
Rationally Designed Pentapeptide Analogs of Aβ19–23 Fragment as Potent Inhibitors of Aβ42 Aggregation

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Result and Discussion
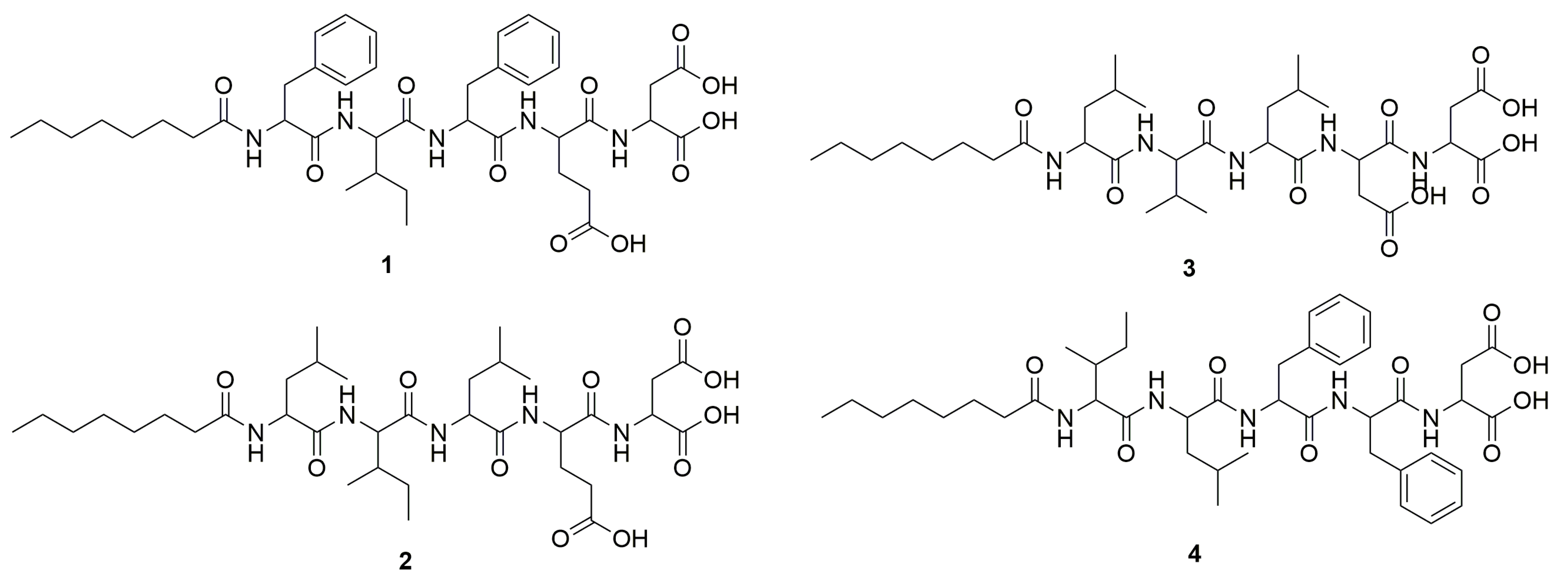
2.1. Peptide Design and Synthesis
2.2. The Inhibition of Aβ42 Aggregation by Pentapeptides Using Thioflavin T (ThT) Fluorescence-Based Kinetic Assay
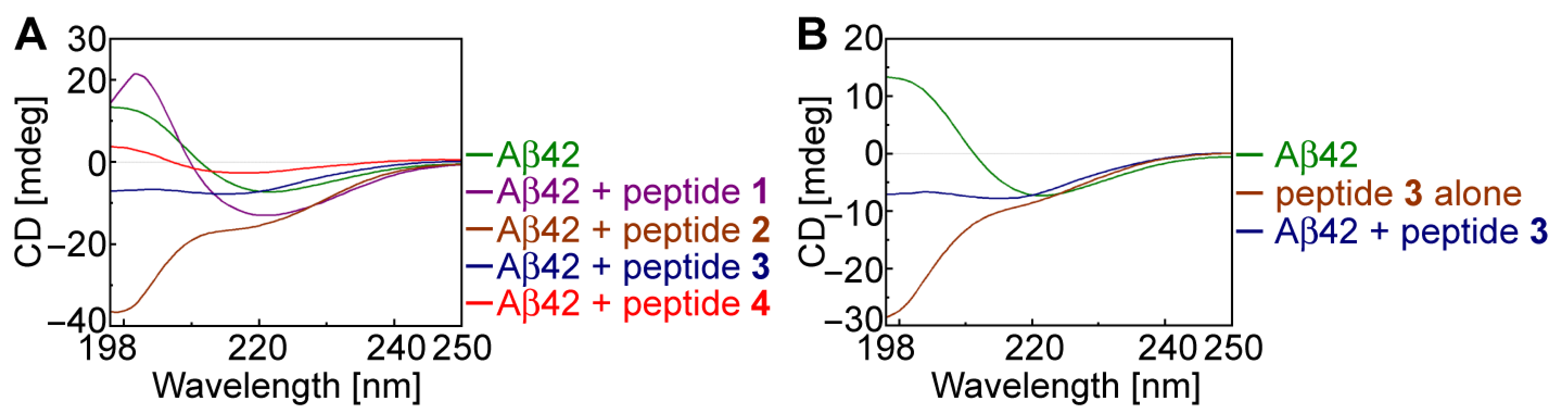
2.3. Secondary Structural Changes in Aβ42 Using CD Spectroscopy
2.4. Investigation of Aβ42 Aggregation and Its Inhibition Using Transmission Electron Microscopy (TEM) Analysis
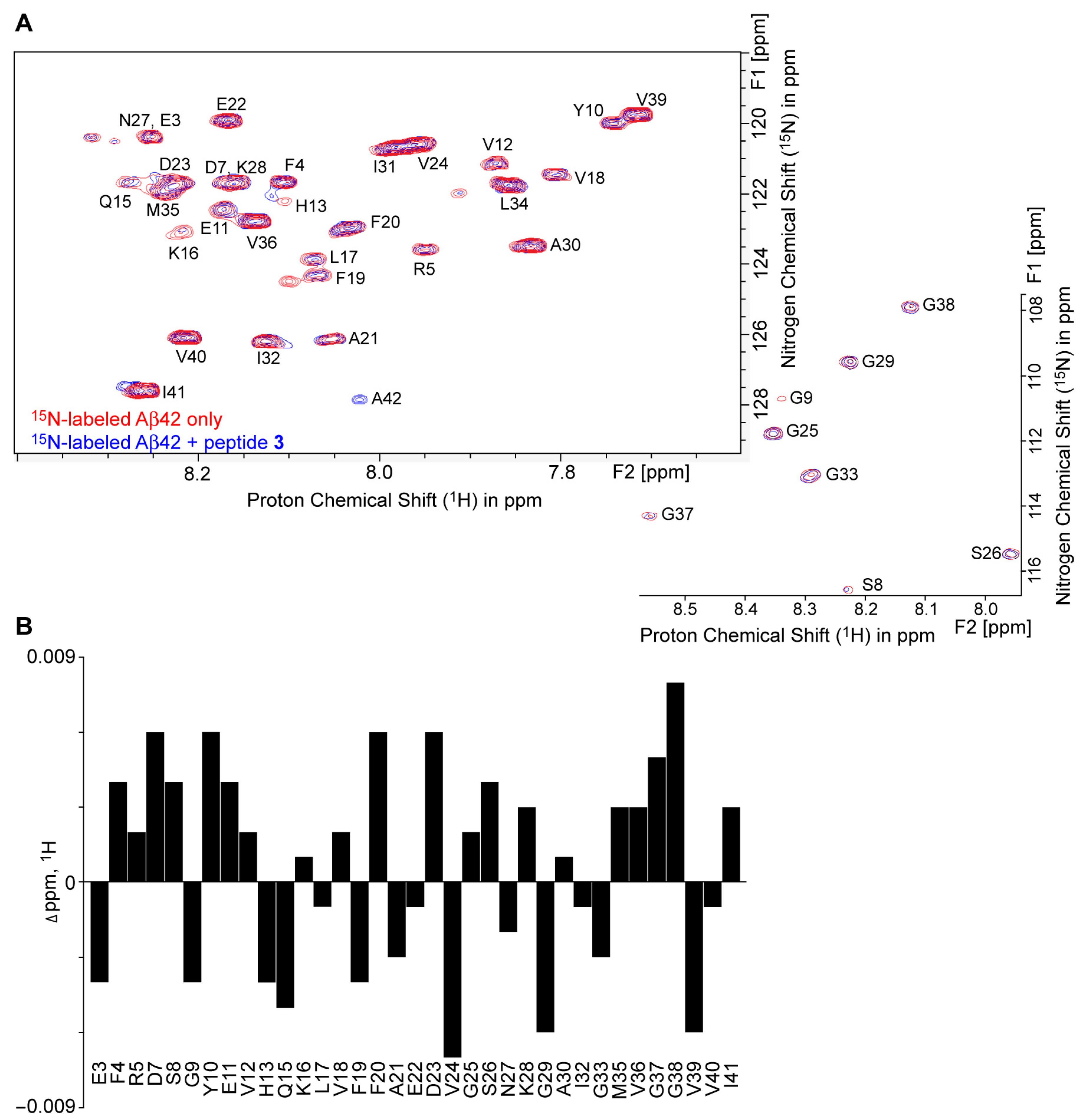
2.5. 1H–15N HSQC NMR Revealed Conformational Changes of Aβ42 by Peptide 3
3. Materials and Methods
3.1. Materials
3.2. Solid-Phase Synthesis of Peptides (SPPS)
3.2.1. General Procedure for SPPS of Peptides 1–4 (Example of SPPS of Peptide 3)
3.2.2. Procedures to Remove Trifluoracetic Acid Counter-Ions from Fatty Acid–Peptide Conjugates by Counter-Anion Exchange
3.2.3. Determination of the Molecular Structures and Quantities of Compounds
3.3. Preparation of Amyloid β42
3.4. Thioflavin T (ThT) Fluorescence-Based Kinetic Assay
3.5. Circular Dichroism Spectroscopy
3.6. 1H–15N HSQC NMR Spectroscopy
3.7. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alzheimer’s Association. 2018 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2018, 14, 367–429. [Google Scholar] [CrossRef]
- World Health Organization. Newsroom-Facts Sheet-Deatial-Dementia. Available online: https://www.who.int/news-room/fact-sheets/detail/dementia (accessed on 15 March 2023).
- DiBello, J.R.; Lu, Y.; Swartz, J.; Bortnichak, E.A.; Liaw, K.L.; Zhong, W.; Liu, X. Patterns of use of symptomatic treatments for Alzheimer’s disease dementia (AD). BMC Neurol. 2023, 23, 400. [Google Scholar] [CrossRef] [PubMed]
- Kasim, J.K.; Kavianinia, I.; Harris, P.W.R.; Brimble, M.A. Three Decades of Amyloid Beta Synthesis: Challenges and Advances. Front. Chem. 2019, 7, 472. [Google Scholar] [CrossRef]
- Yan, R.; Vassar, R. Targeting the beta secretase BACE1 for Alzheimer’s disease therapy. Lancet Neurol. 2014, 13, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.S.; Bowman, G.R.; Beauchamp, K.A.; Pande, V.S. Investigating how peptide length and a pathogenic mutation modify the structural ensemble of amyloid beta monomer. Biophys. J. 2012, 102, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Korshavn, K.J.; Nam, Y.; Kang, J.; Paul, T.J.; Kerr, R.A.; Youn, I.S.; Ozbil, M.; Kim, K.S.; Ruotolo, B.T.; et al. Structural and Mechanistic Insights into Development of Chemical Tools to Control Individual and Inter-Related Pathological Features in Alzheimer’s Disease. Chemistry 2017, 23, 2706–2715. [Google Scholar] [CrossRef]
- Selkoe, D.J. Alzheimer’s disease: Genes, proteins, and therapy. Physiol. Rev. 2001, 81, 741–766. [Google Scholar] [CrossRef]
- Roher, A.E.; Lowenson, J.D.; Clarke, S.; Wolkow, C.; Wang, R.; Cotter, R.J.; Reardon, I.M.; Zurcher-Neely, H.A.; Heinrikson, R.L.; Ball, M.J.; et al. Structural alterations in the peptide backbone of beta-amyloid core protein may account for its deposition and stability in Alzheimer’s disease. J. Biol. Chem. 1993, 268, 3072–3083. [Google Scholar] [CrossRef]
- Danielsson, J.; Jarvet, J.; Damberg, P.; Graslund, A. The Alzheimer beta-peptide shows temperature-dependent transitions between left-handed 3-helix, beta-strand and random coil secondary structures. FEBS J. 2005, 272, 3938–3949. [Google Scholar] [CrossRef]
- Abelein, A.; Abrahams, J.P.; Danielsson, J.; Graslund, A.; Jarvet, J.; Luo, J.; Tiiman, A.; Warmlander, S.K. The hairpin conformation of the amyloid beta peptide is an important structural motif along the aggregation pathway. J. Biol. Inorg. Chem. 2014, 19, 623–634. [Google Scholar] [CrossRef]
- Roche, J.; Shen, Y.; Lee, J.H.; Ying, J.; Bax, A. Monomeric Abeta(1-40) and Abeta(1-42) Peptides in Solution Adopt Very Similar Ramachandran Map Distributions That Closely Resemble Random Coil. Biochemistry 2016, 55, 762–775. [Google Scholar] [CrossRef] [PubMed]
- Giulian, D.; Haverkamp, L.J.; Yu, J.; Karshin, W.; Tom, D.; Li, J.; Kazanskaia, A.; Kirkpatrick, J.; Roher, A.E. The HHQK domain of beta-amyloid provides a structural basis for the immunopathology of Alzheimer’s disease. J. Biol. Chem. 1998, 273, 29719–29726. [Google Scholar] [CrossRef]
- Tiiman, A.; Krishtal, J.; Palumaa, P.; Tõugu, V. In vitro fibrillization of Alzheimer’s amyloid-b peptide (1-42). AIP Adv. 2015, 5, 092401. [Google Scholar] [CrossRef]
- Hilbich, C.; Kisters-Woike, B.; Reed, J.; Masters, C.L.; Beyreuther, K. Substitutions of hydrophobic amino acids reduce the amyloidogenicity of Alzheimer’s disease beta A4 peptides. J. Mol. Biol. 1992, 228, 460–473. [Google Scholar] [CrossRef] [PubMed]
- Serpell, L.C. Alzheimer’s amyloid fibrils: Structure and assembly. Biochim. Biophys. Acta 2000, 1502, 16–30. [Google Scholar] [CrossRef]
- Gazit, E. A possible role for pi-stacking in the self-assembly of amyloid fibrils. FASEB J. 2002, 16, 77–83. [Google Scholar] [CrossRef]
- de Groot, N.S.; Aviles, F.X.; Vendrell, J.; Ventura, S. Mutagenesis of the central hydrophobic cluster in Abeta42 Alzheimer’s peptide. Side-chain properties correlate with aggregation propensities. FEBS J. 2006, 273, 658–668. [Google Scholar] [CrossRef]
- Citron, M. Alzheimer’s disease: Strategies for disease modification. Nat. Rev. Drug Discov. 2010, 9, 387–398. [Google Scholar] [CrossRef]
- Warmlander, S.; Tiiman, A.; Abelein, A.; Luo, J.; Jarvet, J.; Soderberg, K.L.; Danielsson, J.; Graslund, A. Biophysical studies of the amyloid beta-peptide: Interactions with metal ions and small molecules. Chembiochem 2013, 14, 1692–1704. [Google Scholar] [CrossRef]
- Kumar, S.; Henning-Knechtel, A.; Chehade, I.; Magzoub, M.; Hamilton, A.D. Foldamer-Mediated Structural Rearrangement Attenuates Abeta Oligomerization and Cytotoxicity. J. Am. Chem. Soc. 2017, 139, 17098–17108. [Google Scholar] [CrossRef]
- Kumar, S.; Henning-Knechtel, A.; Magzoub, M.; Hamilton, A.D. Peptidomimetic-Based Multidomain Targeting Offers Critical Evaluation of Abeta Structure and Toxic Function. J. Am. Chem. Soc. 2018, 140, 6562–6574. [Google Scholar] [CrossRef] [PubMed]
- Doody, R.S.; Raman, R.; Farlow, M.; Iwatsubo, T.; Vellas, B.; Joffe, S.; Kieburtz, K.; He, F.; Sun, X.; Thomas, R.G.; et al. A phase 3 trial of semagacestat for treatment of Alzheimer’s disease. N. Engl. J. Med. 2013, 369, 341–350. [Google Scholar] [CrossRef]
- Goyal, D.; Shuaib, S.; Mann, S.; Goyal, B. Rationally Designed Peptides and Peptidomimetics as Inhibitors of Amyloid-beta (Abeta) Aggregation: Potential Therapeutics of Alzheimer’s Disease. ACS Comb. Sci. 2017, 19, 55–80. [Google Scholar] [CrossRef]
- Deike, S.; Rothemund, S.; Voigt, B.; Samantray, S.; Strodel, B.; Binder, W.H. beta-Turn mimetic synthetic peptides as amyloid-beta aggregation inhibitors. Bioorg. Chem. 2020, 101, 104012. [Google Scholar] [CrossRef] [PubMed]
- Kapadia, A.; Sharma, K.K.; Maurya, I.K.; Singh, V.; Khullar, M.; Jain, R. Structural and mechanistic insights into the inhibition of amyloid-beta aggregation by Abeta(39-42) fragment derived synthetic peptides. Eur. J. Med. Chem. 2021, 212, 113126. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Srivastav, S.; Fatima, M.; Giri, R.S.; Mandal, B.; Mondal, A.C. A Synthetic Pro-Drug Peptide Reverses Amyloid-beta-Induced Toxicity in the Rat Model of Alzheimer’s Disease. J. Alzheimer’s Dis. 2019, 69, 499–512. [Google Scholar] [CrossRef]
- Shea, D.; Hsu, C.C.; Bi, T.M.; Paranjapye, N.; Childers, M.C.; Cochran, J.; Tomberlin, C.P.; Wang, L.; Paris, D.; Zonderman, J.; et al. alpha-Sheet secondary structure in amyloid beta-peptide drives aggregation and toxicity in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2019, 116, 8895–8900. [Google Scholar] [CrossRef] [PubMed]
- Baine, M.; Georgie, D.S.; Shiferraw, E.Z.; Nguyen, T.P.; Nogaj, L.A.; Moffet, D.A. Inhibition of Abeta42 aggregation using peptides selected from combinatorial libraries. J. Pept. Sci. 2009, 15, 499–503. [Google Scholar] [CrossRef]
- Rajasekhar, K.; Madhu, C.; Govindaraju, T. Natural Tripeptide-Based Inhibitor of Multifaceted Amyloid beta Toxicity. ACS Chem. Neurosci. 2016, 7, 1300–1310. [Google Scholar] [CrossRef]
- Henning-Knechtel, A.; Kuma, S.; Wallin, C.; Król, S.; Wärmländer, S.K.T.S.; Jarvet, J.; Esposito, G.; Kirmizialtin, S.; Gräslund, A.; Hamilton, A.D.; et al. Designed Cell-Penetrating Peptide Inhibitors of Amyloid-beta Aggregation and Cytotoxicity. Cell Rep. Phys. Sci. 2020, 1, 100014. [Google Scholar] [CrossRef]
- Rajasekhar, K.; Suresh, S.N.; Manjithaya, R.; Govindaraju, T. Rationally designed peptidomimetic modulators of abeta toxicity in Alzheimer’s disease. Sci. Rep. 2015, 5, 8139. [Google Scholar] [CrossRef] [PubMed]
- Konar, M.; Ghosh, D.; Samanta, S.; Govindaraju, T. Combating amyloid-induced cellular toxicity and stiffness by designer peptidomimetics. RSC Chem. Biol. 2022, 3, 220–226. [Google Scholar] [CrossRef] [PubMed]
- Bollati, M.; Peqini, K.; Barone, L.; Natale, C.; Beeg, M.; Gobbi, M.; Diomede, L.; Trucchi, M.; de Rosa, M.; Pellegrino, S. Rational Design of a Peptidomimetic Inhibitor of Gelsolin Amyloid Aggregation. Int. J. Mol. Sci. 2022, 23, 13973. [Google Scholar] [CrossRef]
- Ramesh, M.; Govindaraju, T. Multipronged diagnostic and therapeutic strategies for Alzheimer’s disease. Chem. Sci. 2022, 13, 13657–13689. [Google Scholar] [CrossRef] [PubMed]
- De Lorenzi, E.; Chiari, M.; Colombo, R.; Cretich, M.; Sola, L.; Vanna, R.; Gagni, P.; Bisceglia, F.; Morasso, C.; Lin, J.S.; et al. Evidence that the Human Innate Immune Peptide LL-37 may be a Binding Partner of Amyloid-beta and Inhibitor of Fibril Assembly. J. Alzheimer’s Dis. 2017, 59, 1213–1226. [Google Scholar] [CrossRef]
- Iqbal, U.H.; Zeng, E.; Pasinetti, G.M. The Use of Antimicrobial and Antiviral Drugs in Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 4920. [Google Scholar] [CrossRef]
- Armiento, V.; Hille, K.; Naltsas, D.; Lin, J.S.; Barron, A.E.; Kapurniotu, A. The Human Host-Defense Peptide Cathelicidin LL-37 is a Nanomolar Inhibitor of Amyloid Self-Assembly of Islet Amyloid Polypeptide (IAPP). Angew. Chem. Int. Ed. Engl. 2020, 59, 12837–12841. [Google Scholar] [CrossRef]
- Bruno, F.; Malvaso, A.; Canterini, S.; Bruni, A.C. Antimicrobial Peptides (AMPs) in the Pathogenesis of Alzheimer’s Disease: Implications for Diagnosis and Treatment. Antibiotics 2022, 11, 726. [Google Scholar] [CrossRef]
- Cheng, P.N.; Liu, C.; Zhao, M.; Eisenberg, D.; Nowick, J.S. Amyloid beta-sheet mimics that antagonize protein aggregation and reduce amyloid toxicity. Nat. Chem. 2012, 4, 927–933. [Google Scholar] [CrossRef]
- Seither, K.M.; McMahon, H.A.; Singh, N.; Wang, H.; Cushman-Nick, M.; Montalvo, G.L.; DeGrado, W.F.; Shorter, J. Specific aromatic foldamers potently inhibit spontaneous and seeded Abeta42 and Abeta43 fibril assembly. Biochem. J. 2014, 464, 85–98. [Google Scholar] [CrossRef]
- Kumar, S.; Birol, M.; Miranker, A.D. Foldamer scaffolds suggest distinct structures are associated with alternative gains-of-function in a preamyloid toxin. Chem. Commun. 2016, 52, 6391–6394. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Song, Y.; Cheng, P.N.; Moore, J.S. Molecular Design for Dual Modulation Effect of Amyloid Protein Aggregation. J. Am. Chem. Soc. 2015, 137, 8062–8068. [Google Scholar] [CrossRef]
- Tjernberg, L.O.; Naslund, J.; Lindqvist, F.; Johansson, J.; Karlstrom, A.R.; Thyberg, J.; Terenius, L.; Nordstedt, C. Arrest of beta-amyloid fibril formation by a pentapeptide ligand. J. Biol. Chem. 1996, 271, 8545–8548. [Google Scholar] [CrossRef] [PubMed]
- Tjernberg, L.O.; Lilliehook, C.; Callaway, D.J.; Naslund, J.; Hahne, S.; Thyberg, J.; Terenius, L.; Nordstedt, C. Controlling amyloid beta-peptide fibril formation with protease-stable ligands. J. Biol. Chem. 1997, 272, 12601–12605. [Google Scholar] [CrossRef] [PubMed]
- Horsley, J.R.; Jovcevski, B.; Wegener, K.L.; Yu, J.; Pukala, T.L.; Abell, A.D. Rationally designed peptide-based inhibitor of Abeta42 fibril formation and toxicity: A potential therapeutic strategy for Alzheimer’s disease. Biochem. J. 2020, 477, 2039–2054. [Google Scholar] [CrossRef]
- Soto, C.; Kindy, M.S.; Baumann, M.; Frangione, B. Inhibition of Alzheimer’s amyloidosis by peptides that prevent beta-sheet conformation. Biochem. Biophys. Res. Commun. 1996, 226, 672–680. [Google Scholar] [CrossRef]
- Minicozzi, V.; Chiaraluce, R.; Consalvi, V.; Giordano, C.; Narcisi, C.; Punzi, P.; Rossi, G.C.; Morante, S. Computational and experimental studies on beta-sheet breakers targeting Abeta1-40 fibrils. J. Biol. Chem. 2014, 289, 11242–11252. [Google Scholar] [CrossRef]
- Datki, Z.; Papp, R.; Zadori, D.; Soos, K.; Fulop, L.; Juhasz, A.; Laskay, G.; Hetenyi, C.; Mihalik, E.; Zarandi, M.; et al. In vitro model of neurotoxicity of Abeta 1-42 and neuroprotection by a pentapeptide: Irreversible events during the first hour. Neurobiol. Dis. 2004, 17, 507–515. [Google Scholar] [CrossRef]
- Jagota, S.; Rajadas, J. Synthesis of D-amino acid peptides and their effect on beta-amyloid aggregation and toxicity in transgenic Caenorhabditis elegans. Med. Chem. Res. 2013, 22, 3991–4000. [Google Scholar] [CrossRef]
- Gilead, S.; Gazit, E. Inhibition of amyloid fibril formation by peptide analogues modified with alpha-aminoisobutyric acid. Angew. Chem. Int. Ed. Engl. 2004, 43, 4041–4044. [Google Scholar] [CrossRef]
- Mishra, A.; Misra, A.; Vaishnavi, T.S.; Thota, C.; Gupta, M.; Ramakumar, S.; Chauhan, V.S. Conformationally restricted short peptides inhibit human islet amyloid polypeptide (hIAPP) fibrillization. Chem. Commun. 2013, 49, 2688–2690. [Google Scholar] [CrossRef]
- Loureiro, J.A.; Crespo, R.; Borner, H.; Martins, P.M.; Rocha, F.A.; Coelho, M.; Pereira, M.C.; Rocha, S. Fluorinated beta-sheet breaker peptides. J. Mater. Chem. B 2014, 2, 2259–2264. [Google Scholar] [CrossRef] [PubMed]
- Sinopoli, A.; Giuffrida, A.; Tomasello, M.F.; Giuffrida, M.L.; Leone, M.; Attanasio, F.; Caraci, F.; De Bona, P.; Naletova, I.; Saviano, M.; et al. Ac-LPFFD-Th: A Trehalose-Conjugated Peptidomimetic as a Strong Suppressor of Amyloid-beta Oligomer Formation and Cytotoxicity. Chembiochem 2016, 17, 1541–1549. [Google Scholar] [CrossRef] [PubMed]
- Findeis, M.A.; Lee, J.J.; Kelley, M.; Wakefield, J.D.; Zhang, M.H.; Chin, J.; Kubasek, W.; Molineaux, S.M. Characterization of cholyl-leu-val-phe-phe-ala-OH as an inhibitor of amyloid beta-peptide polymerization. Amyloid 2001, 8, 231–241. [Google Scholar] [CrossRef]
- Wei, C.W.; Peng, Y.; Zhang, L.; Huang, Q.; Cheng, M.; Liu, Y.N.; Li, J. Synthesis and evaluation of ferrocenoyl pentapeptide (Fc-KLVFF) as an inhibitor of Alzheimer’s Abeta(1)-(4)(2) fibril formation in vitro. Bioorg. Med. Chem. Lett. 2011, 21, 5818–5821. [Google Scholar] [CrossRef] [PubMed]
- Arai, T.; Araya, T.; Sasaki, D.; Taniguchi, A.; Sato, T.; Sohma, Y.; Kanai, M. Rational design and identification of a non-peptidic aggregation inhibitor of amyloid-beta based on a pharmacophore motif obtained from cyclo[-Lys-Leu-Val-Phe-Phe-]. Angew. Chem. Int. Ed. Engl. 2014, 53, 8236–8239. [Google Scholar] [CrossRef]
- Ikenoue, T.; Aprile, F.A.; Sormanni, P.; Ruggeri, F.S.; Perni, M.; Heller, G.T.; Haas, C.P.; Middel, C.; Limbocker, R.; Mannini, B.; et al. A rationally designed bicyclic peptide remodels Abeta42 aggregation in vitro and reduces its toxicity in a worm model of Alzheimer’s disease. Sci. Rep. 2020, 10, 15280. [Google Scholar] [CrossRef]
- Puneeth Kumar, D.R.; Reja, R.M.; Senapati, D.K.; Singh, M.; Nalawade, S.A.; George, G.; Kaul, G.; Akhir, A.; Chopra, S.; Raghothama, S.; et al. A cationic amphiphilic peptide chaperone rescues Abeta(42) aggregation and cytotoxicity. RSC Med. Chem. 2023, 14, 332–340. [Google Scholar] [CrossRef]
- Puneeth Kumar, D.R.; Nalawade, S.A.; Pahan, S.; Singh, M.; Senapati, D.K.; Roy, S.; Dey, S.; Toraskar, S.U.; Raghothama, S.; Gopi, H.N. Proteolytically Stable alphaalphagamma-Hybrid Peptides Inhibit the Aggregation and Cytotoxicity of Abeta(42). ACS Chem. Neurosci. 2023, 14, 3398–3408. [Google Scholar] [CrossRef]
- Pariary, R.; Shome, G.; Kalita, S.; Kalita, S.; Roy, A.; Harikishore, A.; Jana, K.; Senapati, D.; Mandal, B.; Mandal, A.K.; et al. Peptide-Based Strategies: Combating Alzheimer’s Amyloid beta Aggregation through Ergonomic Design and Fibril Disruption. Biochemistry 2024, 63, 2397–2413. [Google Scholar] [CrossRef]
- Bodenhausen, G.; Ruben, D.J. Natural abundance nitrogen-15 NMR by enhanced heteronuclear spectroscopy. Chem. Phys. Lett. 1980, 69, 185–189. [Google Scholar] [CrossRef]
- Khandogin, J.; Brooks, C.L., 3rd. Linking folding with aggregation in Alzheimer’s beta-amyloid peptides. Proc. Natl. Acad. Sci. USA 2007, 104, 16880–16885. [Google Scholar] [CrossRef]
- Thirumalai, D.; Reddy, G.; Straub, J.E. Role of water in protein aggregation and amyloid polymorphism. Acc. Chem. Res. 2012, 45, 83–92. [Google Scholar] [CrossRef]
- Xue, C.; Lin, T.Y.; Chang, D.; Guo, Z. Thioflavin T as an amyloid dye: Fibril quantification, optimal concentration and effect on aggregation. R. Soc. Open Sci. 2017, 4, 160696. [Google Scholar] [CrossRef]
- Vadukul, D.M.; Gbajumo, O.; Marshall, K.E.; Serpell, L.C. Amyloidogenicity and toxicity of the reverse and scrambled variants of amyloid-beta 1-42. FEBS Lett. 2017, 591, 822–830. [Google Scholar] [CrossRef]
- Keskitalo, S.; Farkas, M.; Hanenberg, M.; Szodorai, A.; Kulic, L.; Semmler, A.; Weller, M.; Nitsch, R.M.; Linnebank, M. Reciprocal modulation of Abeta42 aggregation by copper and homocysteine. Front. Aging Neurosci. 2014, 6, 237. [Google Scholar] [CrossRef] [PubMed]
- Ultsch, M.; Li, B.; Maurer, T.; Mathieu, M.; Adolfsson, O.; Muhs, A.; Pfeifer, A.; Pihlgren, M.; Bainbridge, T.W.; Reichelt, M.; et al. Structure of Crenezumab Complex with Abeta Shows Loss of beta-Hairpin. Sci. Rep. 2016, 6, 39374. [Google Scholar] [CrossRef] [PubMed]
- Fawzi, N.L.; Ying, J.; Ghirlando, R.; Torchia, D.A.; Clore, G.M. Atomic-resolution dynamics on the surface of amyloid-beta protofibrils probed by solution NMR. Nature 2011, 480, 268–272. [Google Scholar] [CrossRef]
- Hong, S.; Baravkar, S.B.; Lu, Y.; Masoud, A.R.; Zhao, Q.; Zhou, W. Molecular Modification of Queen Bee Acid and 10-Hydroxydecanoic Acid with Specific Tripeptides: Rational Design, Organic Synthesis, and Assessment for Prohealing and Antimicrobial Hydrogel Properties. Molecules 2025, 30, 615. [Google Scholar] [CrossRef]
- Baravkar, S.B.; Lu, Y.; Masoud, A.R.; Zhao, Q.; He, J.; Hong, S. Development of a Novel Covalently Bonded Conjugate of Caprylic Acid Tripeptide (Isoleucine-Leucine-Aspartic Acid) for Wound-Compatible and Injectable Hydrogel to Accelerate Healing. Biomolecules 2024, 14, 94. [Google Scholar] [CrossRef]
- Hong, S.; Baravkar, S.B.; Lu, Y. Amphiphilic Conjugates of Fatty Acids or their Derivatives. US Provisional Patent No. 63617303, 3 January 2024. [Google Scholar]
- Oakley, H.; Cole, S.L.; Logan, S.; Maus, E.; Shao, P.; Craft, J.; Guillozet-Bongaarts, A.; Ohno, M.; Disterhoft, J.; Van Eldik, L.; et al. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: Potential factors in amyloid plaque formation. J. Neurosci. 2006, 26, 10129–10140. [Google Scholar] [CrossRef] [PubMed]
- Drummond, E.; Wisniewski, T. Alzheimer’s disease: Experimental models and reality. Acta Neuropathol. 2017, 133, 155–175. [Google Scholar] [CrossRef]
- Myers, A.; McGonigle, P. Overview of Transgenic Mouse Models for Alzheimer’s Disease. Curr. Protoc. Neurosci. 2019, 89, e81. [Google Scholar] [CrossRef]
- Jacob, H.J.; Kwitek, A.E. Rat genetics: Attaching physiology and pharmacology to the genome. Nat. Rev. Genet. 2002, 3, 33–42. [Google Scholar] [CrossRef]
- Cohen, R.M.; Rezai-Zadeh, K.; Weitz, T.M.; Rentsendorj, A.; Gate, D.; Spivak, I.; Bholat, Y.; Vasilevko, V.; Glabe, C.G.; Breunig, J.J.; et al. A transgenic Alzheimer rat with plaques, tau pathology, behavioral impairment, oligomeric abeta, and frank neuronal loss. J. Neurosci. 2013, 33, 6245–6256. [Google Scholar] [CrossRef]
- Do Carmo, S.; Cuello, A.C. Modeling Alzheimer’s disease in transgenic rats. Mol. Neurodegener. 2013, 8, 37. [Google Scholar] [CrossRef]
- Berkowitz, L.E.; Harvey, R.E.; Drake, E.; Thompson, S.M.; Clark, B.J. Progressive impairment of directional and spatially precise trajectories by TgF344-Alzheimer’s disease rats in the Morris Water Task. Sci. Rep. 2018, 8, 16153. [Google Scholar] [CrossRef] [PubMed]
- Lochhead, J.J.; Thorne, R.G. Intranasal delivery of biologics to the central nervous system. Adv. Drug Deliv. Rev. 2012, 64, 614–628. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, M.B.; Chauhan, N.B. Brain Uptake of Neurotherapeutics after Intranasal versus Intraperitoneal Delivery in Mice. J. Neurol. Neurosurg. 2015, 2, 009. [Google Scholar] [CrossRef]
- Erdo, F.; Bors, L.A.; Farkas, D.; Bajza, A.; Gizurarson, S. Evaluation of intranasal delivery route of drug administration for brain targeting. Brain Res. Bull. 2018, 143, 155–170. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baravkar, S.B.; Lu, Y.; Zhao, Q.; Peng, H.; Zhou, W.; Hong, S. Rationally Designed Pentapeptide Analogs of Aβ19–23 Fragment as Potent Inhibitors of Aβ42 Aggregation. Molecules 2025, 30, 2071. https://doi.org/10.3390/molecules30092071
Baravkar SB, Lu Y, Zhao Q, Peng H, Zhou W, Hong S. Rationally Designed Pentapeptide Analogs of Aβ19–23 Fragment as Potent Inhibitors of Aβ42 Aggregation. Molecules. 2025; 30(9):2071. https://doi.org/10.3390/molecules30092071
Chicago/Turabian StyleBaravkar, Sachin B., Yan Lu, Qi Zhao, Hongying Peng, Weilie Zhou, and Song Hong. 2025. "Rationally Designed Pentapeptide Analogs of Aβ19–23 Fragment as Potent Inhibitors of Aβ42 Aggregation" Molecules 30, no. 9: 2071. https://doi.org/10.3390/molecules30092071
APA StyleBaravkar, S. B., Lu, Y., Zhao, Q., Peng, H., Zhou, W., & Hong, S. (2025). Rationally Designed Pentapeptide Analogs of Aβ19–23 Fragment as Potent Inhibitors of Aβ42 Aggregation. Molecules, 30(9), 2071. https://doi.org/10.3390/molecules30092071