.L.A.; El-Hafidi, M. Protection Strategies Against Palmitic Acid-Induced Lipotoxicity in Metabolic Syndrome and Related Diseases. Int. J. Mol. Sci. 2025, 26, 788. [Google Scholar] [CrossRef]
- Congur, I.; Mingrone, G.; Guan, K. Targeting Endoplasmic Reticulum Stress as a Potential Therapeutic Strategy for Diabetic Cardiomyopathy. Metabolism 2025, 162, 156062. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Liu, N.; Zhang, D.; Guo, L.; Shang, Q.; Liu, Y.; Ren, G.; Ma, X. Mitochondria-Associated Endoplasmic Reticulum Membranes as a Therapeutic Target for Cardiovascular Diseases. Front. Pharmacol. 2024, 15, 1398381. [Google Scholar] [CrossRef]
- Olzmann, J.A.; Carvalho, P. Dynamics and Functions of Lipid Droplets. Nat. Rev. Mol. Cell Biol. 2019, 20, 137–155. [Google Scholar] [CrossRef] [PubMed]
- Prevost, C.T.; Gansereit, W.B.; Kashatus, D.F. NRF2 Regulates Lipid Droplet Dynamics to Prevent Lipotoxicity. iScience 2025, 28, 112925. [Google Scholar] [CrossRef]
- Petan, T.; Jarc, E.; Jusović, M. Lipid Droplets in Cancer: Guardians of Fat in a Stressful World. Molecules 2018, 23, 1941. [Google Scholar] [CrossRef]
- Zhang, R.; Meng, J.; Yang, S.; Liu, W.; Shi, L.; Zeng, J.; Chang, J.; Liang, B.; Liu, N.; Xing, D. Recent Advances on the Role of ATGL in Cancer. Front. Oncol. 2022, 12, 944025. [Google Scholar] [CrossRef]
- Rieusset, J. The Role of Endoplasmic Reticulum-Mitochondria Contact Sites in the Control of Glucose Homeostasis: An Update. Cell Death Dis. 2018, 9, 388. [Google Scholar] [CrossRef]
- Zhang, P.; Konja, D.; Zhang, Y.; Wang, Y. Communications between Mitochondria and Endoplasmic Reticulum in the Regulation of Metabolic Homeostasis. Cells 2021, 10, 2195. [Google Scholar] [CrossRef] [PubMed]
- Alonso, A.; Goñi, F.M. The Physical Properties of Ceramides in Membranes. Annu. Rev. Biophys. 2018, 47, 633–654. [Google Scholar] [CrossRef]
- Bharath, L.P.; Ruan, T.; Li, Y.; Ravindran, A.; Wan, X.; Nhan, J.K.; Walker, M.L.; Deeter, L.; Goodrich, R.; Johnson, E.; et al. Ceramide-Initiated Protein Phosphatase 2A Activation Contributes to Arterial Dysfunction In Vivo. Diabetes 2015, 64, 3914–3926. [Google Scholar] [CrossRef]
- Sokolowska, E.; Blachnio-Zabielska, A. The Role of Ceramides in Insulin Resistance. Front. Endocrinol. 2019, 10, 577. [Google Scholar] [CrossRef]
- Eaton, S.; Bartlett, K.B.; Pourfarzam, M. Mammalian Mitochondrial β-Oxidation. Biochem. J. 1996, 320, 345–357. [Google Scholar] [CrossRef]
- Nakamura, S.; Takamura, T.; Matsuzawa-Nagata, N.; Takayama, H.; Misu, H.; Noda, H.; Nabemoto, S.; Kurita, S.; Ota, T.; Ando, H.; et al. Palmitate Induces Insulin Resistance in H4IIEC3 Hepatocytes through Reactive Oxygen Species Produced by Mitochondria. J. Biol. Chem. 2009, 284, 14809–14818. [Google Scholar] [CrossRef]
- Schon, E.A.; Przedborski, S. Mitochondria: The Next (Neurode)Generation. Neuron 2011, 70, 1033–1053. [Google Scholar] [CrossRef]
- Tait, S.W.G.; Green, D.R. Mitochondria and Cell Death: Outer Membrane Permeabilization and Beyond. Nat. Rev. Mol. Cell Biol. 2010, 11, 621–632. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, P.; Gerle, C.; Halestrap, A.P.; Jonas, E.A.; Karch, J.; Mnatsakanyan, N.; Pavlov, E.; Sheu, S.-S.; Soukas, A.A. Identity, Structure, and Function of the Mitochondrial Permeability Transition Pore: Controversies, Consensus, Recent Advances, and Future Directions. Cell Death Differ. 2023, 30, 1869–1885. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; Strasser, A. The BCL-2 Protein Family: Opposing Activities That Mediate Cell Death. Nat. Rev. Mol. Cell Biol. 2008, 9, 47–59. [Google Scholar] [CrossRef]
- Zhang, J.; Ney, P.A. Role of BNIP3 and NIX in Cell Death, Autophagy, and Mitophagy. Cell Death Differ. 2009, 16, 939–946. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial Fission, Fusion, and Stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef]
- Jheng, H.-F.; Tsai, P.-J.; Guo, S.-M.; Kuo, L.-H.; Chang, C.-S.; Su, I.-J.; Chang, C.-R.; Tsai, Y.-S. Mitochondrial Fission Contributes to Mitochondrial Dysfunction and Insulin Resistance in Skeletal Muscle. Mol. Cell. Biol. 2012, 32, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Jarocki, M.; Turek, K.; Saczko, J.; Tarek, M.; Kulbacka, J. Lipids Associated with Autophagy: Mechanisms and Therapeutic Targets. Cell Death Discov. 2024, 10, 460. [Google Scholar] [CrossRef] [PubMed]
- Sharma, G.; Chaurasia, S.S.; Carlson, M.A.; Mishra, P.K. Recent Advances Associated with Cardiometabolic Remodeling in Diabetes-Induced Heart Failure. Am. J. Physiol.-Heart Circ. Physiol. 2024, 327, H1327–H1342. [Google Scholar] [CrossRef] [PubMed]
- Pickles, S.; Vigié, P.; Youle, R.J. Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr. Biol. 2018, 28, R170–R185. [Google Scholar] [CrossRef]
- Ernst, R.; Renne, M.F.; Jain, A.; von der Malsburg, A. Endoplasmic Reticulum Membrane Homeostasis and the Unfolded Protein Response. Cold Spring Harb. Perspect. Biol. 2024, 16, a041400. [Google Scholar] [CrossRef]
- Halbleib, K.; Pesek, K.; Covino, R.; Hofbauer, H.F.; Wunnicke, D.; Hänelt, I.; Hummer, G.; Ernst, R. Activation of the Unfolded Protein Response by Lipid Bilayer Stress. Mol. Cell 2017, 67, 673–684.e8. [Google Scholar] [CrossRef]
- Kettel, P.; Karagöz, G.E. Endoplasmic Reticulum: Monitoring and Maintaining Protein and Membrane Homeostasis in the Endoplasmic Reticulum by the Unfolded Protein Response. Int. J. Biochem. Cell Biol. 2024, 172, 106598. [Google Scholar] [CrossRef]
- Hetz, C.; Zhang, K.; Kaufman, R.J. Mechanisms, Regulation and Functions of the Unfolded Protein Response. Nat. Rev. Mol. Cell Biol. 2020, 21, 421–438. [Google Scholar] [CrossRef]
- Ron, D.; Walter, P. Signal Integration in the Endoplasmic Reticulum Unfolded Protein Response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef]
- Haze, K.; Yoshida, H.; Yanagi, H.; Yura, T.; Mori, K. Mammalian Transcription Factor ATF6 Is Synthesized as a Transmembrane Protein and Activated by Proteolysis in Response to Endoplasmic Reticulum Stress. Mol. Biol. Cell 1999, 10, 3787–3799. [Google Scholar] [CrossRef]
- Tirasophon, W.; Lee, K.; Callaghan, B.; Welihinda, A.; Kaufman, R.J. The Endoribonuclease Activity of Mammalian IRE1 Autoregulates Its MRNA and Is Required for the Unfolded Protein Response. Genes Dev. 2000, 14, 2725–2736. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 MRNA Is Induced by ATF6 and Spliced by IRE1 in Response to ER Stress to Produce a Highly Active Transcription Factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Papa, F.R. The Unfolded Protein Response and Cell Fate Control. Mol. Cell 2018, 69, 169–181. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Endoplasmic Reticulum Stress and the Inflammatory Basis of Metabolic Disease. Cell 2010, 140, 900–917. [Google Scholar] [CrossRef]
- Sharma, S.; Sharma, P.; Bailey, T.; Bhattarai, S.; Subedi, U.; Miller, C.; Ara, H.; Kidambi, S.; Sun, H.; Panchatcharam, M.; et al. Electrophilic Aldehyde 4-Hydroxy-2-Nonenal Mediated Signaling and Mitochondrial Dysfunction. Biomolecules 2022, 12, 1555. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Zhang, Q.; Pan, Y.; Chen, H.; Zhou, K.; Cai, H.; Huang, P. Roles and Prospective Applications of Ferroptosis Suppressor Protein 1 (FSP1) in Malignant Tumor Treatment. Curr. Oncol. 2025, 32, 456. [Google Scholar] [CrossRef]
- Zhang, M.; Guo, M.; Gao, Y.; Wu, C.; Pan, X.; Huang, Z. Mechanisms and Therapeutic Targets of Ferroptosis: Implications for Nanomedicine Design. J. Pharm. Anal. 2024, 14, 100960. [Google Scholar] [CrossRef]
- Wu, Z.; Zhang, Y.; Zhong, W.; Wu, K.; Zhong, T.; Jiang, T. Targeting Ferroptosis: A Promising Approach for Treating Lung Carcinoma. Cell Death Discov. 2025, 11, 33. [Google Scholar] [CrossRef] [PubMed]
- Study Details|NCT03247088|Sorafenib, Busulfan and Fludarabine in Treating Patients with Recurrent or Refractory Acute Myeloid Leukemia Undergoing Donor Stem Cell Transplant|ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/study/NCT03247088 (accessed on 6 June 2026).


{kind=link}
{kind=link}
| Mechanism | Key Cellular Events | Biological Consequences | Relevance to Lipid-Induced Cell Damage | References |
|---|---|---|---|---|
| Lipid overload/lipotoxicity | Excessive uptake and intracellular accumulation of fatty acids in non-adipose tissues | Metabolic imbalance, activation of stress pathways, impaired cellular homeostasis | Initiates metabolic disturbance leading to organelle dysfunction and cell damage | [119,120] |
| Ceramide and lipid intermediate accumulation | Increased levels of bioactive lipids, including ceramides and diacylglycerols | Disrupted signaling, mitochondrial dysfunction, and insulin resistance | Links altered lipid metabolism to stress signaling and apoptosis susceptibility | [119] |
| Mitochondrial dysfunction | Enhanced β-oxidation and mitochondrial stress responses | ROS generation, mitochondrial depolarization, impaired ATP production, apoptosis | Central driver of oxidative damage and energy failure | [131,132,133] |
| Endoplasmic Reticulum Stress | Disruption of protein folding due to altered ER membrane composition | Activation of unfolded protein response (UPR), proteostasis imbalance | Reflects failure of adaptive stress responses and promotes cell death pathways | [133,134] |
| Oxidative stress | Increased production of reactive oxygen species (ROS) | Damage to proteins, lipids, and mitochondrial DNA | Amplifies lipid-induced damage and promotes irreversible cellular injury | [135] |
| Membrane destabilization | Altered phospholipid and sphingolipid composition | Impaired membrane integrity and signaling, organelle dysfunction | Disrupts membrane structure and cellular compartmentalization | [119] |
| Mechanism | Key Molecular Mediators | Cellular Consequences | Relevance to Lipid-Induced Cell Damage | References |
|---|---|---|---|---|
| Enhanced fatty acid β-oxidation | Mitochondrial respiratory chain enzymes | Increased electron flux and mitochondrial stress | Promotes electron leakage and predisposes mitochondria to oxidative injury | [13,14] |
| Reactive oxygen species (ROS) generation | Mitochondrial ROS | Oxidative damage to mitochondrial proteins, lipids, and DNA | Amplifies mitochondrial dysfunction and contributes to irreversible cellular injury | [135] |
| Mitochondrial membrane depolarization | Loss of mitochondrial membrane potential (ΔΨm) | Activation of mitochondrial cell death pathways | Indicates bioenergetic failure and increased susceptibility to apoptosis | [138] |
| Mitochondrial fragmentation | DRP1-mediated mitochondrial fission | Impaired mitochondrial respiration and increased apoptotic susceptibility | Reflects structural remodeling associated with mitochondrial stress and dysfunction | [142,143] |
| Activation of pro-apoptotic proteins | BAX, BNIP3 | Mitochondrial outer membrane permeabilization (MOMP), mitophagy | Links mitochondrial injury to apoptosis and selective organelle turnover | [140,141] |
| Mitophagy | Autophagy machinery | Removal of damaged mitochondria | Represents an adaptive quality-control mechanism that may limit mitochondrial damage | [146] |
| UPR Branch | Key Molecular Components | Core Mechanism | Cellular Consequences | Functional Relevance in ER Stress | References |
|---|---|---|---|---|---|
| PERK pathway | PERK, eIF2α, ATF4 | PERK phosphorylates eIF2α, attenuating global protein translation while promoting ATF4 translation | Reduced ER protein load, stress adaptation | Limits protein overload and promotes adaptive stress responses under ER stress | [150,151] |
| ATF6 pathway | ATF6, GRP78/BiP | ATF6 translocates to the Golgi apparatus, where it undergoes proteolytic activation | Increased expression of ER chaperones and folding-related proteins | Enhances ER protein-folding capacity and supports restoration of proteostasis | [150,151] |
| IRE1α pathway | IRE1α, XBP1 | IRE1α mediates unconventional splicing of XBP1 mRNA | Upregulation of genes involved in protein folding, lipid biosynthesis, and ER-associated degradation (ERAD) | Coordinates adaptive remodeling of ER function and quality-control pathways | [152,153] |
| Agent | Principal Target | Effect on LPO Axis | Status (Indicative) |
|---|---|---|---|
| Sorafenib | Multikinase (RAF/VEGFR/PDGFR); indirectly system xc− | Promotes lipid ROS via xc− inhibition (context-dependent) | Approved (HCC, RCC); ferroptosis role investigational |
| Cisplatin | DNA cross-links; mitochondrial ROS; GPX4 (context) | Secondary lipid peroxidation downstream of oxidative stress | Approved (many solid tumors) |
| Olaparib | PARP; p53-dependent SLC7A11 down-regulation | Sensitizes to ferroptosis; enhances inducers | Approved (BRCA/HRD cancers) |
| Erastin/IKE | SLC7A11 (system xc−) | Depletes GSH → ferroptosis | Preclinical (tool/IKE optimized) |
| RSL3/ML210/ML162 | GPX4 | Direct GPX4 inhibition → lipid-peroxide accumulation | Preclinical tool compounds |
| iFSP1/FSeIII | FSP1 (AIFM2) | Blocks CoQ10-based radical trapping; synergy with GPX4 inhibition | Preclinical |
| Brequinar | DHODH | Restores mitochondrial lipid peroxidation in GPX4-low tumors | Repurposing/preclinical for ferroptosis |
| SCD1 inhibitors: rapamycin (mTOR) | SCD1/SREBP1 axis | Lower MUFA, restores peroxidizable substrate; reverse resistance | Preclinical/clinical (rapamycin approved other indications) |
| ML385 | NRF2 | Inhibits oncogenic NRF2; re-sensitizes to ferroptosis | Preclinical |
| Elesclomol | Copper ionophore (FDX1) | ROS/cuproptosis; crosstalk with ferroptosis | Clinically tested (other endpoints) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.
Share and Cite
Andryszkiewicz, W.; Cichowska, Z.; Filipski, M.; Szyda, K.; Wietrzyk, A.; Szpak, P.; Kulbacka, J. Lipid Peroxidation in Cancer Therapy: Molecular Mechanisms Involving Oxidative Stress, Cell Death, and Therapeutic Response. Molecules 2026, 31, 2072. https://doi.org/10.3390/molecules31122072
Andryszkiewicz W, Cichowska Z, Filipski M, Szyda K, Wietrzyk A, Szpak P, Kulbacka J. Lipid Peroxidation in Cancer Therapy: Molecular Mechanisms Involving Oxidative Stress, Cell Death, and Therapeutic Response. Molecules. 2026; 31(12):2072. https://doi.org/10.3390/molecules31122072
Chicago/Turabian StyleAndryszkiewicz, Wiktoria, Zuzanna Cichowska, Michał Filipski, Kamila Szyda, Anna Wietrzyk, Piotr Szpak, and Julita Kulbacka. 2026. "Lipid Peroxidation in Cancer Therapy: Molecular Mechanisms Involving Oxidative Stress, Cell Death, and Therapeutic Response" Molecules 31, no. 12: 2072. https://doi.org/10.3390/molecules31122072
APA StyleAndryszkiewicz, W., Cichowska, Z., Filipski, M., Szyda, K., Wietrzyk, A., Szpak, P., & Kulbacka, J. (2026). Lipid Peroxidation in Cancer Therapy: Molecular Mechanisms Involving Oxidative Stress, Cell Death, and Therapeutic Response. Molecules, 31(12), 2072. https://doi.org/10.3390/molecules31122072