

Synthesis of Diversely Substituted Diethyl (Pyrrolidin-2-Yl)Phosphonates
 , , ,
, , ,
Abstract

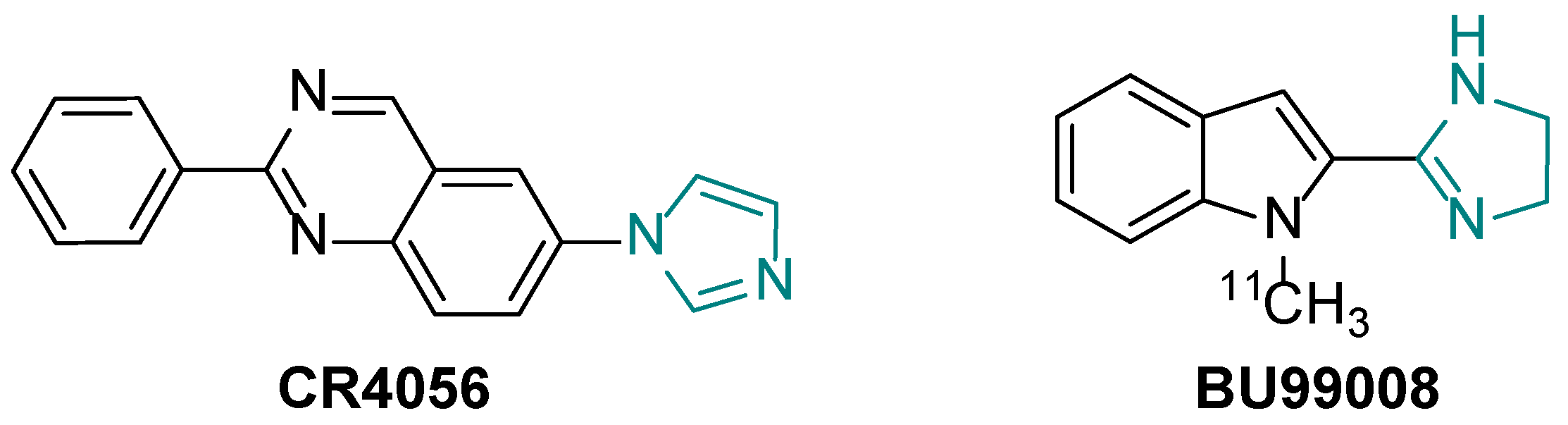
1. Introduction
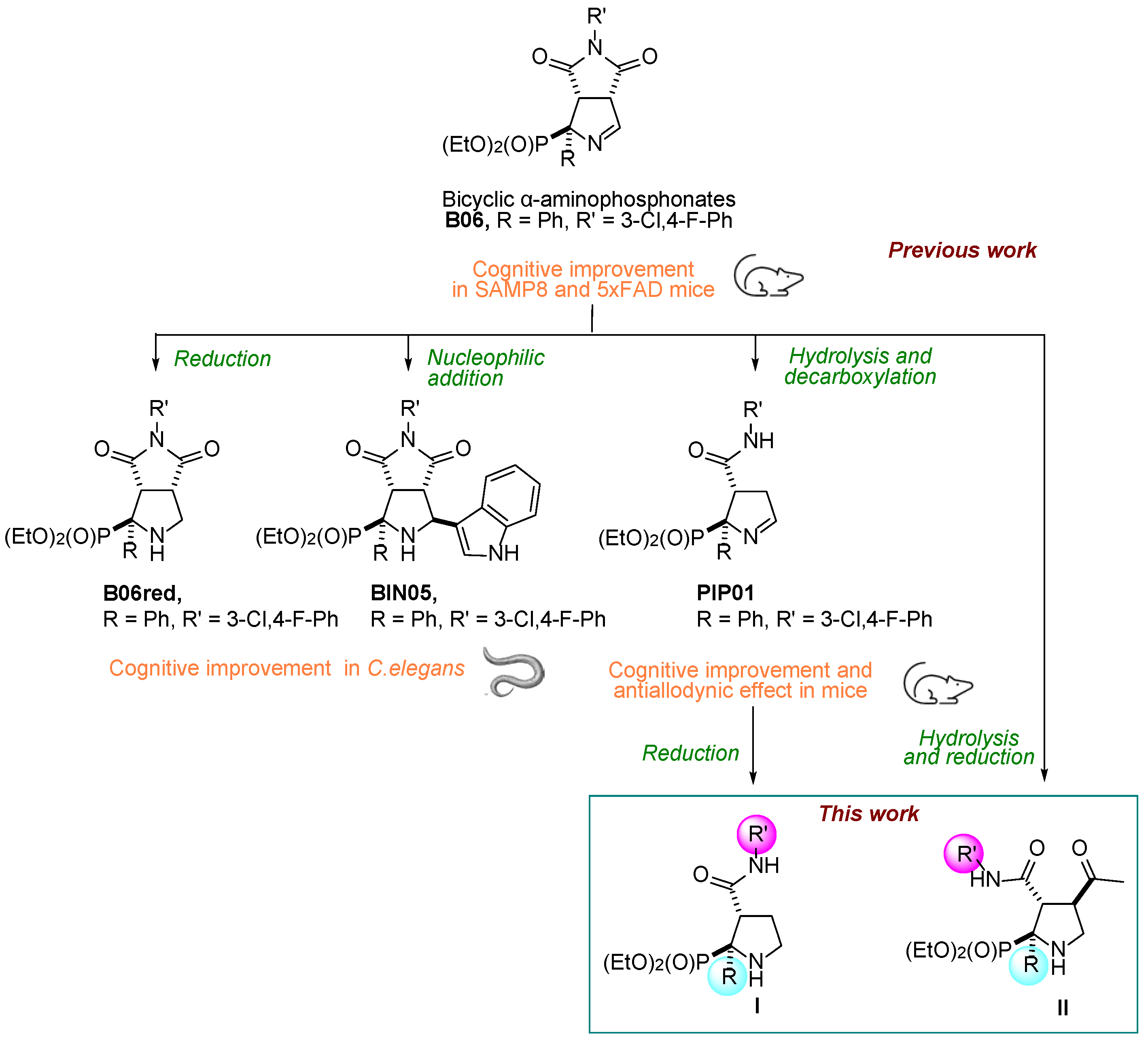
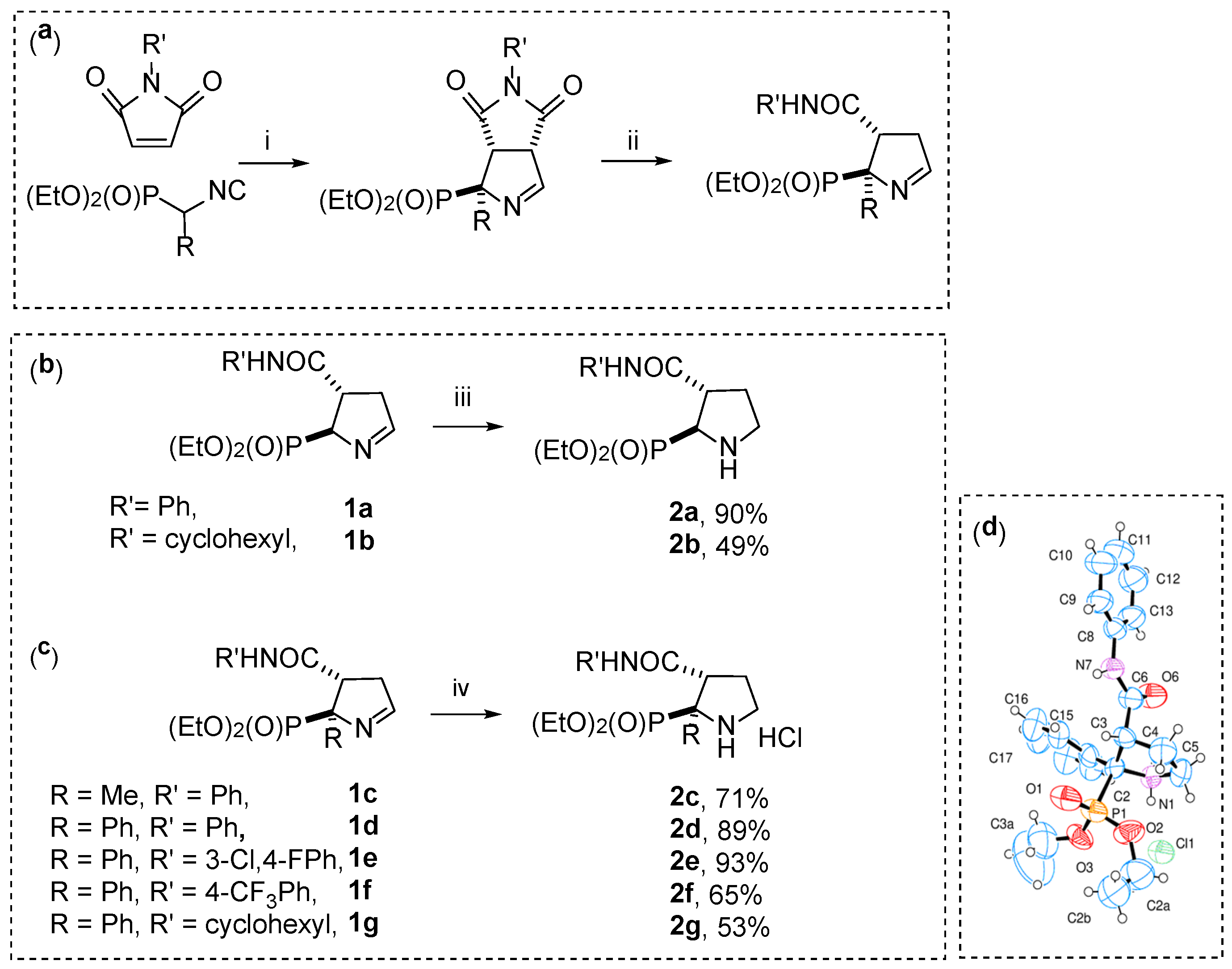
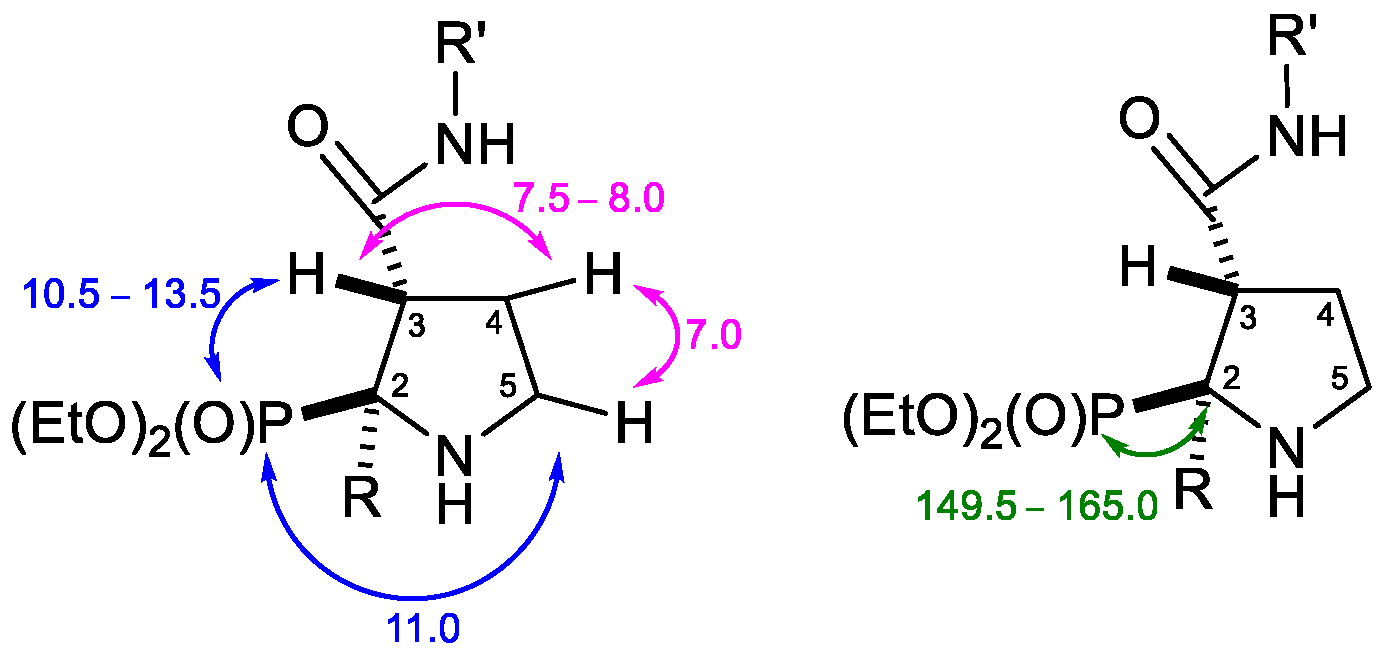
2. Results and Discussion
3. Materials and Methods
3.1. Chemistry
3.1.1. General Information
3.1.2. General Procedure for the Reduction of 1a and 1b
3.1.3. General Procedure for the Reduction of 1c, 1d, 1e, 1f and 1g
3.1.4. General Procedure for the Treatment with Resin and Hydrogenation of 3a, 3b, 3c, 3d, and 3e
3.1.5. Reductive Amination Reaction
3.2. X-Ray Crystallographic Analysis of PIPred02, CPP06
3.3. Binding Studies
3.3.1. Preparation of Cellular Membranes
3.3.2. Competition Binding Assays
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Abás, S.; Rodríguez-Arévalo, S.; Bagán, A.; Griñán-Ferré, C.; Vasilopoulou, F.; Brocos-Mosquera, I.; Muguruza, C.; Pérez, B.; Molins, E.; Luque, F.J.; et al. Bicyclic α-Iminophosphonates as High Affinity Imidazoline I2 Receptor Ligands for Alzheimer’s Disease. J. Med. Chem. 2020, 63, 3610–3633. [Google Scholar] [CrossRef] [PubMed]
- Li, J.X. Imidazoline I2 Receptors: An Update. Pharmacol. Ther. 2017, 178, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Bousquet, P.; Hudson, A.; García-Sevilla, J.A.; Li, J.X. Imidazoline Receptor System: The Past, the Present, and the Future. Pharmacol. Rev. 2020, 72, 50–79. [Google Scholar] [CrossRef] [PubMed]
- Dardonville, C.; Rozas, I. Imidazoline Binding Sites and Their Ligands: An Overview of the Different Chemical Structures. Med. Res. Rev. 2004, 24, 639–661. [Google Scholar] [CrossRef]
- Rovati, L.C.; Brambilla, N.; Blicharski, T.; Connell, J.; Vitalini, C.; Bonazzi, A.; Giacovelli, G.; Girolami, F.; D’Amato, M. Efficacy and Safety of the First-in-Class Imidazoline-2 Receptor Ligand CR4056 in Pain from Knee Osteoarthritis and Disease Phenotypes: A Randomized, Double-Blind, Placebo-Controlled Phase 2 Trial. Osteoarthr. Cartil. 2020, 28, 22–30. [Google Scholar] [CrossRef]
- Wilson, H.; Dervenoulas, G.; Pagano, G.; Tyacke, R.J.; Polychronis, S.; Myers, J.; Gunn, R.N.; Rabiner, E.A.; Nutt, D.; Politis, M. Imidazoline 2 Binding Sites Reflecting Astroglia Pathology in Parkinson’s Disease: An in Vivo 11C-BU99008 PET Study. Brain 2019, 142, 3116–3128. [Google Scholar] [CrossRef]
- Hernández-Hernández, E.; Ledesma-Corvi, S.; Yáñez-Gómez, F.; Garau, C.; Gálvez-Melero, L.; Bagán, A.; Escolano, C.; García-Fuster, M.J. Sex Differences in the Antidepressant-like Response and Molecular Events Induced by the Imidazoline-2 Receptor Agonist CR4056 in Rats. Pharmacol. Biochem. Behav. 2023, 223, 73527. [Google Scholar] [CrossRef]
- Vasilopoulou, F.; Griñán-Ferré, C.; Rodríguez-Arévalo, S.; Bagán, A.; Abás, S.; Escolano, C.; Pallàs, M. I2 Imidazoline Receptor Modulation Protects Aged SAMP8 Mice against Cognitive Decline by Suppressing the Calcineurin Pathway. Geroscience 2021, 43, 965–983. [Google Scholar] [CrossRef]
- Bagán, A.; Abás, S.; Palà-Pujadas, J.; Irisarri, A.; Griñán-Ferré, C.; Pallàs, M.; Muneta-Arrate, I.; Muguruza, C.; Callado, L.F.; Pérez, B.; et al. Exploring the Reactivity of Bicyclic α-Iminophosphonates to Access New Imidazoline I2 Receptor Ligands. Bioorg. Chem. 2024, 142, 106935. [Google Scholar] [CrossRef]
- Li, J.X.; Zhang, Y. Imidazoline I2 Receptors: Target for New Analgesics? Eur. J. Pharmacol. 2011, 658, 49–56. [Google Scholar] [CrossRef]
- Bagán, A.; López-Ruiz, A.; Abás, S.; Ruiz-Cantero, M.C.; Vasilopoulou, F.; Taboada-Jara, T.; Griñán-Ferré, C.; Pallàs, M.; Muguruza, C.; Diez-Alarcia, R.; et al. Discovery of (3-Phenylcarbamoyl-3,4-Dihydro-2H-Pyrrol-2-Yl)Phosphonates as Imidazoline I2 Receptor Ligands with Anti-Alzheimer and Analgesic Properties. J. Med. Chem. 2025, 68, 2551–2573. [Google Scholar] [CrossRef] [PubMed]
- Mayorquín-Torres, M.C.; Simoens, A.; Bonneure, E.; Stevens, C.V. Synthetic Methods for Azaheterocyclic Phosphonates and Their Biological Activity: An Update 2004–2024. Chem. Rev. 2024, 124, 7907–7975. [Google Scholar] [CrossRef] [PubMed]
- Vitaku, E.; Smith, D.T.; Njardarson, J.T. Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals. J. Med. Chem. 2014, 57, 10257–10274. [Google Scholar] [CrossRef] [PubMed]
- Smolobochkin, A.; Gazizov, A.; Appazov, N.; Sinyashin, O.; Burilov, A. Progress in the Stereoselective Synthesis Methods of Pyrrolidine-Containing Drugs and Their Precursors. Int. J. Mol. Sci. 2024, 25, 11158. [Google Scholar] [CrossRef]
- Głowacka, I.E.; Hartwich, A.; Rozpara, I.; Piotrowska, D.G. Synthesis of Functionalized Diethyl (Pyrrolidin-2-Yl)Phosphonate and Diethyl (5-Oxopyrrolidin-2-Yl)Phosphonate. Molecules 2021, 26, 3160. [Google Scholar] [CrossRef]
- Maestro, A.; de Marigorta, E.M.; Palacios, F.; Vicario, J. α-Iminophosphonates: Useful Intermediates for Enantioselective Synthesis of α-Aminophosphonates. Asian J. Org. Chem. 2020, 9, 538–548. [Google Scholar] [CrossRef]
- Ordóñez, M.; Rojas-Cabrera, H.; Cativiela, C. An Overview of Stereoselective Synthesis of α-Aminophosphonic Acids and Derivatives. Tetrahedron 2009, 65, 17–49. [Google Scholar] [CrossRef]
- Varga, P.R.; Keglevich, G. The Last Decade of Optically Active α-Aminophosphonates. Molecules 2023, 28, 6150. [Google Scholar] [CrossRef]
- Elliott, T.S.; Slowey, A.; Ye, Y.; Conway, S.J. The Use of Phosphate Bioisosteres in Medicinal Chemistry and Chemical Biology. Med. Chem. Comm. 2012, 3, 735–751. [Google Scholar] [CrossRef]
- Gluza, K.; Kafarski, P. Transition State Analogues of Enzymatic Reaction as Potential Drugs. In Drug Discovery; InTech: London, UK, 2013; pp. 325–372. [Google Scholar] [CrossRef]
- Orsini, F.; Sello, G.; Sisti, M. Aminophosphonic Acids and Derivatives. Synthesis and Biological Applications. Curr. Med. Chem. 2010, 17, 264–289. [Google Scholar] [CrossRef]
- Amira, A.; Aouf, Z.; K’tir, H.; Chemam, Y.; Ghodbane, R.; Zerrouki, R.; Aouf, N.E. Recent Advances in the Synthesis of α-Aminophosphonates: A Review. ChemistrySelect 2021, 6, 6137–6149. [Google Scholar] [CrossRef]
- Mucha, A.; Kafarski, P.; Berlicki, Ł. Remarkable Potential of the α-Aminophosphonate/Phosphinate Structural Motif in Medicinal Chemistry. J. Med. Chem. 2011, 54, 5955–5980. [Google Scholar] [CrossRef]
- Moonen, K.; Laureyn, I.; Stevens, C.V. Synthetic Methods for Azaheterocyclic Phosphonates and Their Biological Activity. Chem. Rev. 2004, 104, 6177–6215. [Google Scholar] [CrossRef] [PubMed]
- Gazizov, A.S.; Smolobochkin, A.V.; Turmanov, R.A.; Pudovik, M.A.; Burilov, A.R.; Sinyashin, O.G. Synthesis of Phosphaproline Derivatives: A Short Overview. Synthesis 2019, 51, 3397–3409. [Google Scholar] [CrossRef]
- Smolobochkin, A.V.; Gazizov, A.S.; Turmanov, R.A.; Abdullaeva, D.S.; Burilov, A.R.; Pudovik, M.A. N-Phosphorylated Pyrrolidines: An Overview of Synthetic Approaches. Synthesis 2020, 52, 2162–2170. [Google Scholar] [CrossRef]
- Ruggiero, D.A.; Regunathan, S.; Wang, H.; Milner, T.A.; Reis, D.J. Immunocytochemical Localization of an Imidazoline Receptor Protein in the Central Nervous System. Brain Res. 1998, 780, 270–293. [Google Scholar] [CrossRef]
- When the Crude of the Reduction Was Purified Without Treatment with EtOH.HCl (1.25 M) the Product n That Resulted of the Elimination of the Phosphonate Was Isolated. See Supporting Information for Details and NMR Spectra. Available online: https://www.intechopen.com/chapters/41534 (accessed on 29 April 2025).
- Ordóñez, M.; Sayago, F.J.; Cativiela, C. Synthesis of Quaternary α-Aminophosphonic Acids. Tetrahedron 2012, 68, 6369–6412. [Google Scholar] [CrossRef]
- Alix, A.; Lalli, C.; Retailleau, P.; Masson, G. Highly Enantioselective Electrophilic α-Bromination of Enecarbamates: Chiral Phosphoric Acid and Calcium Phosphate Salt Catalysts. J. Am. Chem. Soc. 2012, 134, 10389–10392. [Google Scholar] [CrossRef]
- Bousquet, P.; Feldman, J.; Schwartz, J. Central Cardiovascular Effects of Alpha Adrenergic Drugs: Differences between Catecholamines and Imidazolines. J. Pharmacol. Exp. Ther. 1984, 230, 232–236. [Google Scholar] [CrossRef]
- Giordani, A.; Mandelli, S.; Verpilio, I.; Zanzola, S.; Tarchino, F.; Castelli, G.; Piepoli, T.; Mazzari, S.; Makovec, F.; Rovati, L.C. 6-1H-Imidazo-Quinazoline and Quinolines Derivatives, New Potent Analgesics and Anti-Inflammatory Agents. U.S. Patent US8193353B2, 5 June 2012. [Google Scholar]
- Tyacke, R.J.; Fisher, A.; Robinson, E.S.J.; Grundt, P.; Turner, E.M.; Husbands, S.M.; Hudson, A.L.; Parker, C.A.; Nutt, D.J. Evaluation and Initial in Vitro and Ex Vivo Characterization of the Potential Positron Emission Tomography Ligand, BU99008 (2-(4,5-Dihydro-1H-Imidazol-2-Yl)-1- Methyl-1H-Indole), for the Imidazoline 2 Binding Site. Synapse 2012, 66, 542–551. [Google Scholar] [CrossRef]
- Blessing, R.H. An Empirical Correction for Absorption Anisotropy. Acta Crystallogr. A 1995, 51, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. SHELXT-Integrated Space-Group and Crystal-Structure Determination. Acta Crystallogr. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, L.J. WinGX and ORTEP for Windows: An Update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Di, L.; Kerns, E.H.; Fan, K.; McConnell, O.J.; CarTer, G.T. High throughput artificial membrane permeability assay for blood-brain barrier. Eur. J. Med.Chem. 2003, 38, 223–232. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
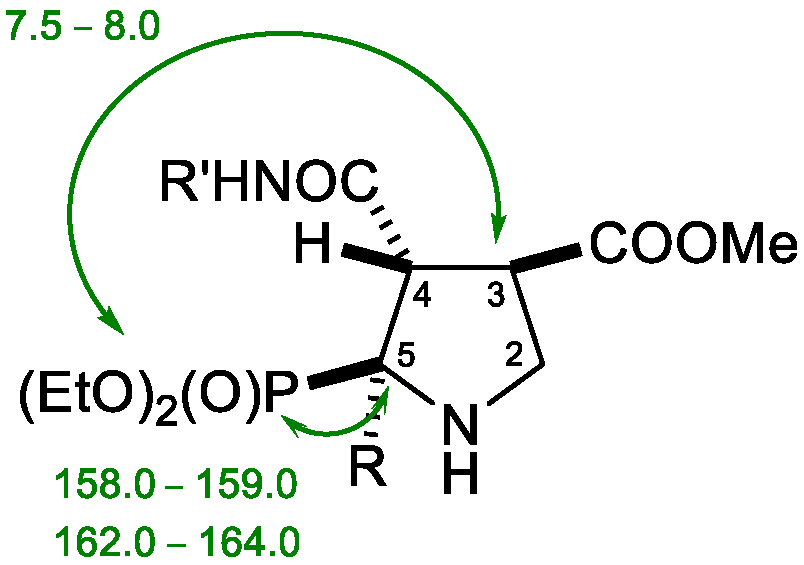
| 1H-NMR Data | 13C-NMR Data | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Comp. | H2 | H3 | H4 | H4 | H5 | H5 | C2 | C3 | C4 | C5 |
| 2a | 3.63 t 1 7.5 | 3.26 ddd 2 24.0, 16.0, 8.0 | 2.06 m 3 | 2.19–2.29 c.s. 4 | 2.99–3.12 c.s. | 57.8 d 5 165.0 | 47.5 d 2.0 | 31.2 d 7.0 | 48.4 d 9.0 | |
| 2b | 3.46 t 7.5 | 3.75 m | 1.97 m | 2.13–2.22 c.s. | 2.91–3.07 c.s. | 58.5 d 165.0 | 48.2 | 30.9 d 6.5 | 48.4 d 9.5 | |
| 2c | - | 3.58 ddd 13.6, 7.6, 6.0 | 2.43–2.53 c.s. | 3.48 ddd 11.2, 8.4, 6.8 | 3.69 ddd 11.2, 8.4, 6.8 | 64.4 d 159.0 | 48.8 | 27.8 d 4.0 | 45.2 d 3.0 | |
| 2d | - | 4.32 dd 6 10.8, 7.6 | 2.42 m | 2.83 m | 3.81–3.89 c.s. | 72.8 d 149.5 | 49.9 | 28.3 | 45.8 | |
| 2e | - | 4.30 dd 10.4, 7.6 | 2.43 m | 2.83 m | 3.79–3.91 c.s. | 72.7 d 150.0 | 50.0 | 28.2 | 45.8 | |
| 2f | - | 4.28 dd 10.5, 7.5 | 2.49 m | 2.84 m | 3.74–3.98 c.s. | 74.2 d 149.5 | 51.6 | 29.7 | 47.2 | |
| 2g | - | 4.06–4.17 c.s. | 2.25 m | 2.73 m | 3.77–3.88 c.s. | 72.5 d 149.5 | 49.1 | 28.1 | 45.9 | |
| 1H-NMR Data | 13C-NMR Data | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Comp. | H2 | H2 | H3 | H4 | H5 | C2 | C3 | C4 | C5 |
| 4a | 3.22–3.33 c.s. 1 | 2.53–3.67 c.s. | 2.53–3.67 c.s. | 2.53–3.67 c.s. | 52.1 d 2 11.5 | 48.2 d 7.5 | 51.2 d 2.5 | 58.9 d 163.0 | |
| 4b | 3.25 m 3 | 3.31 m | 3.48–3.65 c.s. | 3.48–3.65 c.s. | 3.48–3.65 c.s. | 52.1 d 12.0 | 47.6 d 8.0 | 51.1 d 2.2 | 58.8 d 162.0 |
| 4c | 3.19–3.28 c.s. | 3.74 m | 3.34 ddd 4 19.0, 8.0, 6.8 | 3.45 d 8.8 | 51.9 d 10.5 | 48.5 d 7.0 | 50.0 d 2.0 | 59.0 d 164.0 | |
| 4d | 3.31 t 5 9.5 | 3.62–3.81 c.s. | 3.62–3.81 c.s. | 3.95–4.05 c.s. | - | 49.2 d 7.0 | 46.2 d 7.5 | 57.2 d 4.0 | 68.7 d 158.0 |
| 4e | 3.29 m | 3.64–3.70 c.s. | 3.64–3.70 c.s. | 3.94–4.07 c.s. | - | 49.3 d 7.0 | 45.7 d 7.5 | 57.2 d 3.5 | 68.4 d 159.0 |
| 5 | 2.72 dd 6 10.4, 7.2 | 3.27 dd 10.8, 5.6 | 3.68 m | 3.81 m | 3.23 dd 7.6, 2.5 | 56.6 d 13.5 | 44.2 d 7.5 | 49.9 | 63.9 d 169.5 |
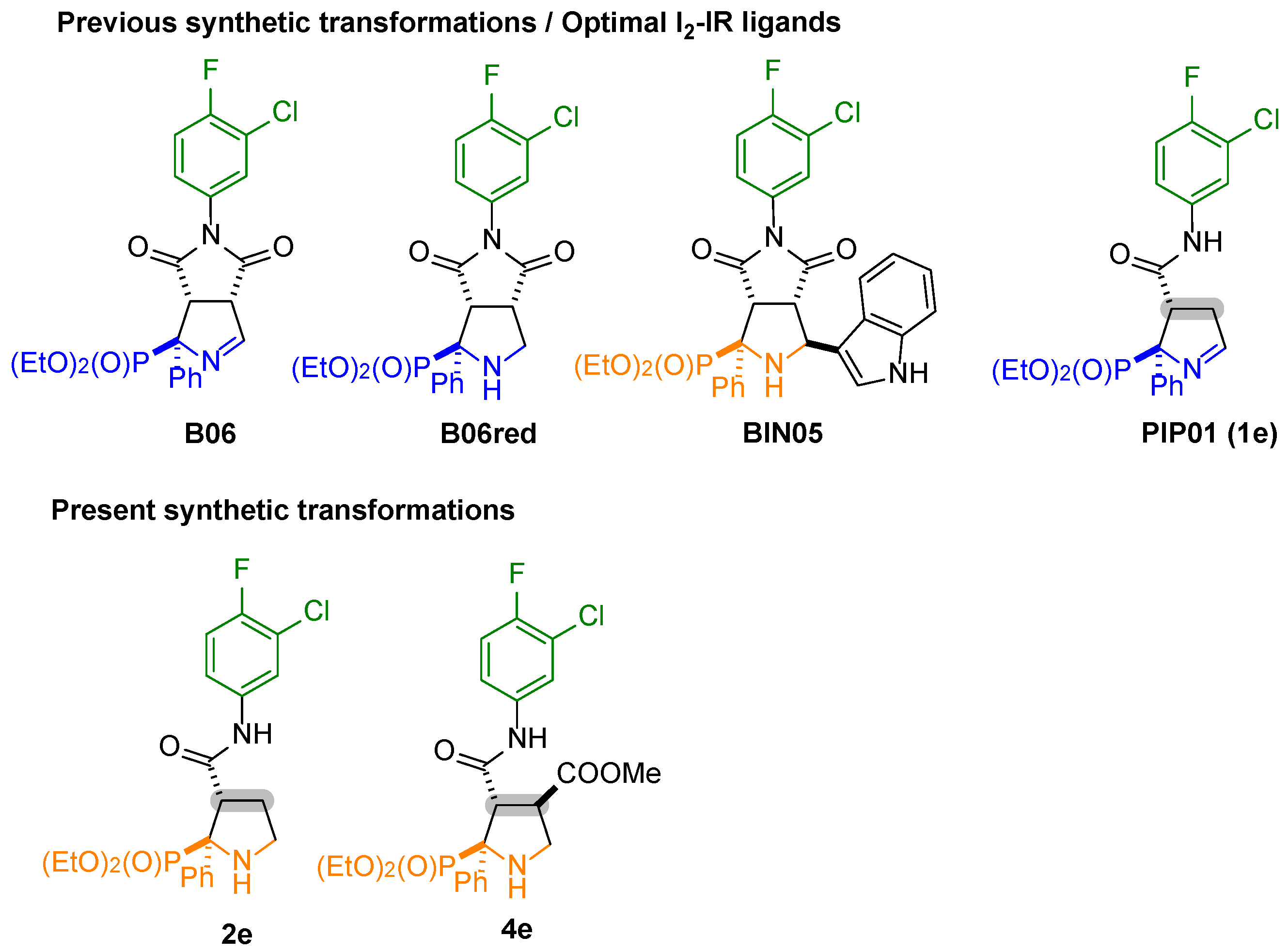
| Entry | Compd. | [3H]-2-BFI, I2 One Site b I2 Two Sites H/L; (pKiH/pKiL) High Affinity Site % | [3H]RX821002, α2 | Selectivity I2/α2 a |
|---|---|---|---|---|
| 1 | CR4056 c | 7.72 ± 0.31/5.45 ± 0.15 29 ± 6 | 2.65 ± 1.24 | 117,490 |
| 2 | BU99008 c | 6.89 ± 0.21/3.82 ± 0.30 51 ± 6 | 4.37 ± 0.17 | 331 |
| 3 | B06 c | 8.56 ± 0.32 8.61 ± 0.28/4.29 ± 0.20 37 ± 4 | 6.27 ± 0.56 | 195 |
| 4 | B06red d | 6.88 ± 0.29 9.81 ± 0.21/< 3 50 ± 4 | 6.66 ± 0.49 | 1.7 |
| 5 | BIN05 d | 3.89 ± 0.19 8.18 ± 0.43/3.56 ± 0.22 21 ± 3 | 5.51 ± 0.24 | - |
| 6 | PIP01 c (1e) | 9.98 ± 0.74 | 9.43 ± 0.22 | 3 |
| 7 | 2e | 4.16 ± 0.15 | 4.07 ± 0.20 | 1.2 |
| 8 | 4e | 7.99 ± 0.39 | 3.22 ± 0.15 | 58,884 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bagán, A.; López-Ruiz, A.; Abás, S.; Molins, E.; Pérez, B.; Muneta-Arrate, I.; Callado, L.F.; Escolano, C. Synthesis of Diversely Substituted Diethyl (Pyrrolidin-2-Yl)Phosphonates. Molecules 2025, 30, 2078. https://doi.org/10.3390/molecules30092078
Bagán A, López-Ruiz A, Abás S, Molins E, Pérez B, Muneta-Arrate I, Callado LF, Escolano C. Synthesis of Diversely Substituted Diethyl (Pyrrolidin-2-Yl)Phosphonates. Molecules. 2025; 30(9):2078. https://doi.org/10.3390/molecules30092078
Chicago/Turabian StyleBagán, Andrea, Alba López-Ruiz, Sònia Abás, Elies Molins, Belén Pérez, Itziar Muneta-Arrate, Luis F. Callado, and Carmen Escolano. 2025. "Synthesis of Diversely Substituted Diethyl (Pyrrolidin-2-Yl)Phosphonates" Molecules 30, no. 9: 2078. https://doi.org/10.3390/molecules30092078
APA StyleBagán, A., López-Ruiz, A., Abás, S., Molins, E., Pérez, B., Muneta-Arrate, I., Callado, L. F., & Escolano, C. (2025). Synthesis of Diversely Substituted Diethyl (Pyrrolidin-2-Yl)Phosphonates. Molecules, 30(9), 2078. https://doi.org/10.3390/molecules30092078