

Exploring the Chemistry and Applications of Thio-, Seleno-, and Tellurosugars

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. S-Containing Carbohydrates
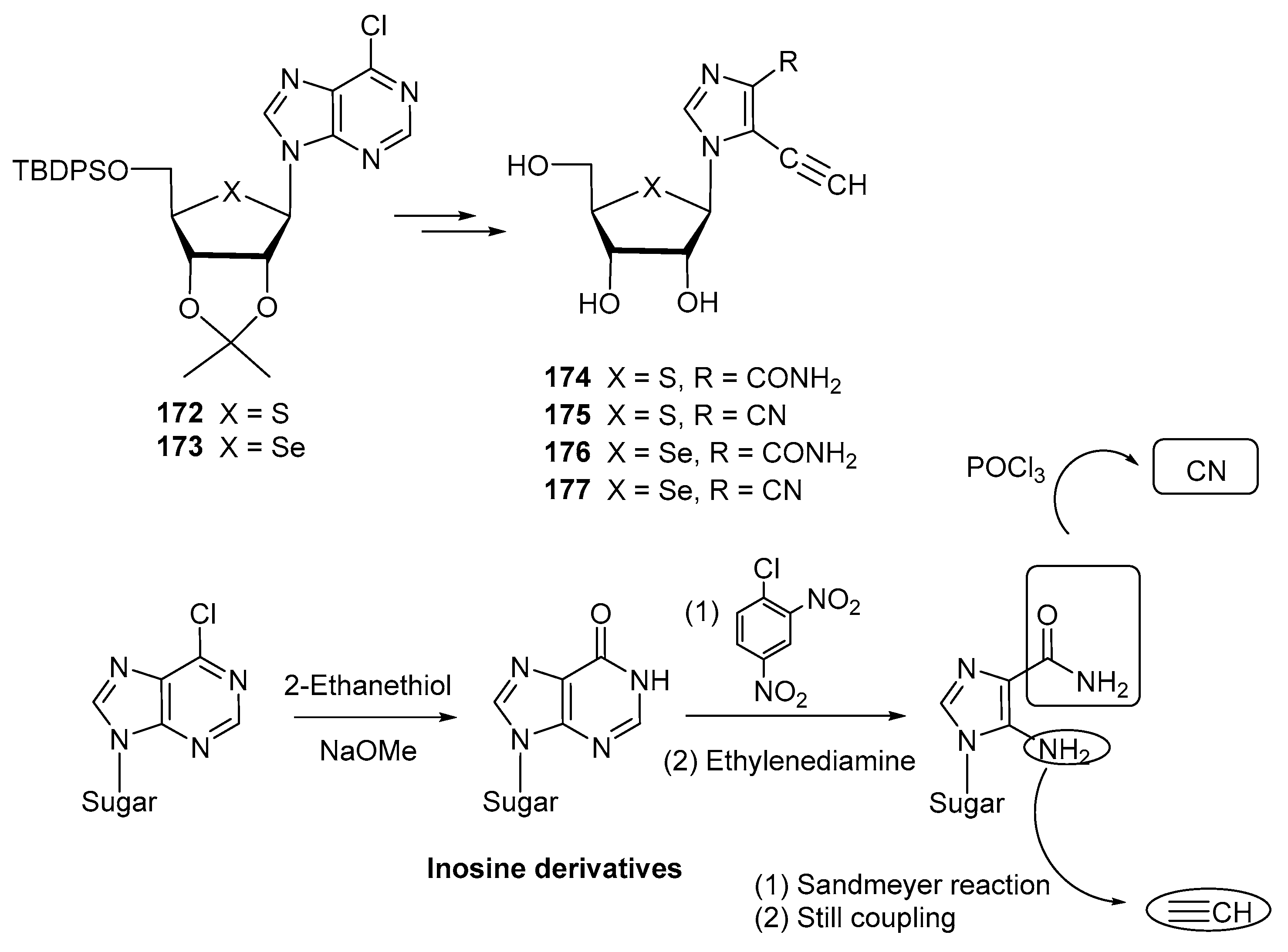
2.1. 4′- and 5′-Thiosugars
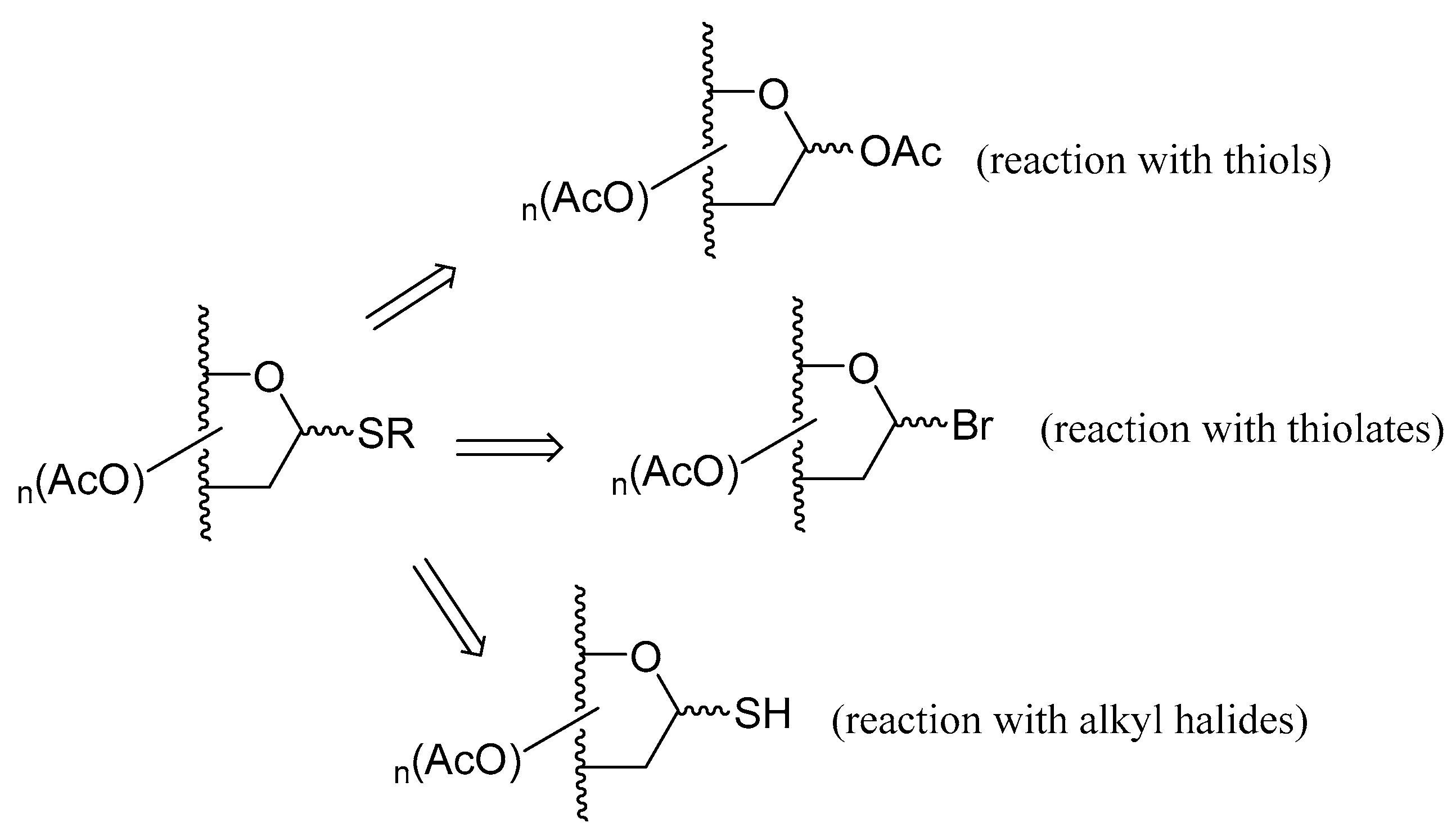
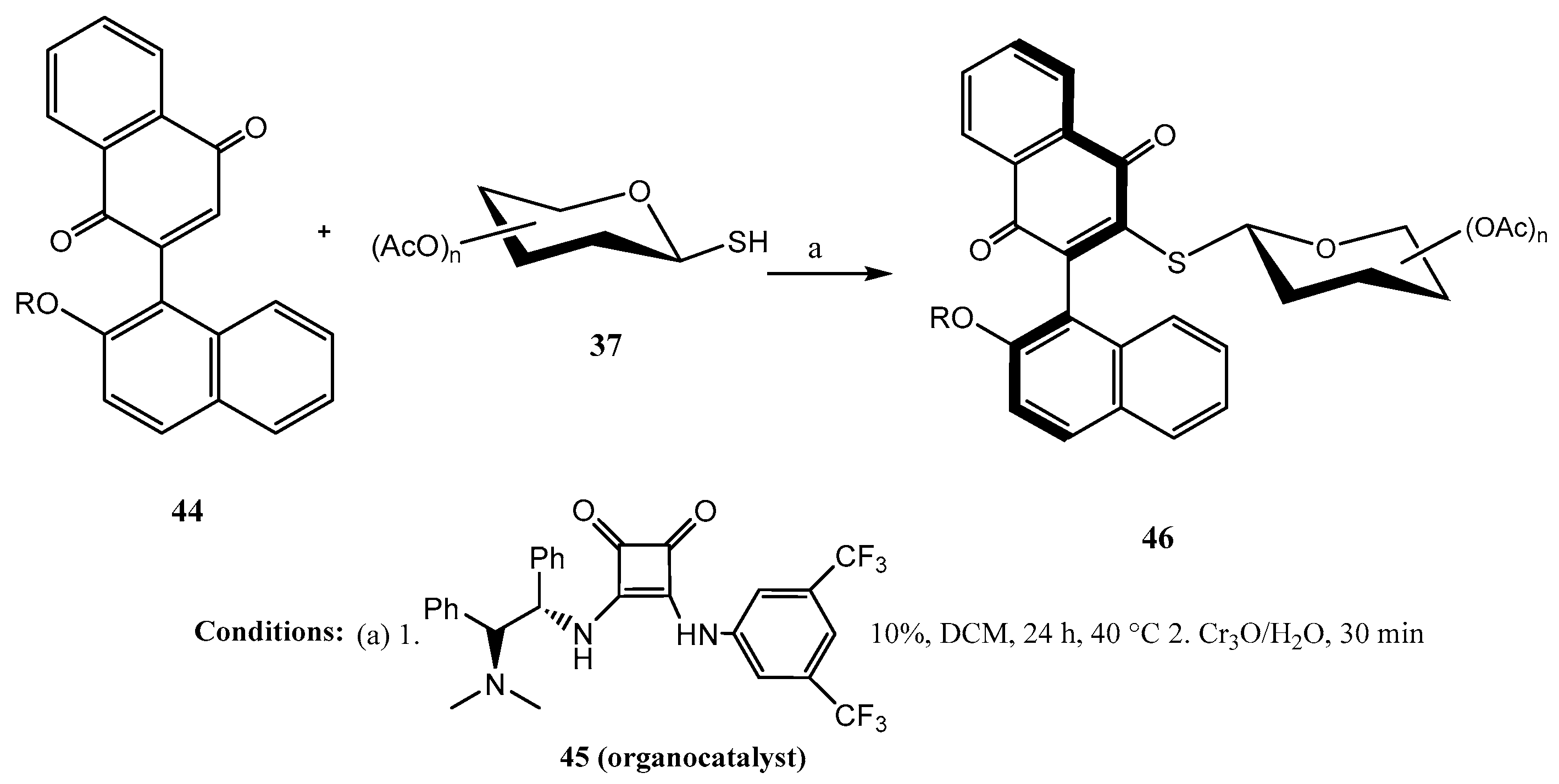
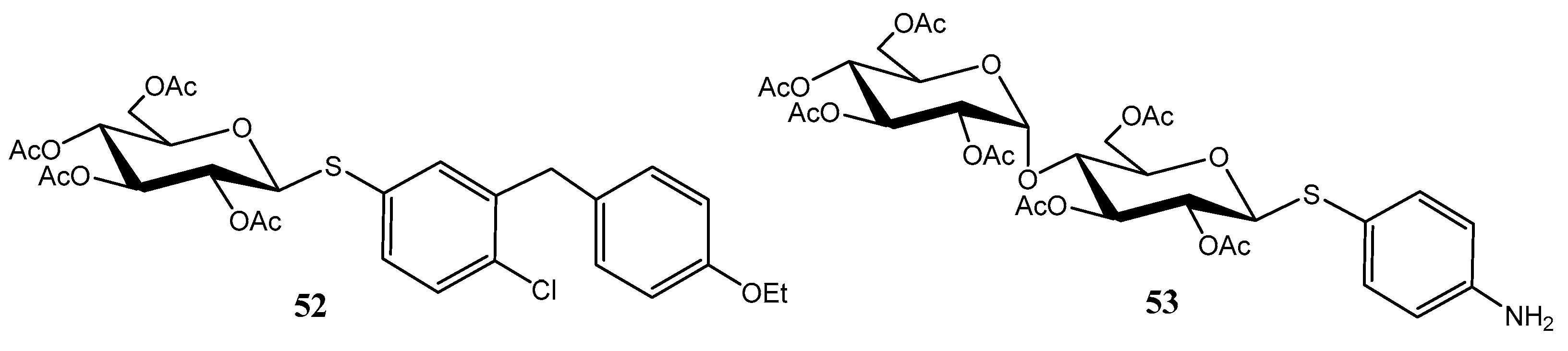
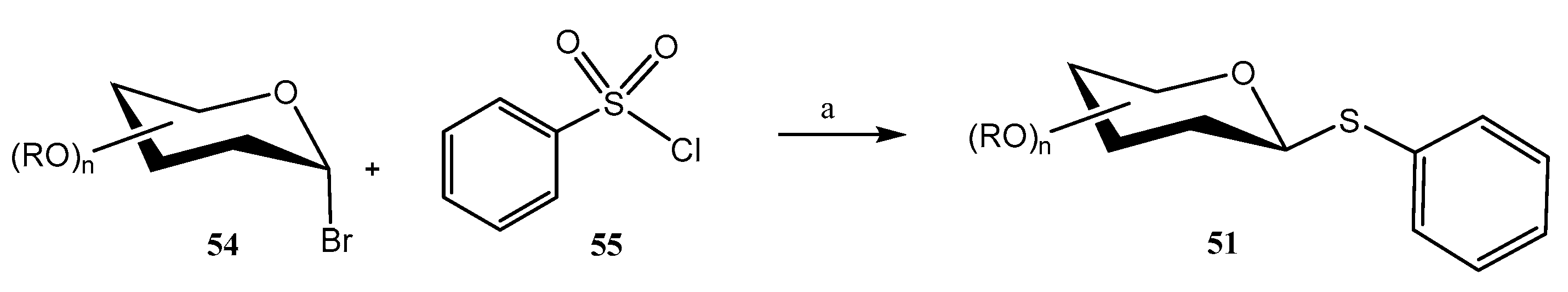
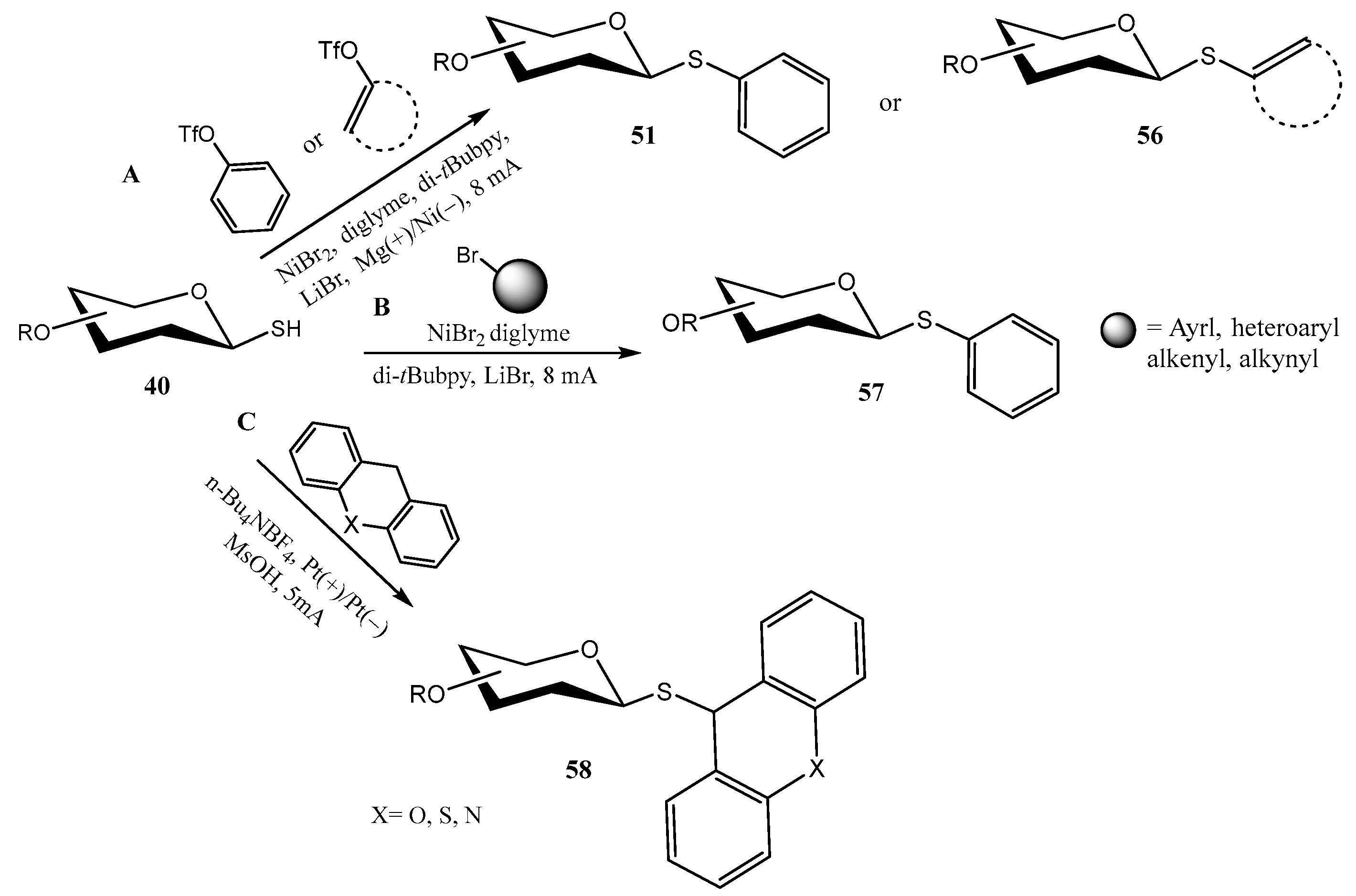
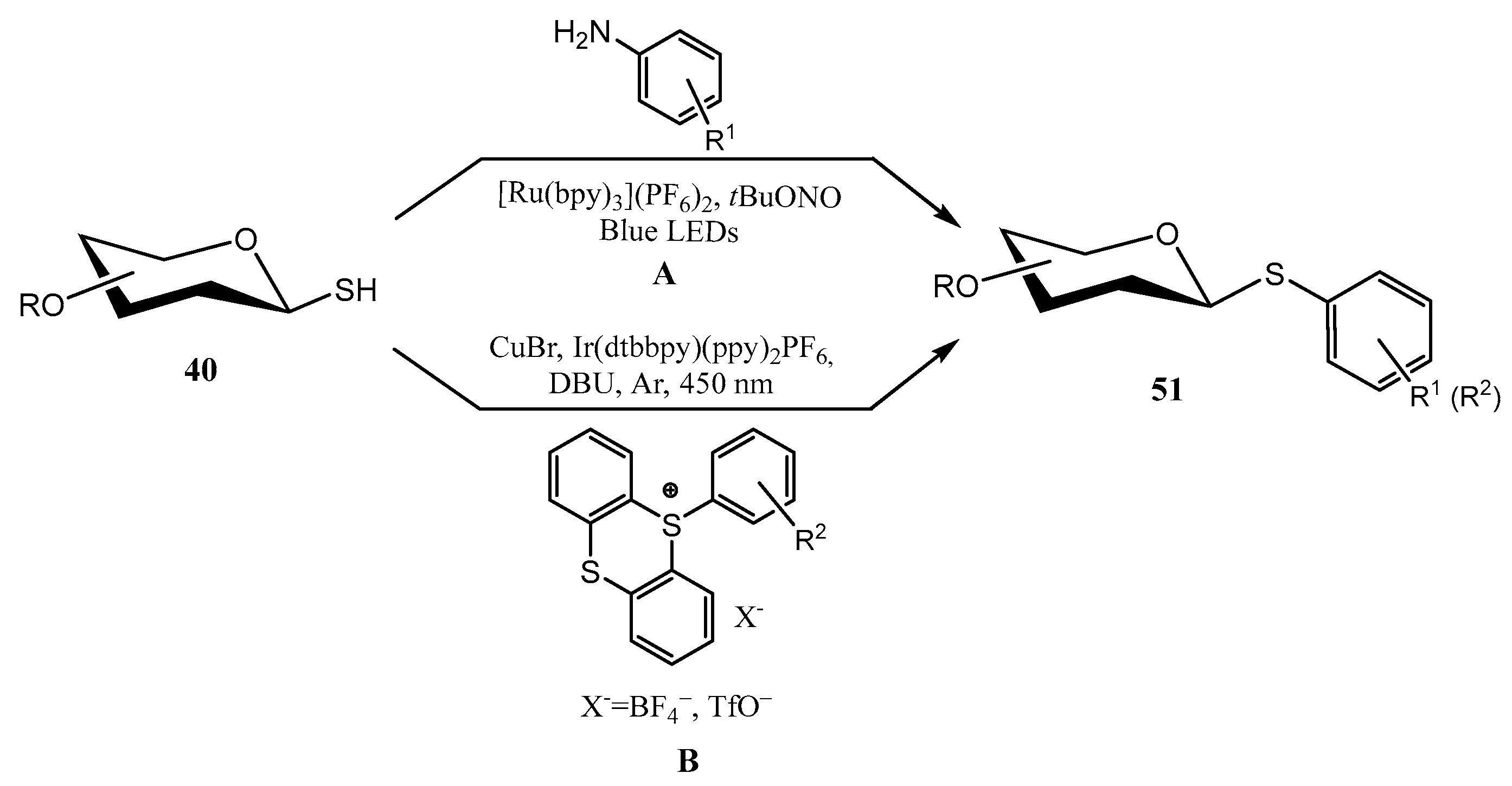
2.2. Thioglycosides
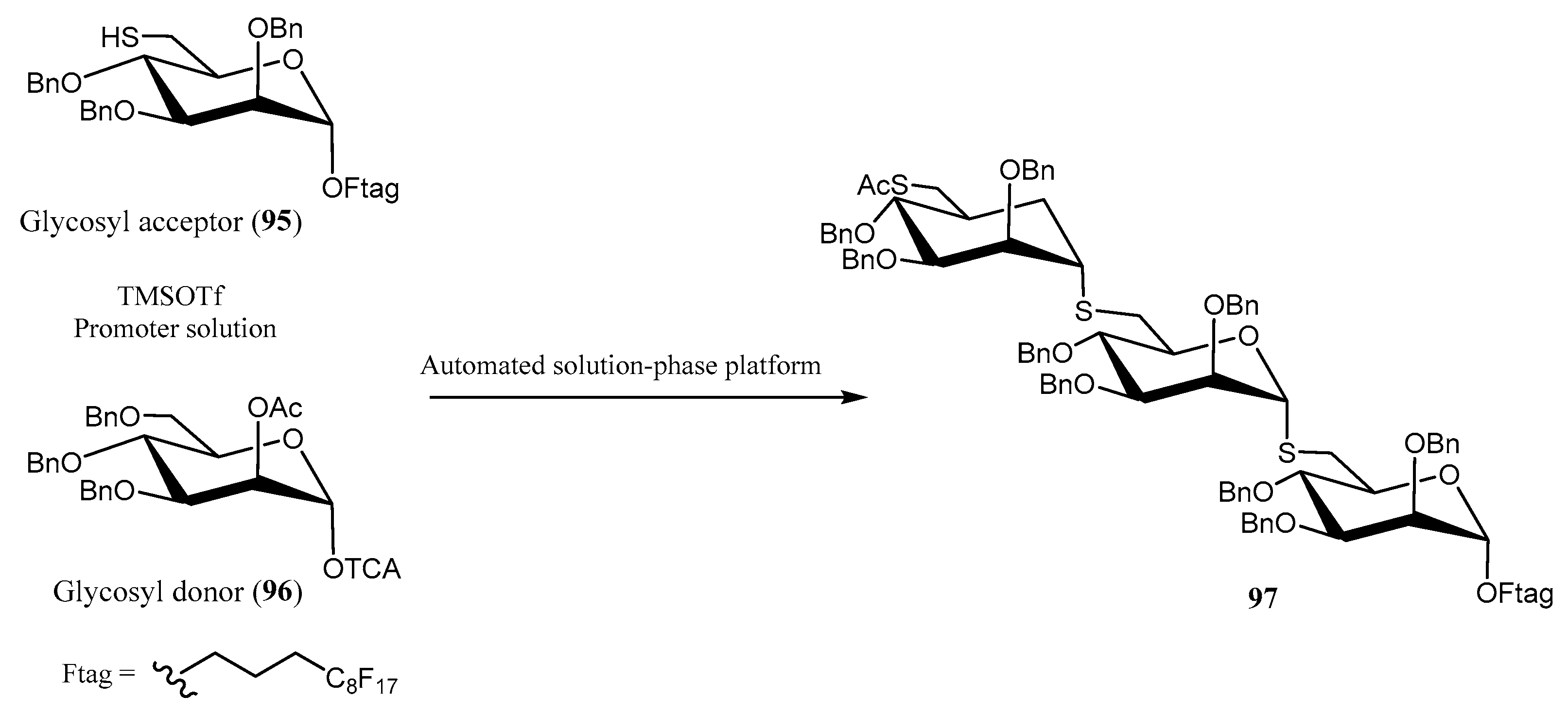
Thioglycosides in Pseudo-Disaccharides and Pseudo-Oligosaccharides
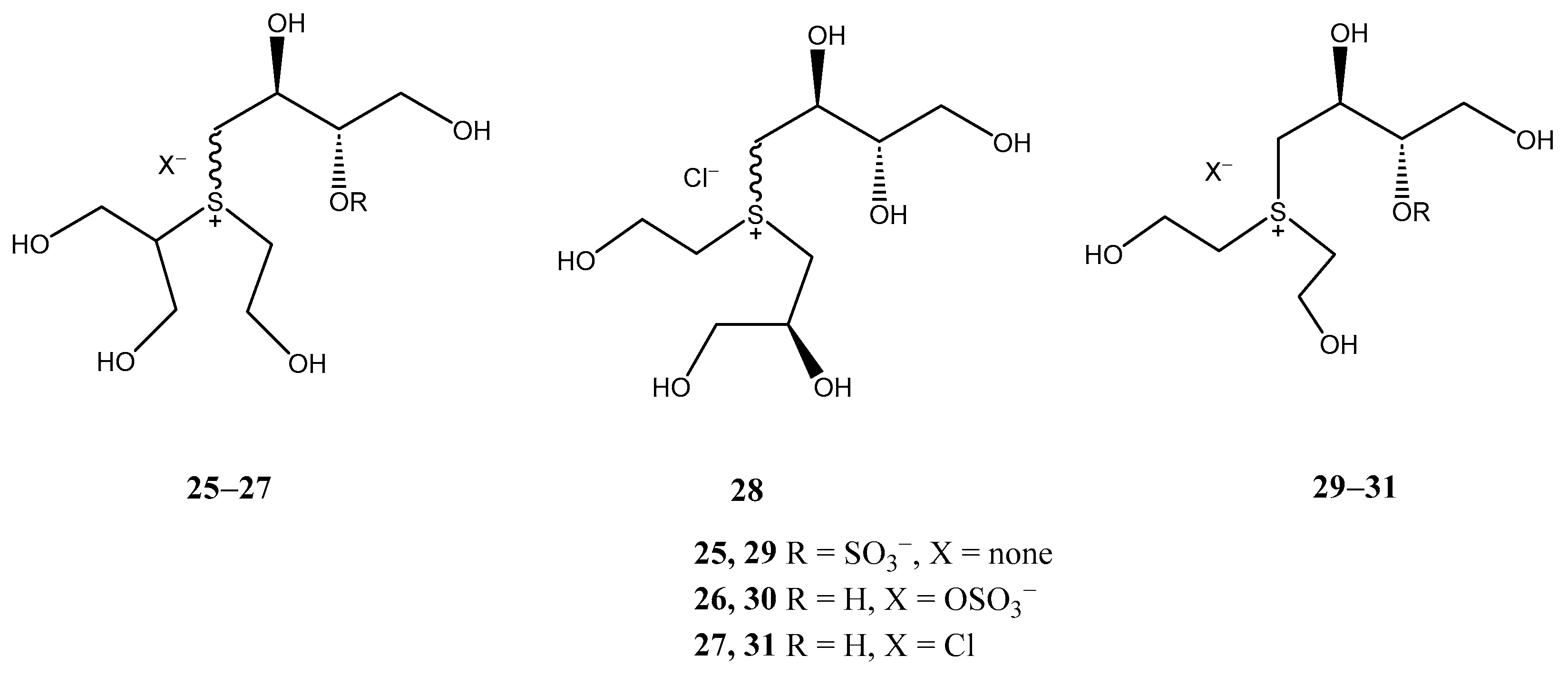
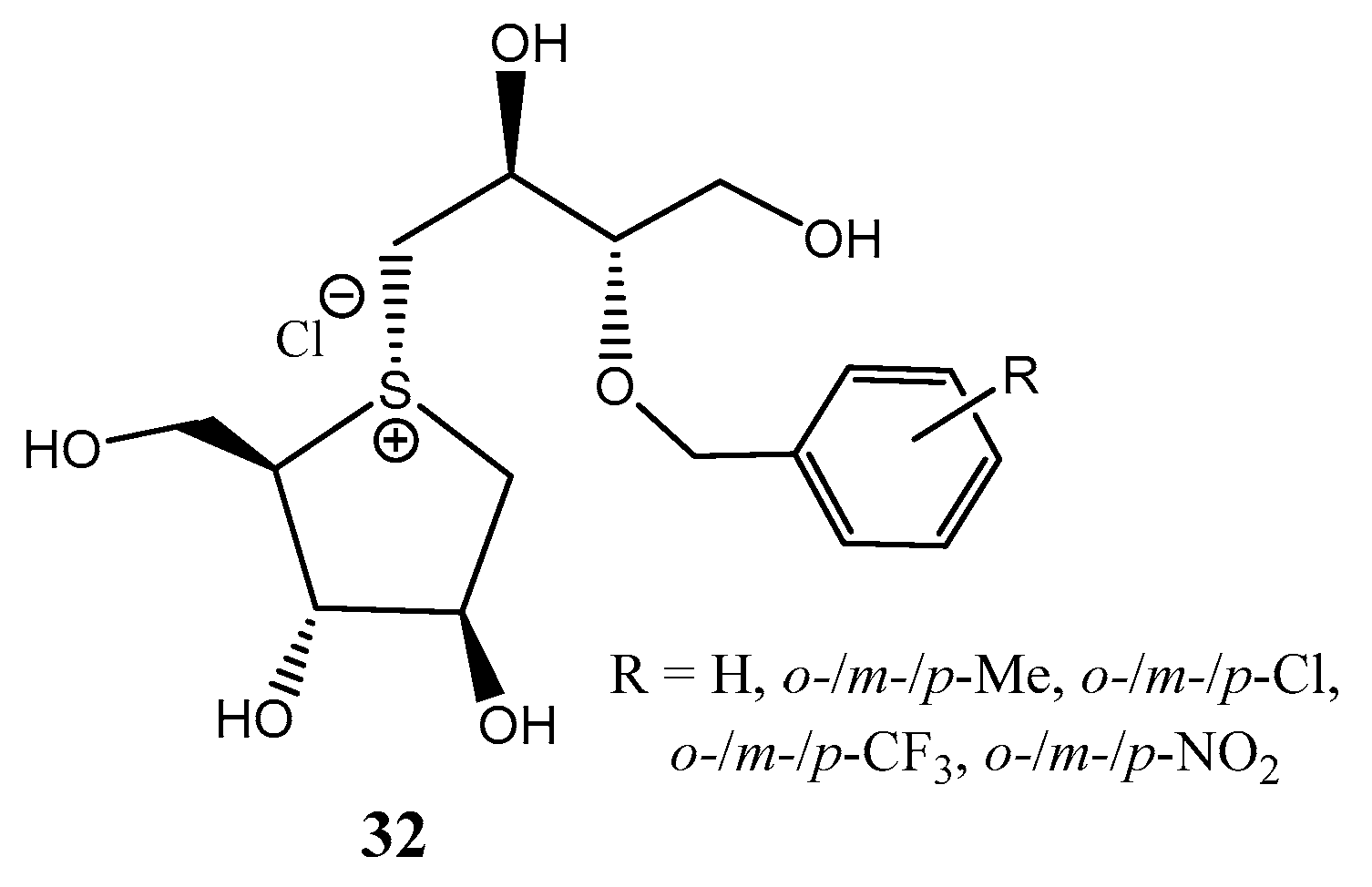
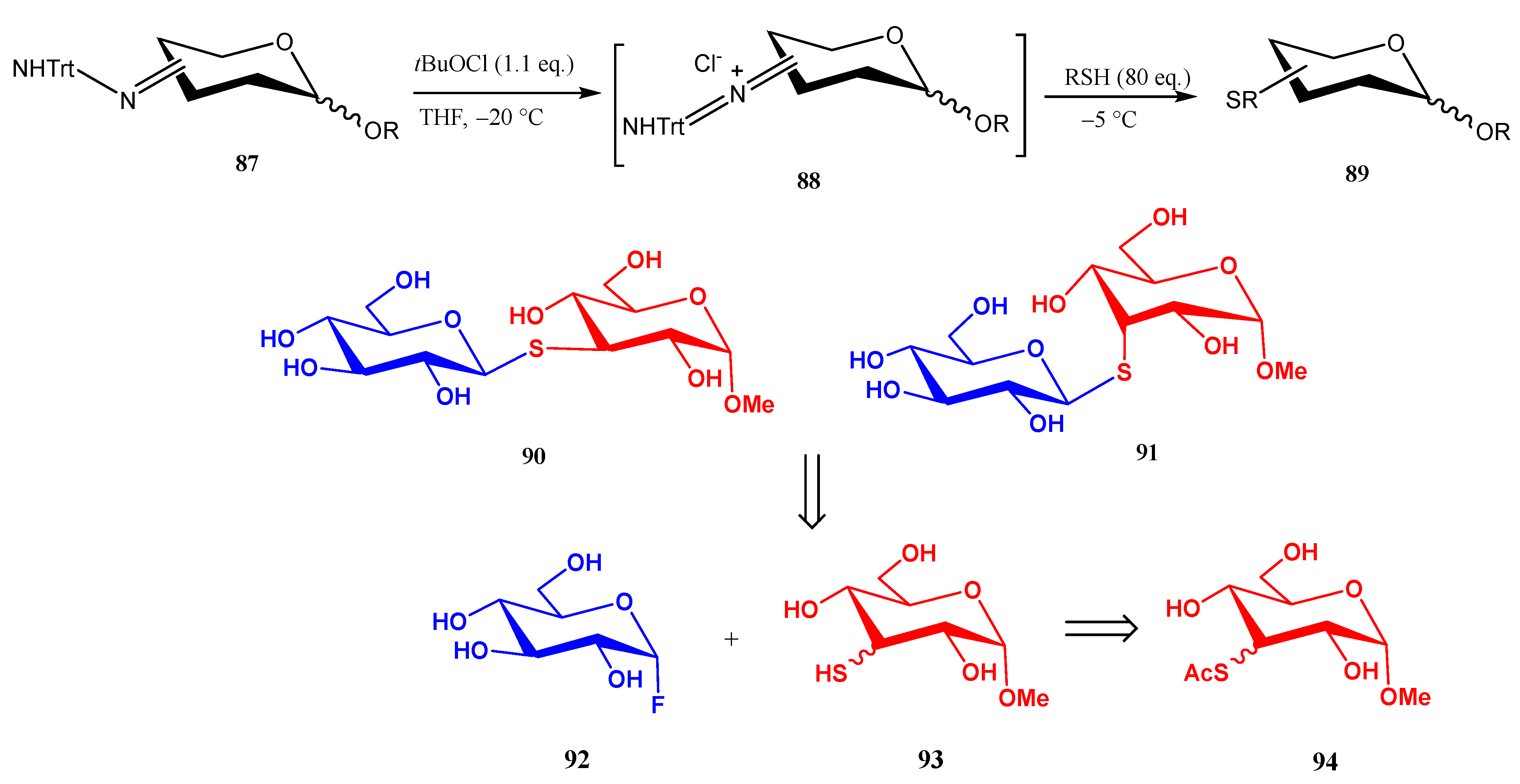
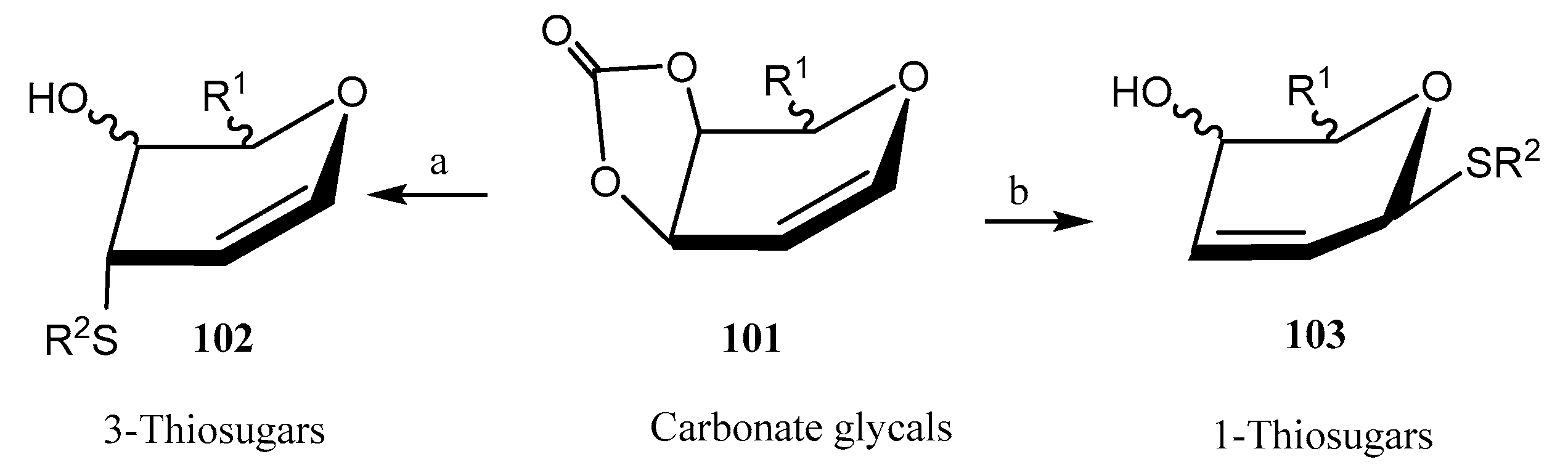
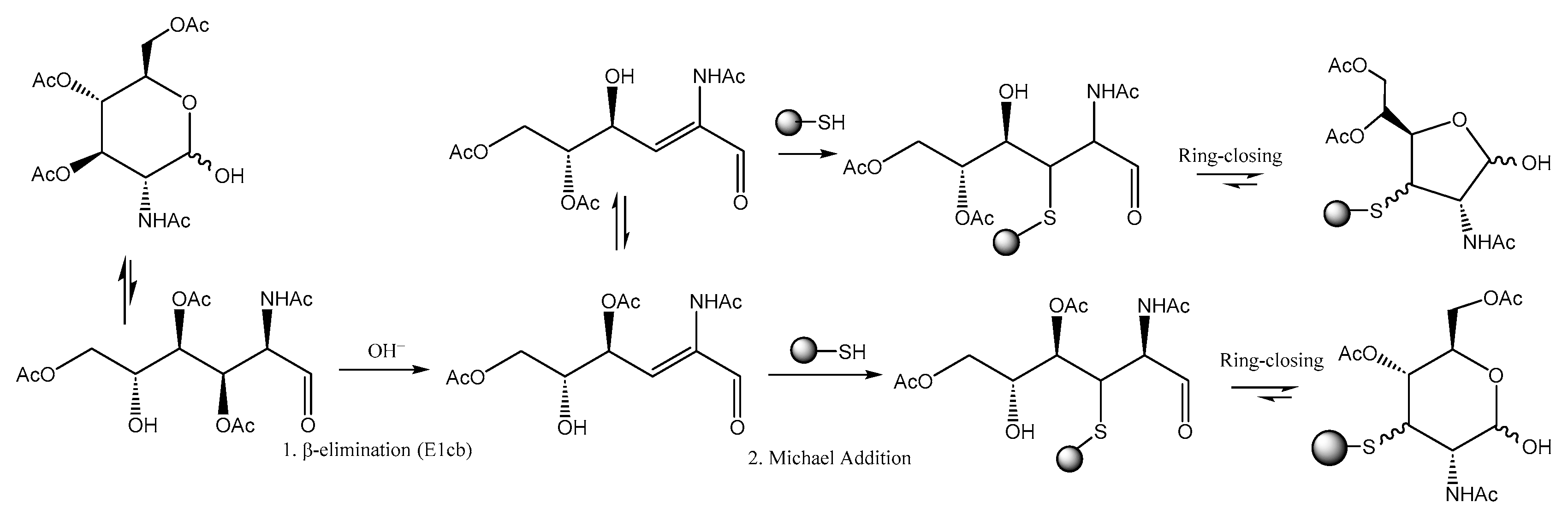
2.3. 3-Thiosugars
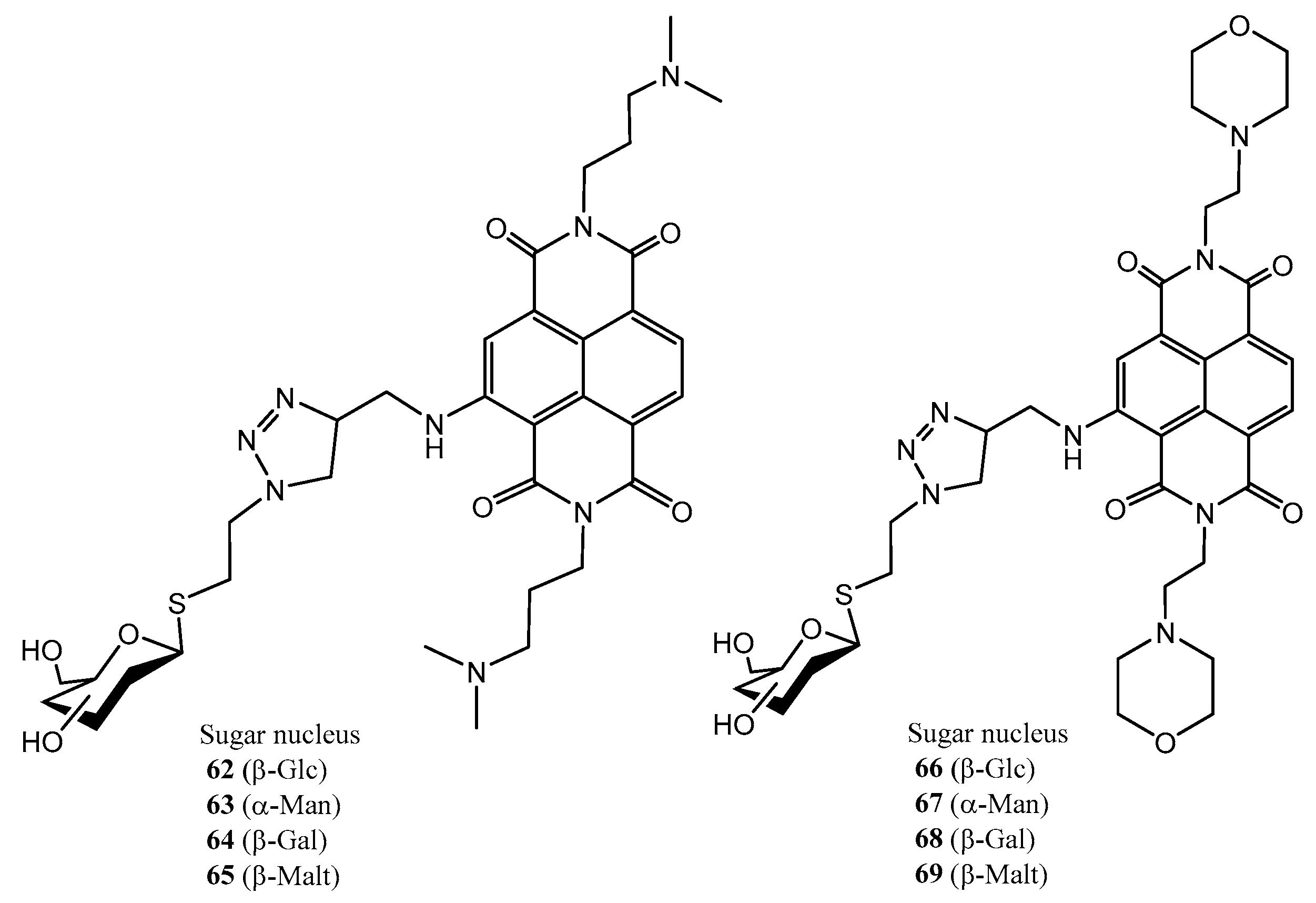
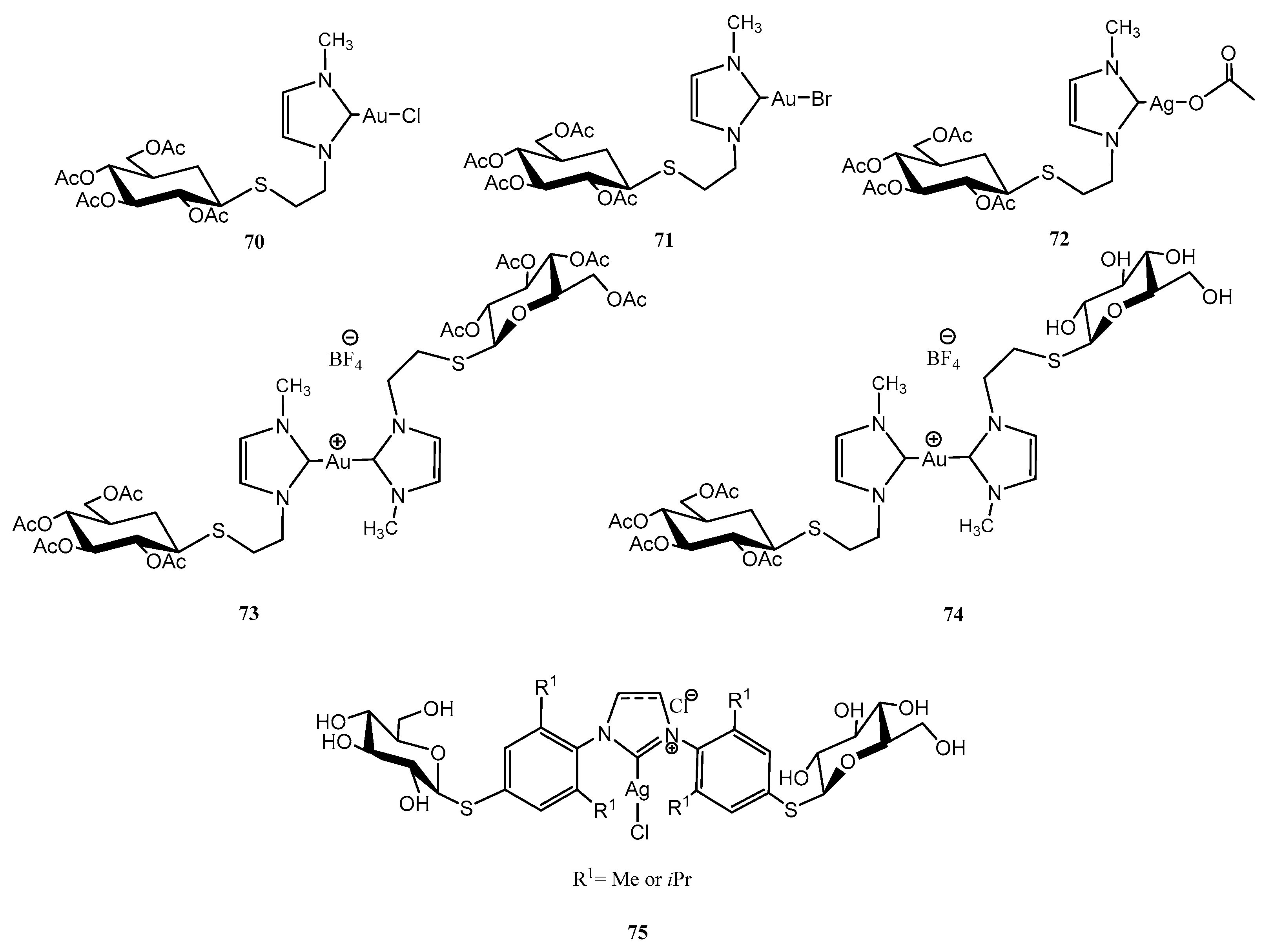
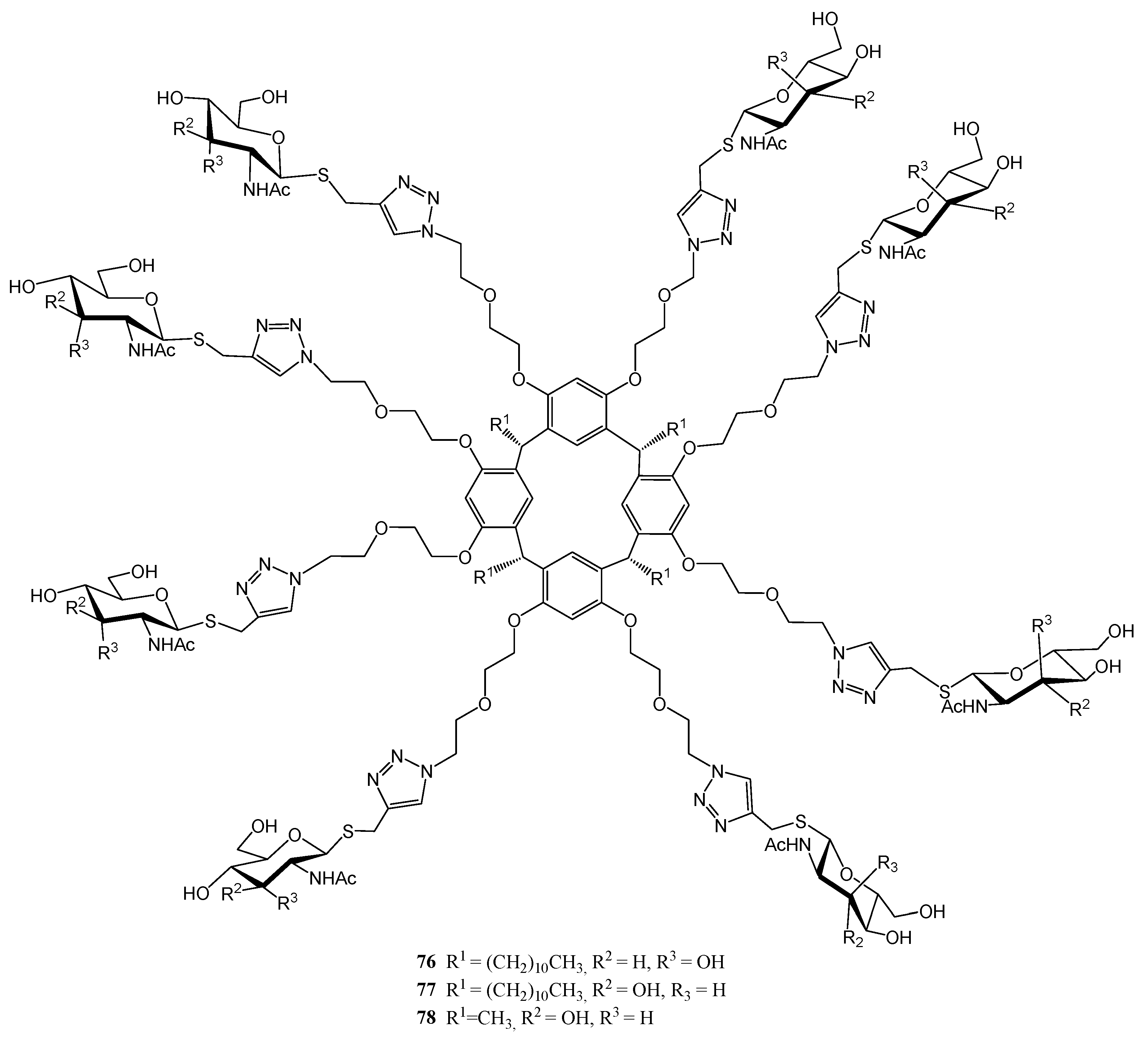
2.4. Other Thiosugars
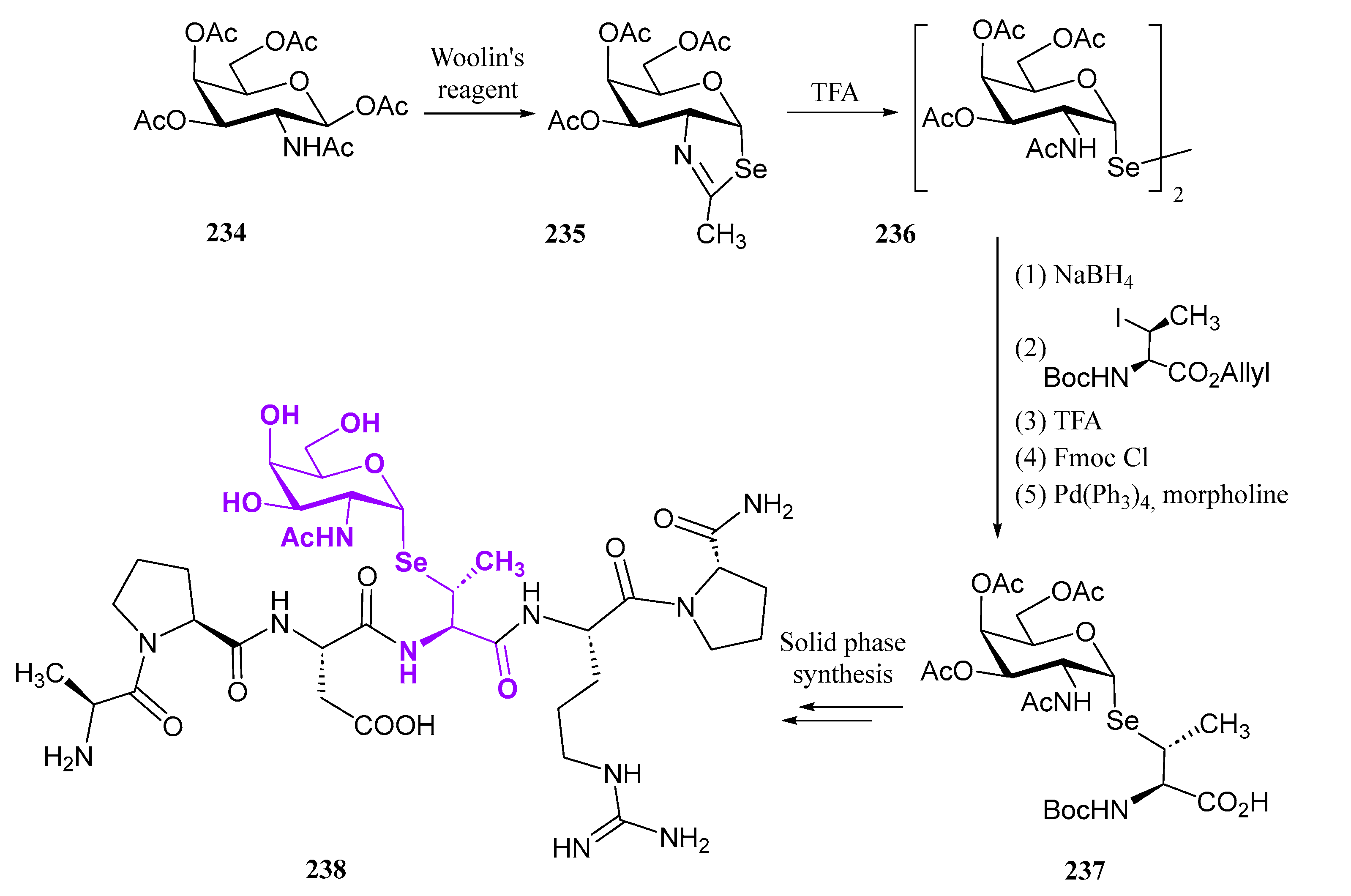
3. Se-Containing Carbohydrates
3.1. 4′- and 5′-Selenosugars
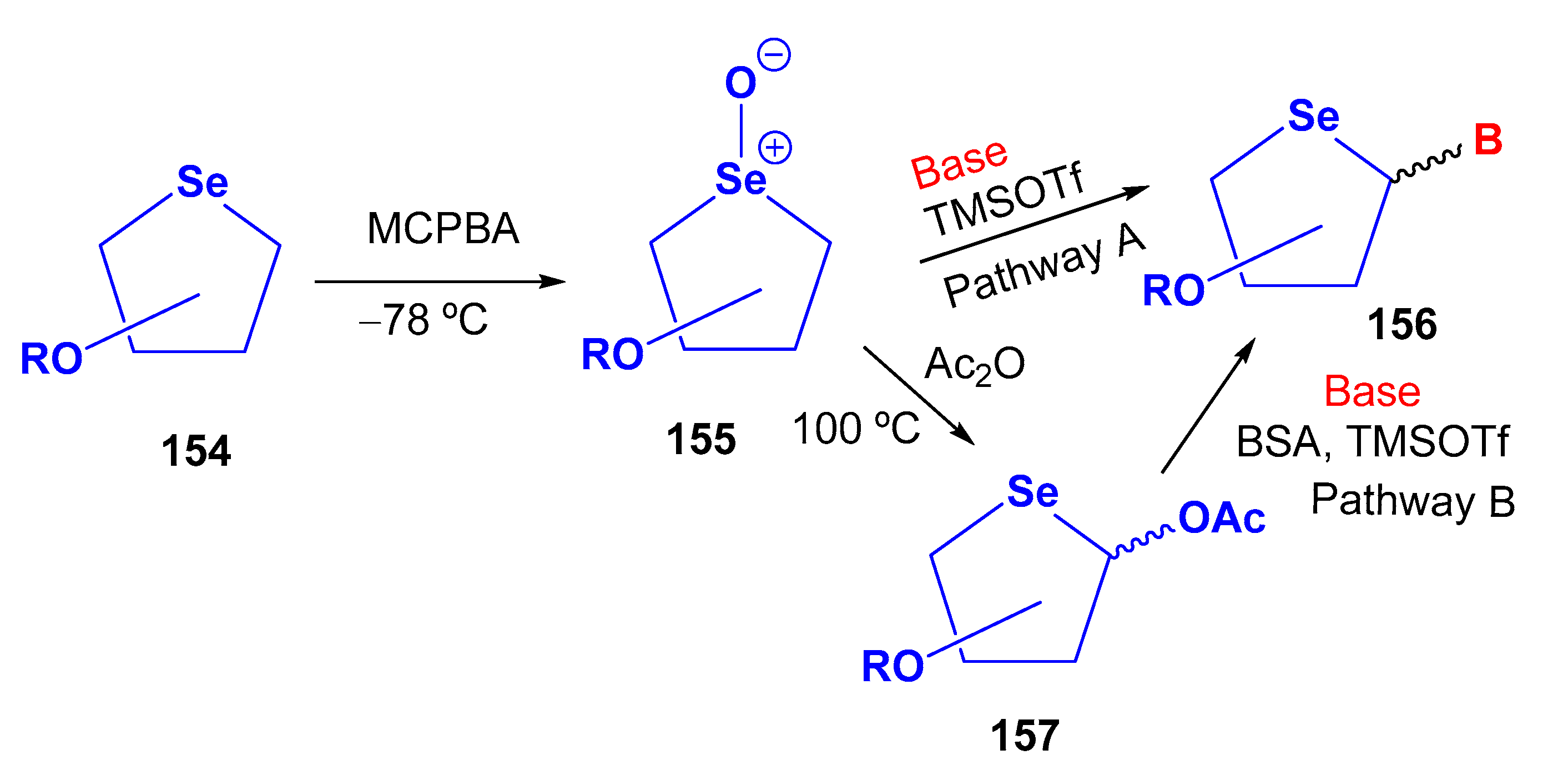
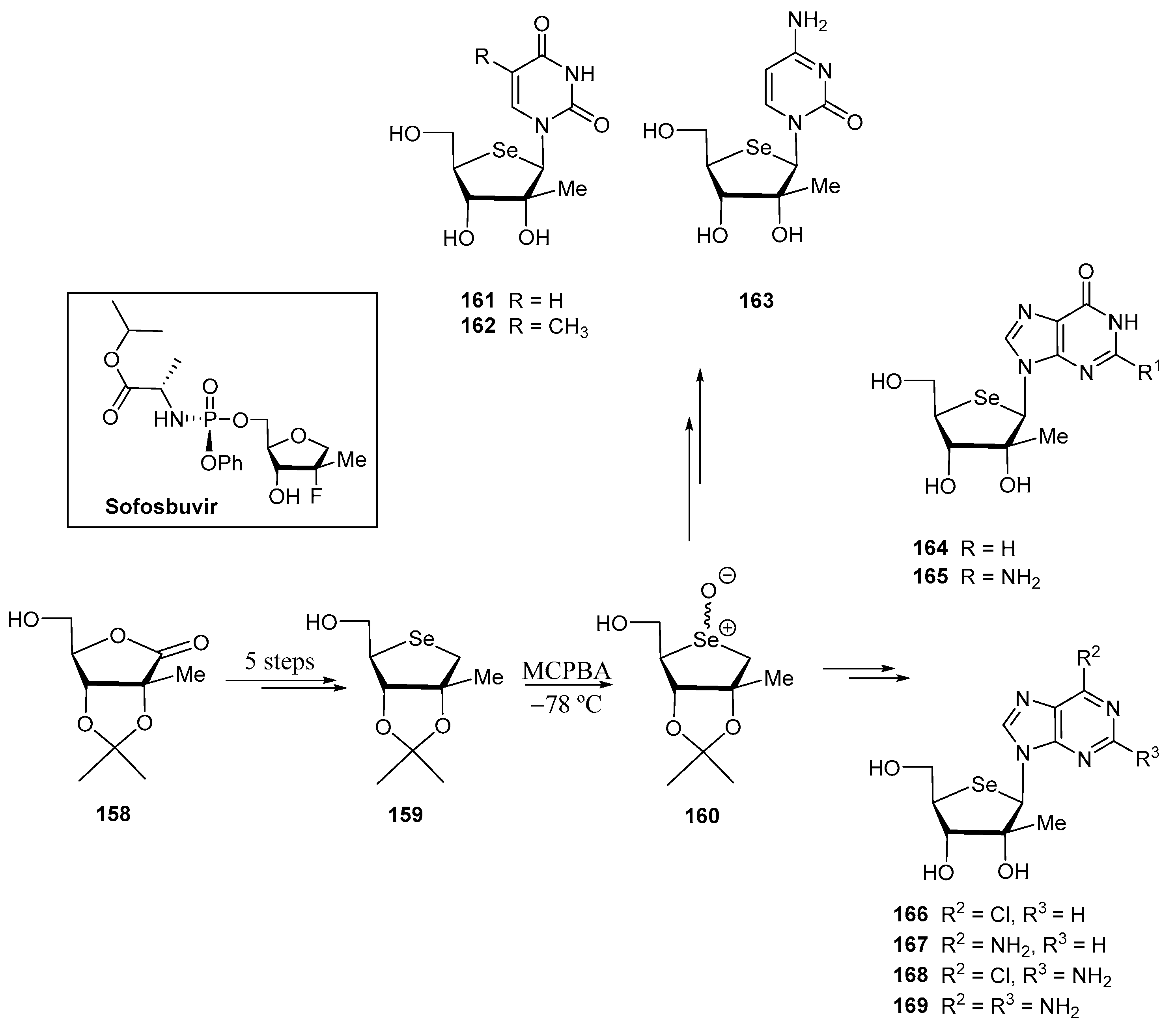
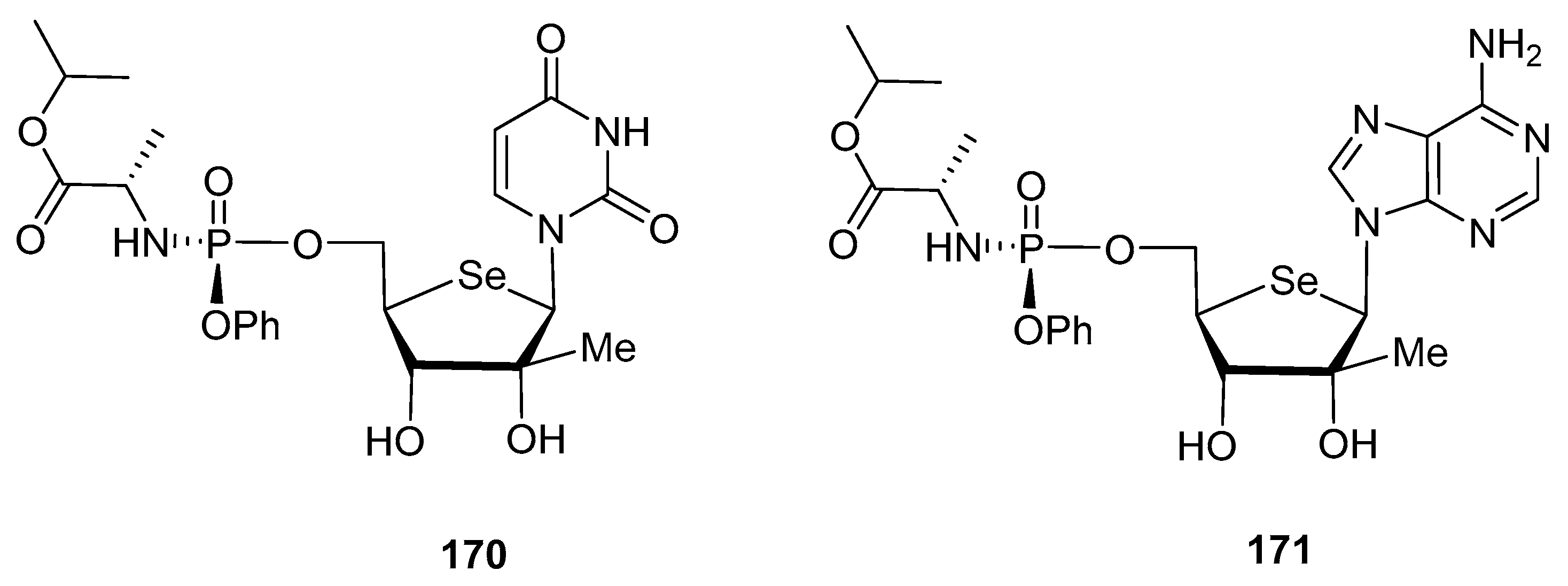
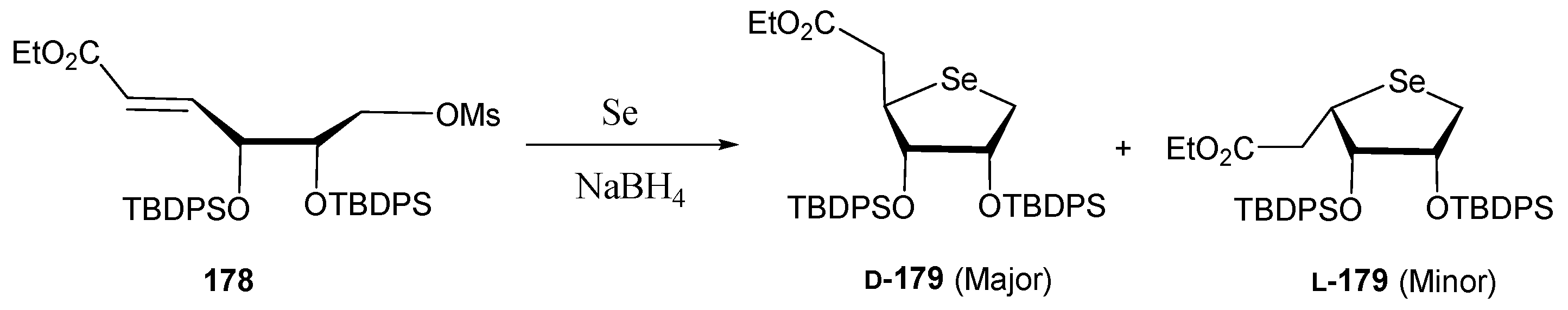
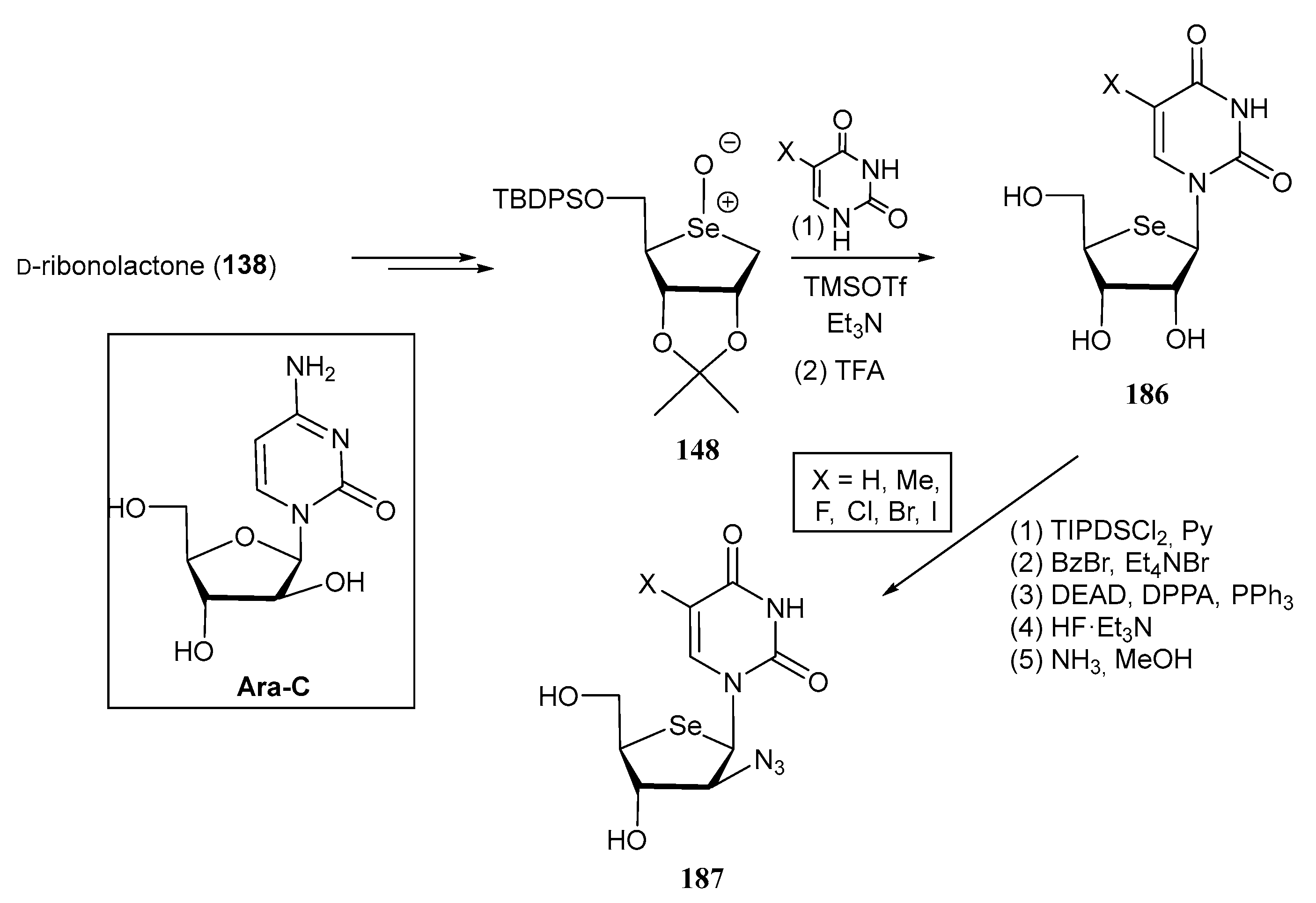
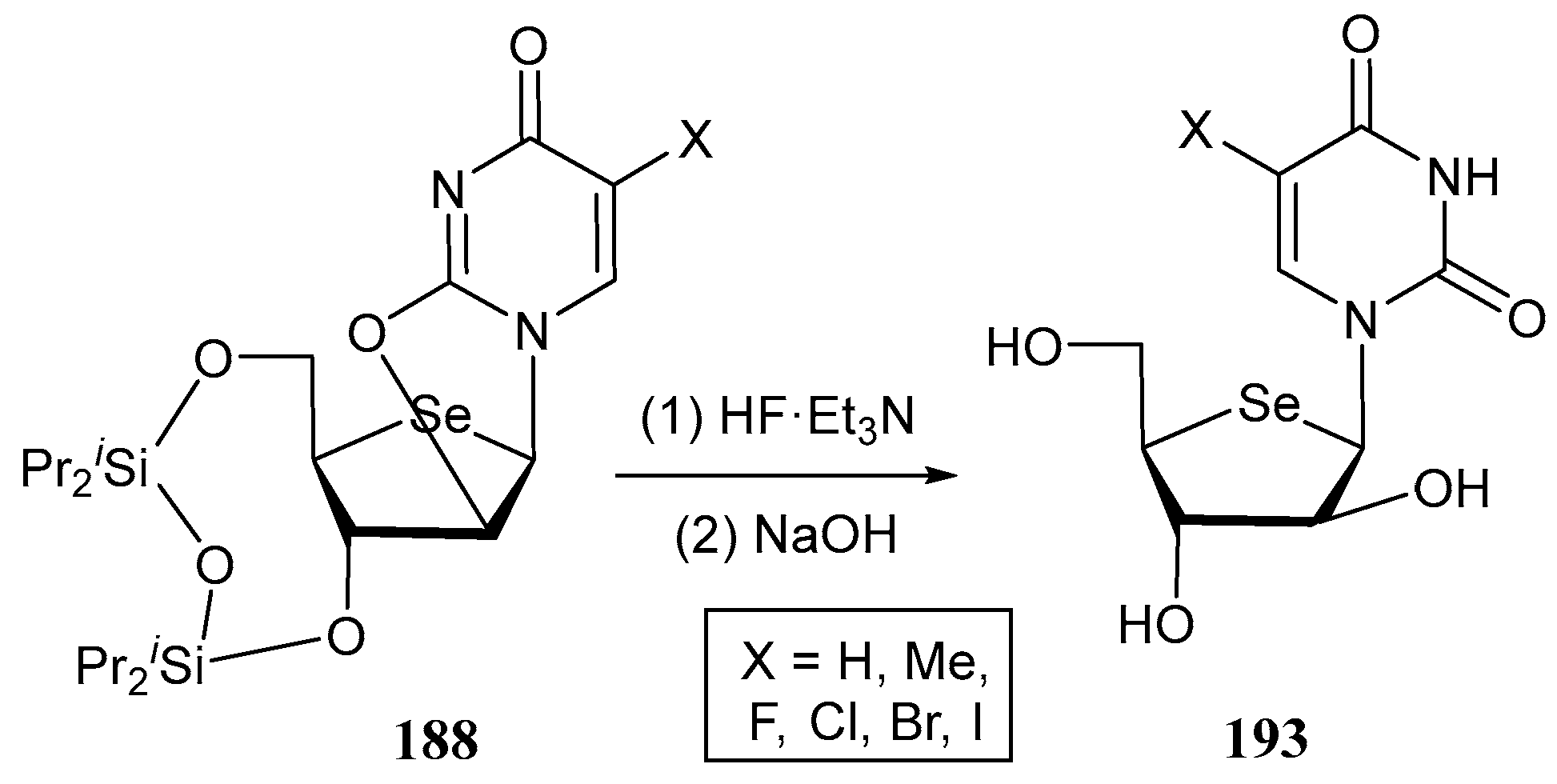
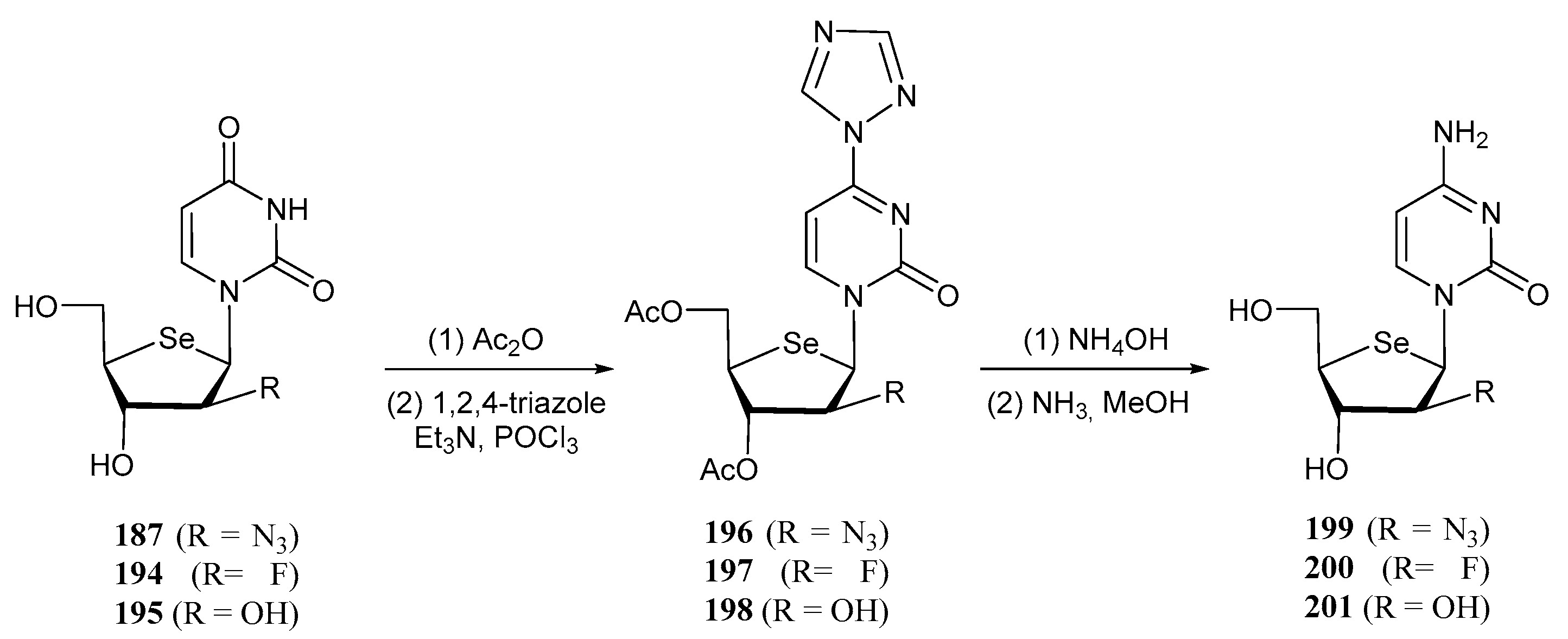
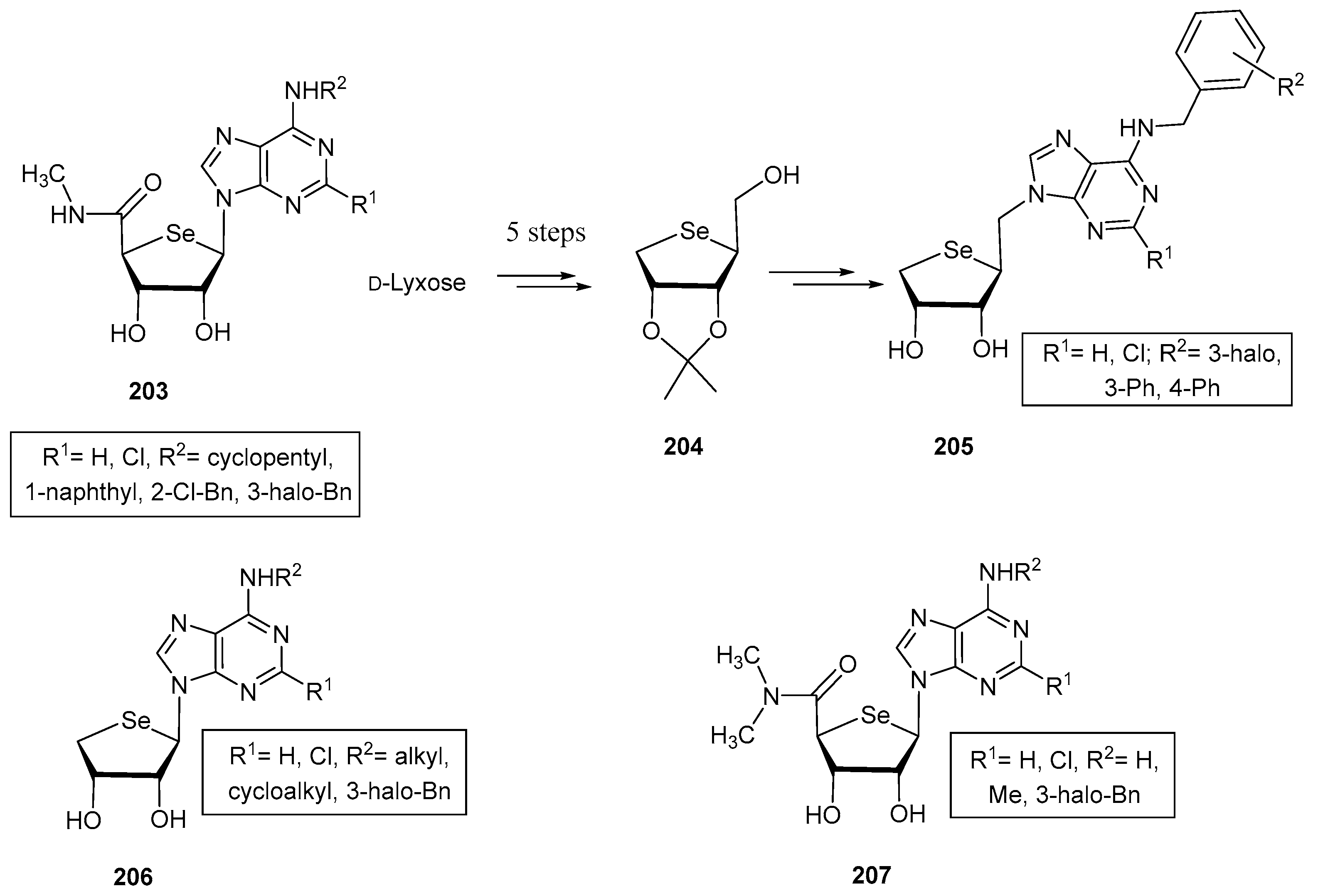
3.2. Selenonucleosides
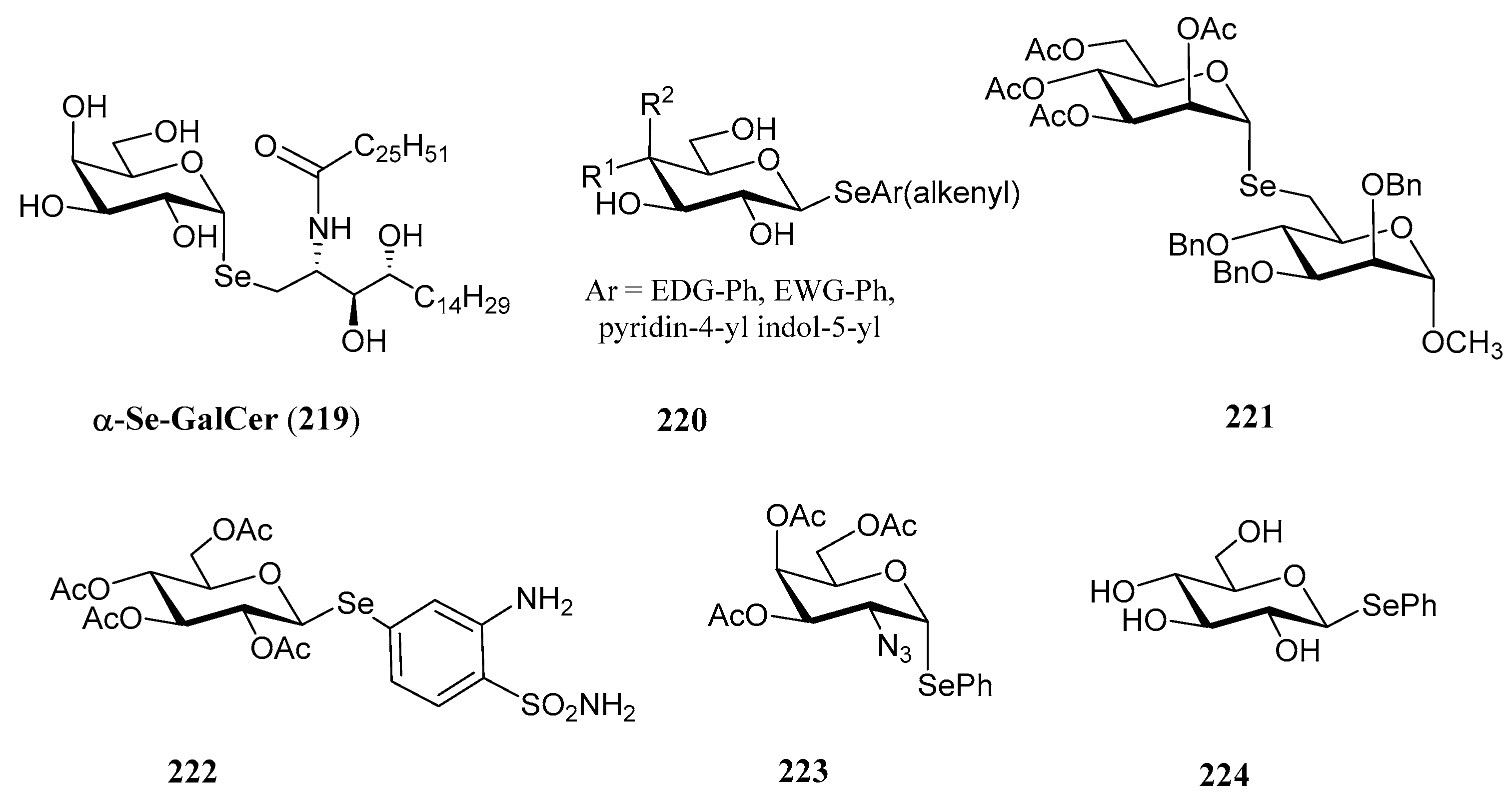
3.3. Selenoglycosides
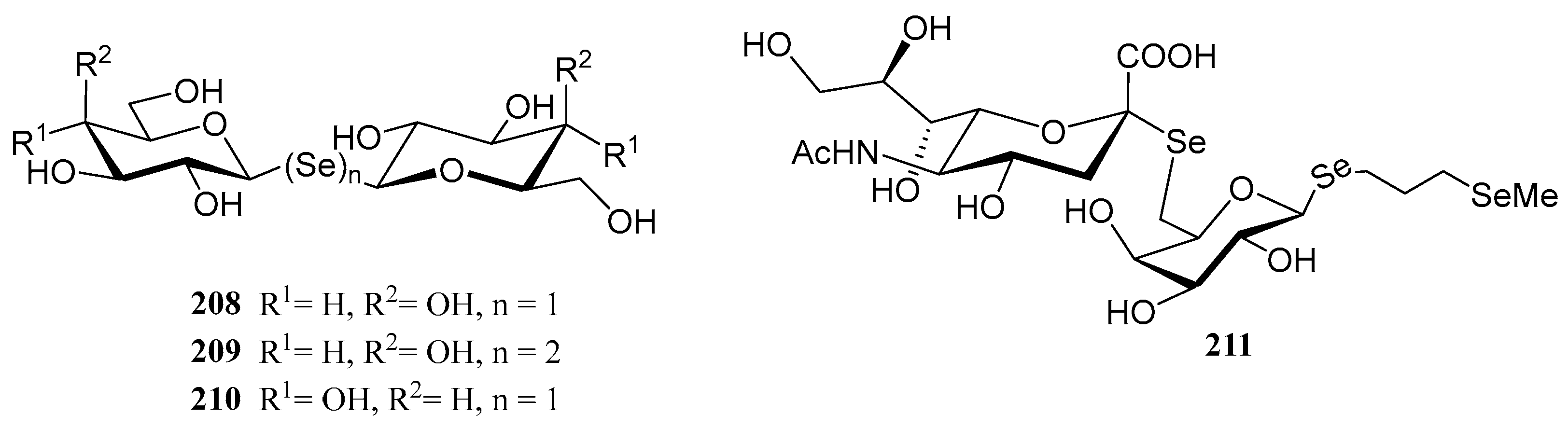
3.4. Miscellaneous Selenosugars
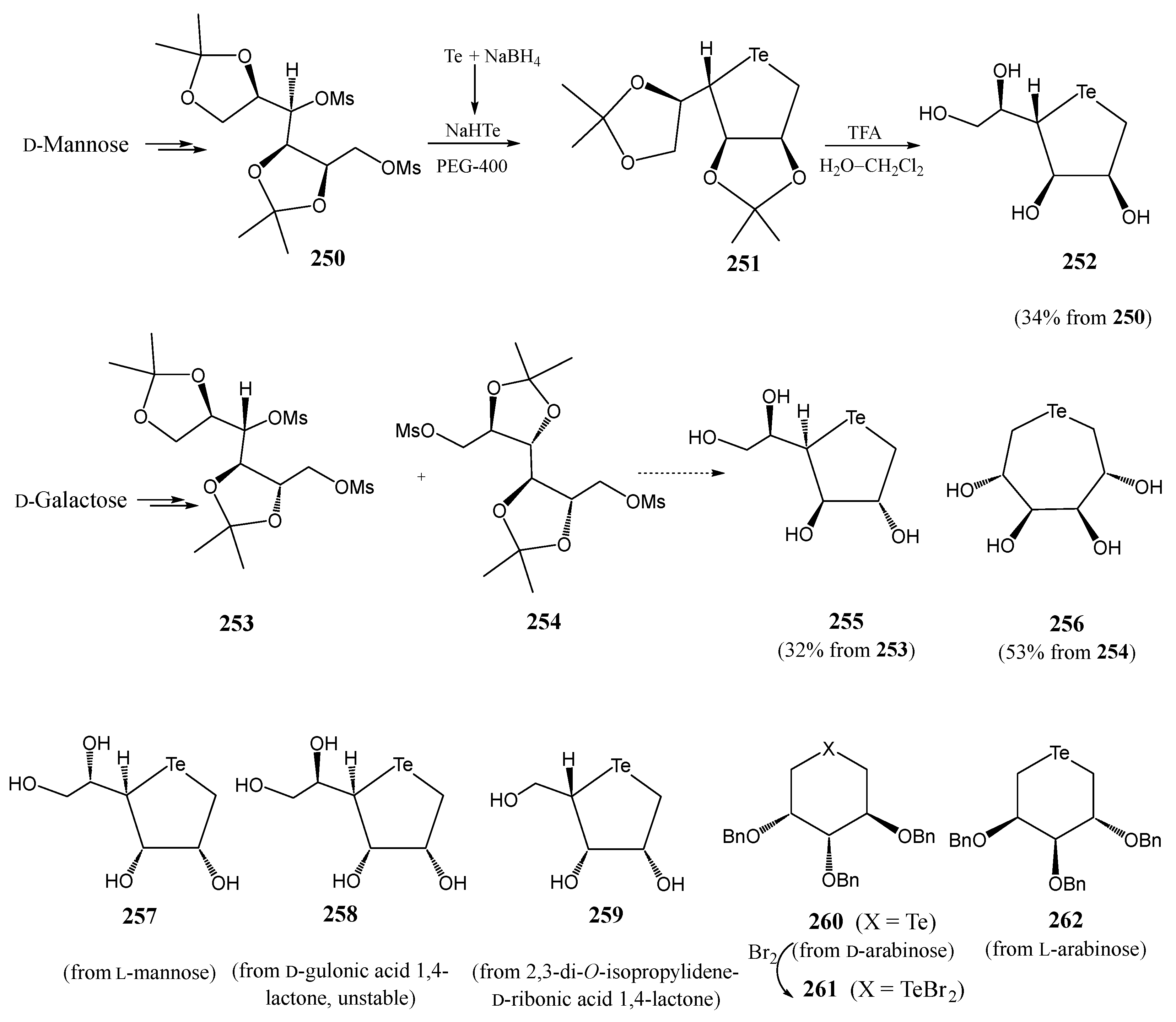
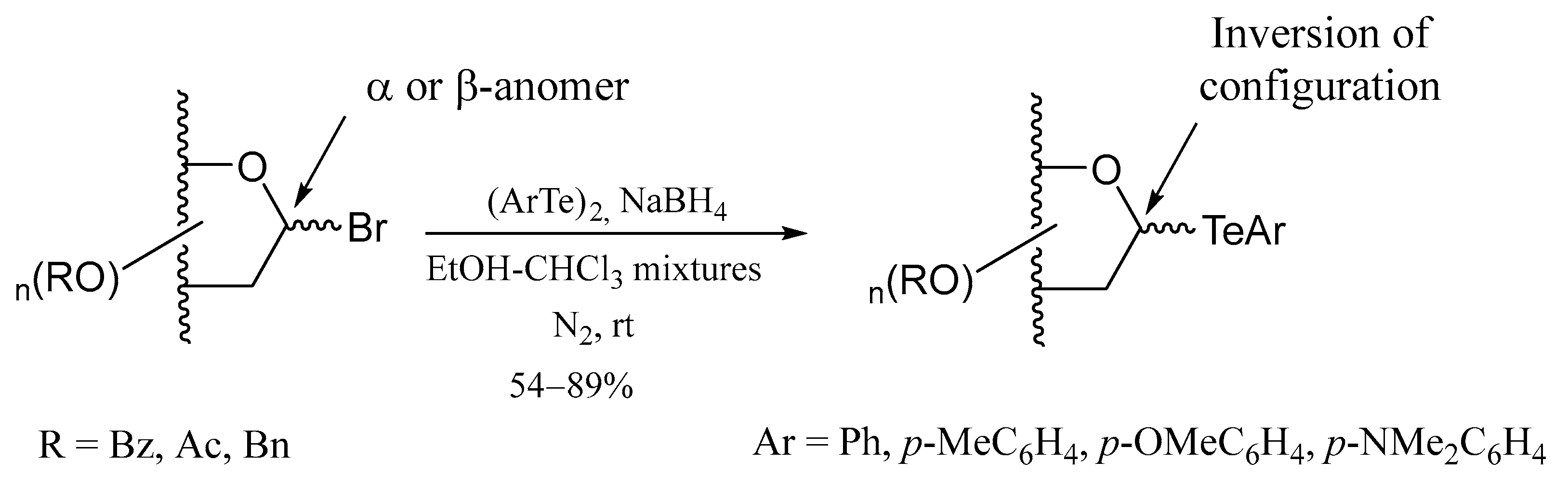
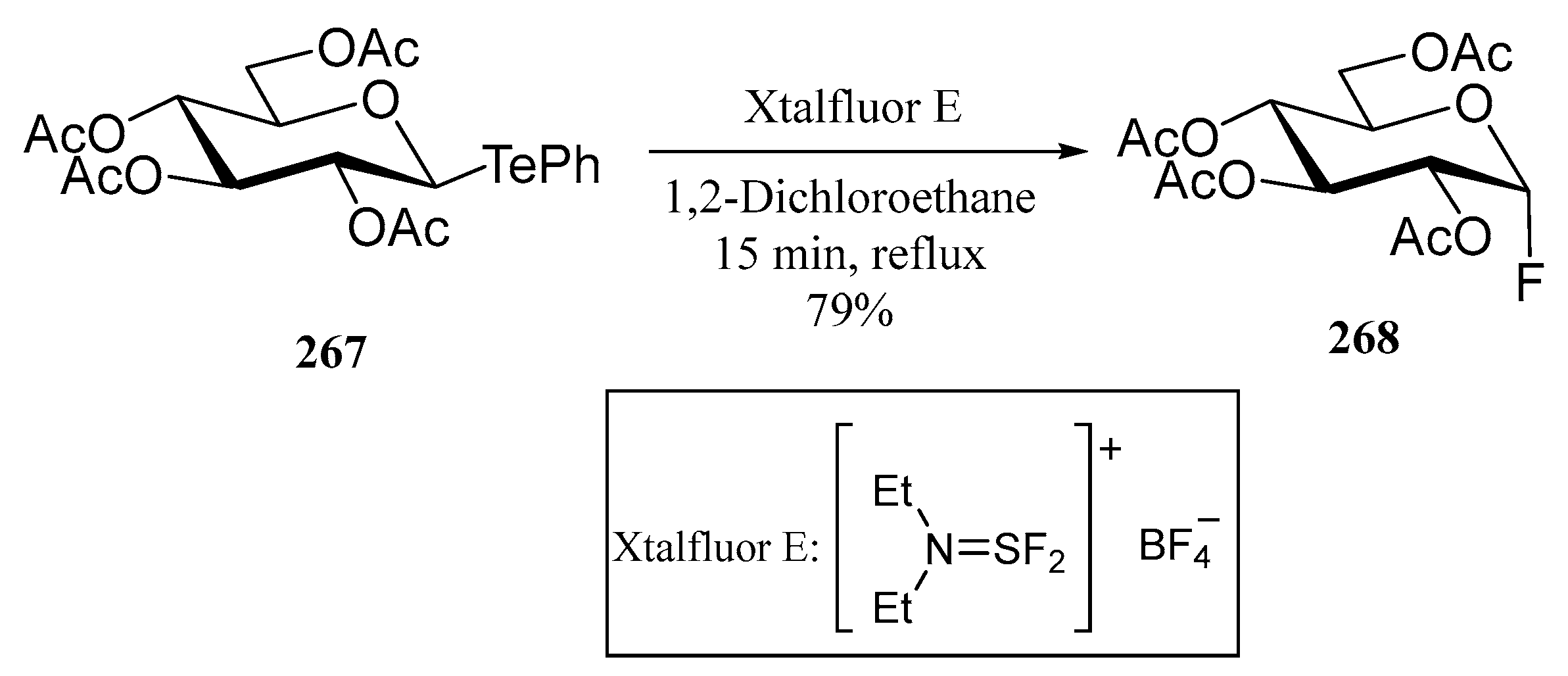
4. Te-Containing Carbohydrates
4.1. Tellurosugars
4.2. Telluroglycosides
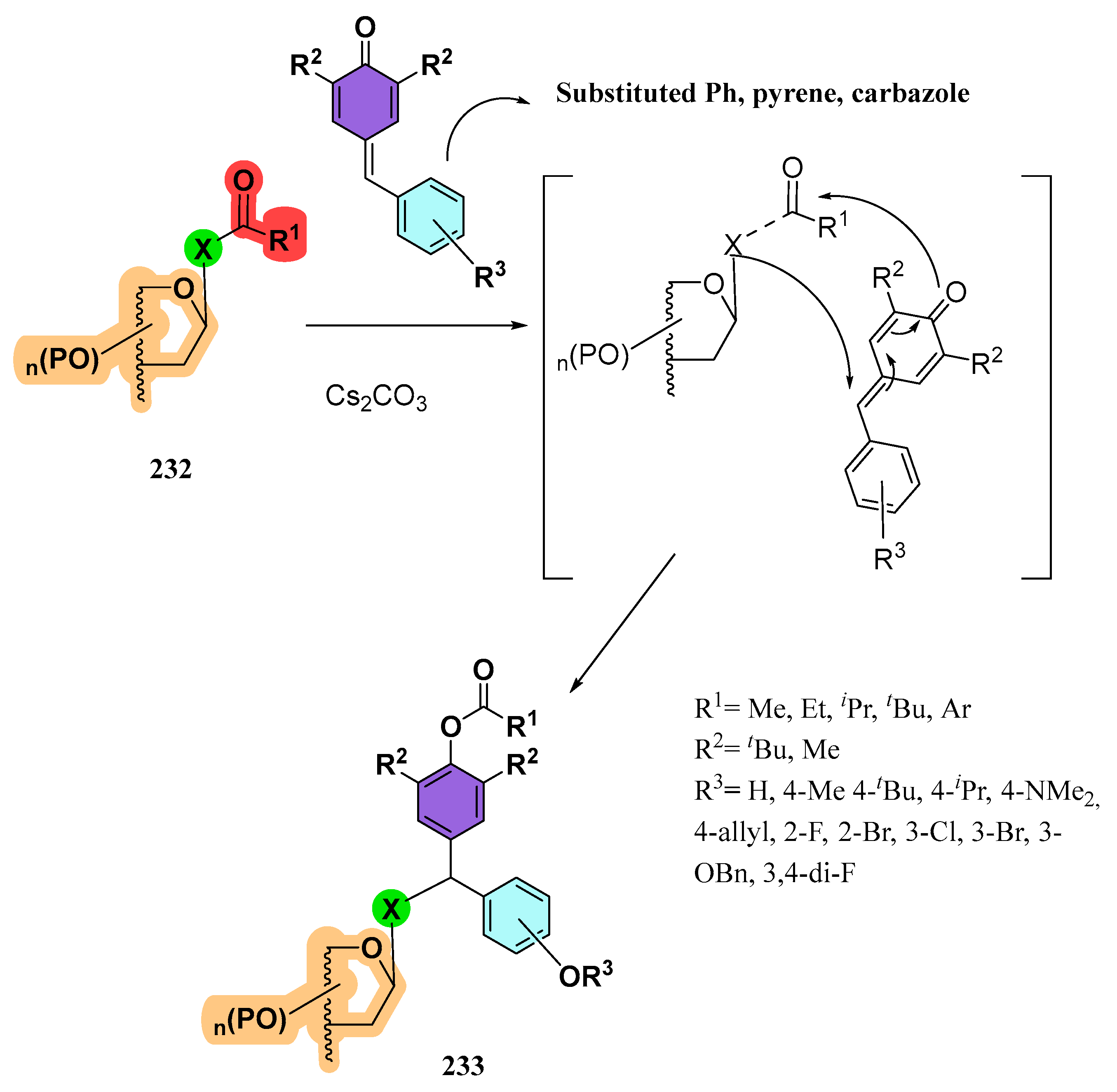
4.3. Sugar-Derived α-Alkoxyacyl Tellurides as Synthetic Intermediates
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| A3AR | A3 Adenosine Receptor |
| AllNAc | N-Acetylallosamine |
| B2pin2 | Bis(pinacolato)diboron |
| BSA | N,O-bis(trimethylsilyl)acetamide |
| BTD | Benzo-2,1,3-thiadiazole |
| C. diff. | Clostridioides difficile |
| Cer | Ceramide |
| CuAAC | Cu(I)-Catalyzed-Azide-Alkyne Cycloaddition |
| DBU | 1,8-Diazabicyclo(5.4.0)undec-7-ene |
| Di-tBubpy | 4,4-Di-tert-butyl-2,2–dipyridyl |
| DAST | Diethylaminosulfur trifluoride |
| DCM | Dichloromethane |
| DENV | Dengue Virus |
| DHS | Dihydroxy selenolane |
| DIAD | Diisopropyl azodicarboxylate |
| DMAP | 4-(N,N-Dimethylamino)pyridine |
| DNsNH | 2,4-(Dinitrobenzenesulfonyl)amino |
| DPAP | 2,2-Dimethoxy-2-phenylacetophenone |
| DPPA | Diphenylphosphoryl azide |
| DTT | Dithiothreitol |
| EDA | Electron-Donor-Aceptor |
| Fdx | Fidaxomicin |
| GHS | Glutahione |
| GLUT | Glucose Transporters |
| GPx | Glutathione peroxidase |
| HCV | Hepatitis C Virus |
| HEMA | Poly(hydroxyethyl methacrylate) |
| HFIP | Hexafluoroisopropanol |
| HOMO | Highest Occupied Molecular Orbital |
| LED | Light Emitting Diode |
| LOOH | Lipid peroxide |
| MCP-1 | Monocyte chemoattractant protein-1 |
| MGL | Metabolic Glycan Labelling |
| MIC | Minimum Inhibitory Concentration |
| MPO | Myeloperoxidase |
| MsOH | Methanesulfonic acid |
| MUC1 | Mucin 1 |
| NBS | N-Bromosuccinimide |
| NDI | Naphthalene diimide |
| NHC | N-Heterocyclic carbene |
| NIS | N-Iodosuccinimide |
| NMM | N-Methylmorpholine |
| OGA | O-GlcNAcase |
| PDI | Protein disulfide isomerases |
| PEGDA | Poly(ethyleneglycol diacrylate) |
| PPAR | Peroxisome Proliferator-Activated Receptor |
| PTA | Phosphotungstic acid |
| p-QMS | p-Quinone methides |
| RBCs | Red Blood Cells |
| ROS | Reactive Oxygen Species |
| SET | Single Electron Transfer |
| Skp2 | S-Phase Kinase Associated Protein 2 |
| TBDPS | Terc-Butyldiphenylsilyl |
| TEMPO | 2,2,6,6-Tetramethylpiperidine-1-oxyl, free radical |
| TIPDS | Tetraisopropyldisiloxanyl |
| TMSOTf | Trimethylsilyl triflate |
| TT | Thianthrenium |
| WGA | Wheat Germ Agglutin |
| XantPhos | 9,9-Dimethyl-9H-xanthene-4,5-diyl)bis(diphenylphosphane |
References
- Cao, X.; Du, X.; Jiao, H.; An, Q.; Chen, R.; Fang, P.; Wang, J.; Yu, B. Carbohydrate-based drugs launched during 2000−2021. Acta Pharm Sin B 2022, 12, 3783–3821. [Google Scholar] [CrossRef]
- Matassini, C.; Clemente, F.; Cardona, F. Carbohydrate-based therapeutics for lysosomal storage disorders. In Carbohydrate-Based Therapeutics; Adamo, R., Lay, L., Eds.; Wiley-VCH: Hoboken, NJ, USA, 2023; pp. 245–292. [Google Scholar] [CrossRef]
- Sorieul, C.; Papi, F.; Carboni, F.; Pecetta, S.; Phogat, S.; Adamo, R. Recent advances and future perspectives on carbohydrate-based cancer vaccines and therapeutics. Pharmacol. Ther. 2022, 235, 108158. [Google Scholar] [CrossRef]
- De Martos, A.M. Recent advances in the development and synthesis of carbohydrate-based molecules with promising antibacterial activity. Eur. J. Org. Chem. 2023, 26, e202200919. [Google Scholar] [CrossRef]
- Mosaiab, T.; Farr, D.C.; Kiefel, M.J.; Houston, T.A. Carbohydrate-based nanocarriers and their application to target macrophages and deliver antimicrobial agents. Adv. Drug Deliv. Rev. 2019, 151–152, 94–129. [Google Scholar] [CrossRef]
- Zorin, A.; Klenk, L.; Mack, T.; Deigner, H.-P.; Schmidt, M.S. Current synthetic approaches to the synthesis of carbasugars from non-carbohydrate sources. Top. Curr. Chem. 2022, 380, 12. [Google Scholar] [CrossRef]
- Clemente, F.; Matassini, C.; Cardona, F. Reductive amination routes in the synthesis of piperidine iminosugars. Eur. J. Org. Chem. 2020, 29, 4447–4462. [Google Scholar] [CrossRef]
- Hernández-Guerra, D.; Kennedy, A.R.; León, E.I.; Martín, Á.; Pérez-Martín, I.; Rodríguez, M.S.; Suárez, E. Synthetic approaches to phosphasugars (2-oxo-1,2-oxaphosphacyclanes) using the anomeric alkoxyl radical β-fragmentation reaction as the key step. J. Org. Chem. 2020, 85, 4861–4880. [Google Scholar] [CrossRef]
- Gupta, S.; Gauthier, C. 1-Thiosugars: From synthesis to applications. Curr. Org. Chem. 2025, 29, 359–401. [Google Scholar] [CrossRef]
- Mangiavacchi, F.; Dias, I.F.C.; Di Lorenzo, I.; Grzes, P.; Palomba, M.; Rosati, O.; Bagnoli, L.; Marini, F.; Santi, C.; Lenardao, E.J.; et al. Sweet selenium: Synthesis and properties of selenium-containing sugars and derivatives. Pharmaceuticals 2020, 13, 211. [Google Scholar] [CrossRef]
- Borges, E.L.; Ignasiak, M.T.; Velichenko, Y.; Perin, G.; Hutton, C.A.; Davies, M.J.; Schiesser, C.H. Synthesis and antioxidant capacity of novel stable 5-tellurofuranose derivatives. Chem. Commun. 2018, 54, 2990–2993. [Google Scholar] [CrossRef]
- Czubatka-Bieńkowska, A.; Sarnik, J.; Poplawski, T. Biological properties of (1–4)-thiodisaccharides. Carbohydr. Res. 2023, 533, 108934. [Google Scholar] [CrossRef]
- Bielski, R.; Mencer, D. New syntheses of thiosaccharides utilizing substitution reactions. Carbohydr. Res. 2023, 532, 108915. [Google Scholar] [CrossRef]
- Ahiadorme, D.; Crich, D. Entropy-enthalpy compensation in the methyl 5-thio-α-D-galactopyranoside–Jacalin interaction. Carbohydr. Res. 2025, 547, 109305. [Google Scholar] [CrossRef]
- Witczak, Z.J.; Culhane, J.M. Thiosugars: New perspectives regarding availability and potential biochemical and medicinal applications. Appl. Microbiol. Biotechnol. 2005, 69, 237–244. [Google Scholar] [CrossRef]
- Al Bujuq, N. Strategies for introducing sulfur atom in a sugar ring: Synthesis of 5-thioaldopyranoses and their NMR data. J. Sulfur Chem. 2019, 40, 664–702. [Google Scholar] [CrossRef]
- Ueda, A.; Pi, J.; Makura, Y.; Tanaka, M.; Uenishi, J. Stereoselective synthesis of (+)-5-thiosucrose and (+)-5-thioisosucrose. RSC Adv. 2020, 10, 9730–9735. [Google Scholar] [CrossRef]
- Díaz-Fernández, M.; Pino-González, S. Thiomonosaccharide derivatives from d-mannose. Chem. Proc. 2021, 3, 28. [Google Scholar] [CrossRef]
- Ma, S.-T.; Lee, C.-W.; Liu, W.-M. Synthesis of 4-thiol-furanosidic uronate via hydrothiolation reaction. RSC Adv. 2021, 11, 18409–18416. [Google Scholar] [CrossRef]
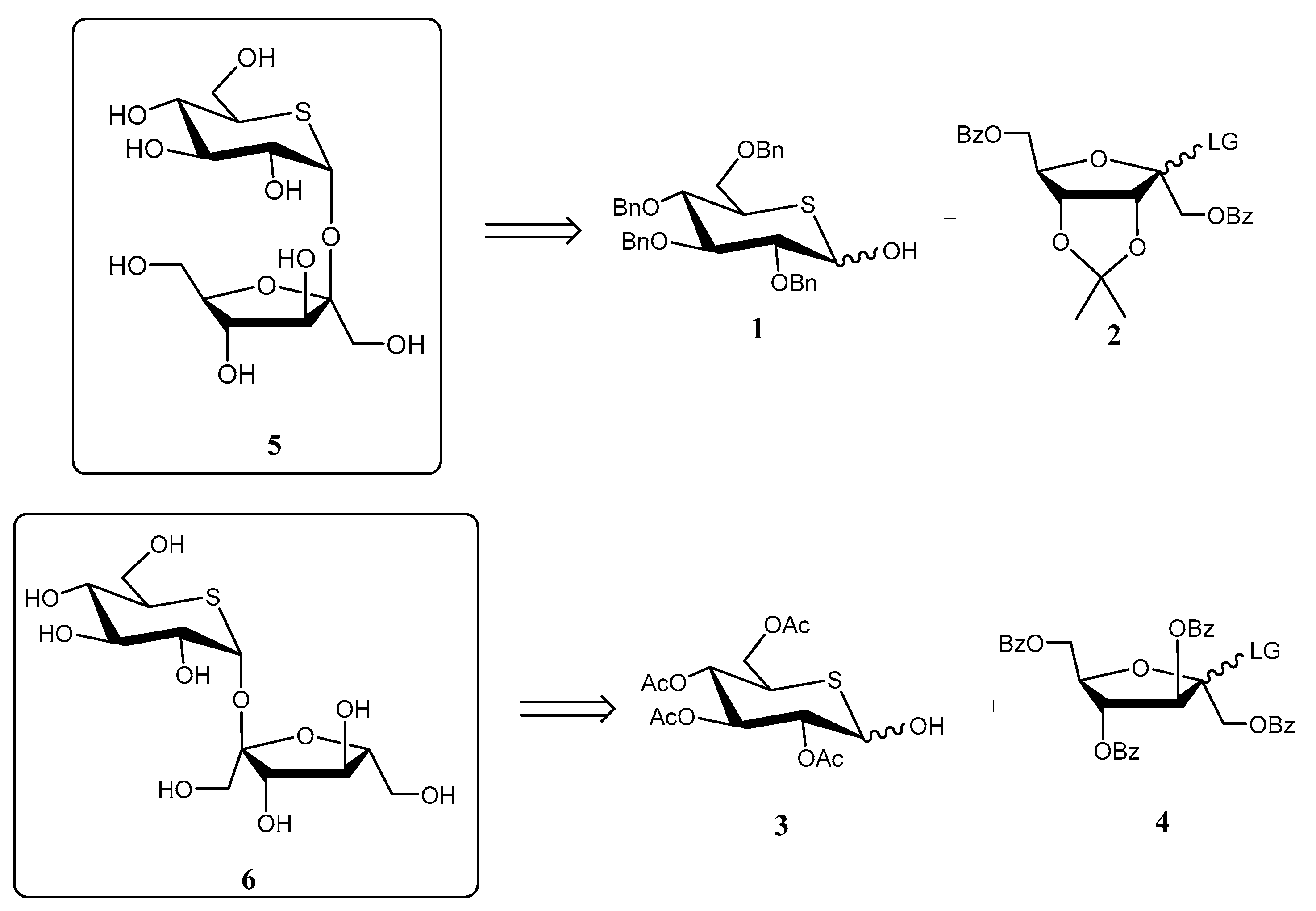
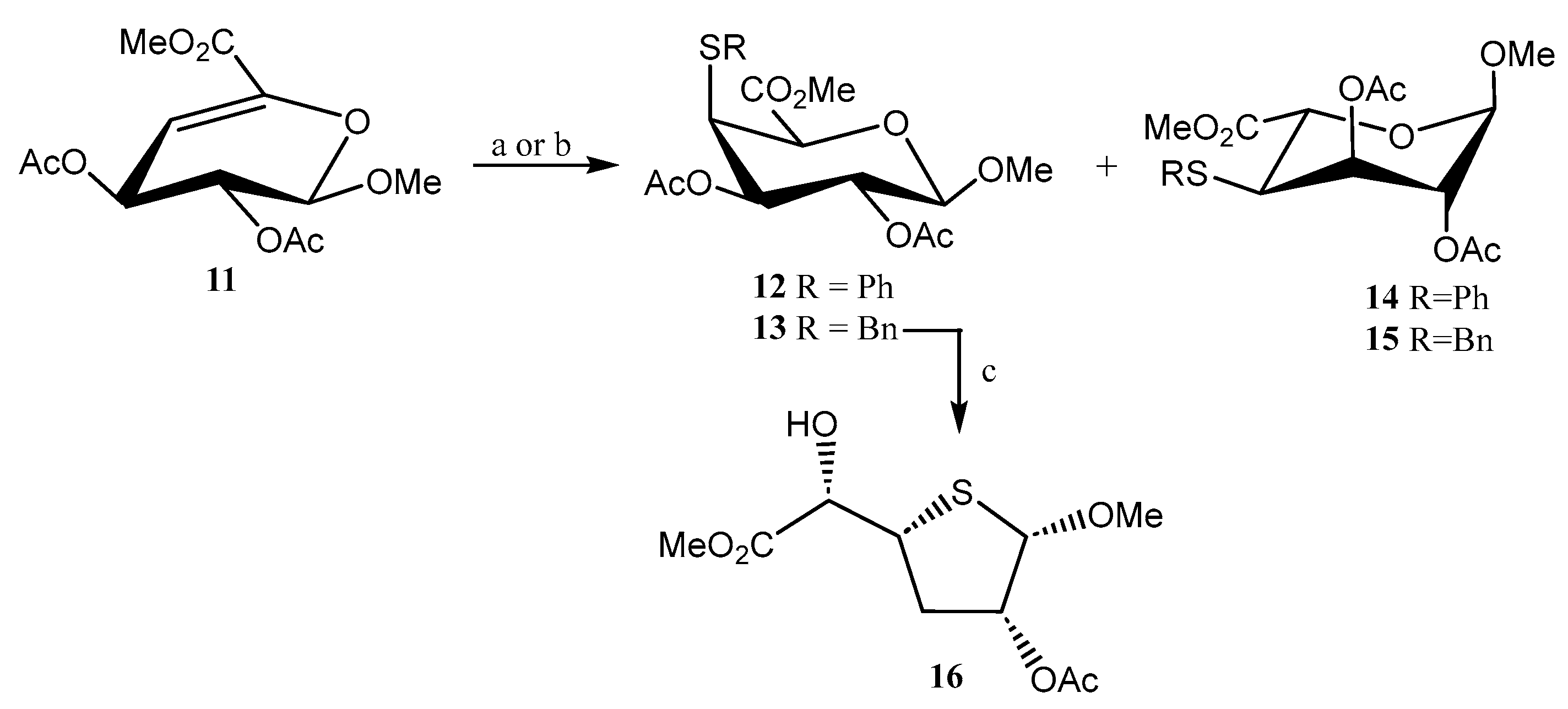
- Johnston, B.D.; Jensen, H.H.; Pinto, B.M. Synthesis of sulfonium sulfate analogues of disaccharides and their conversion to chain-extended homologues of salacinol: New glycosidase inhibitors. J. Org. Chem. 2006, 71, 1111–1118. [Google Scholar] [CrossRef]
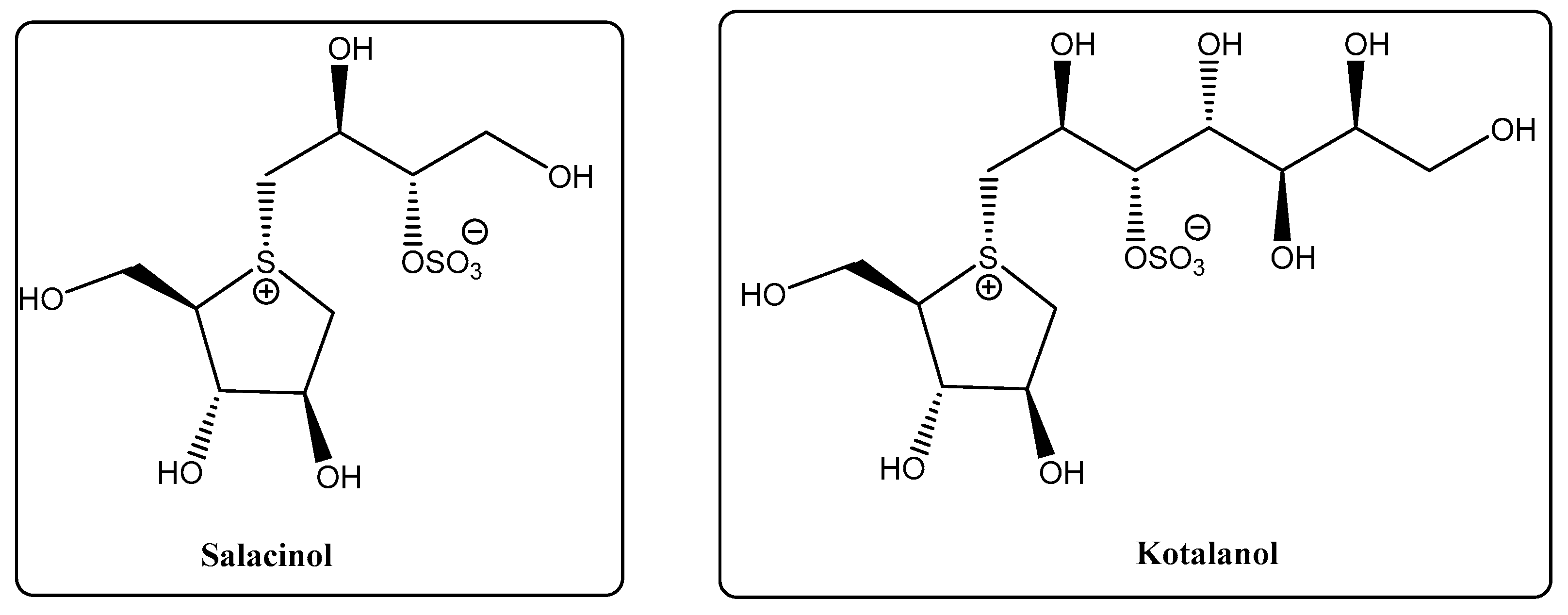
- Morikawa, T.; Ninomiya, K.; Tanabe, G.; Matsuda, H.; Yoshikawa, M.; Muraoka, O. A review of antidiabetic active thiosugar sulfoniums, salacinol and neokotalanol, from plants of the genus Salacia. J. Nat. Med. 2021, 75, 449–466. [Google Scholar] [CrossRef]
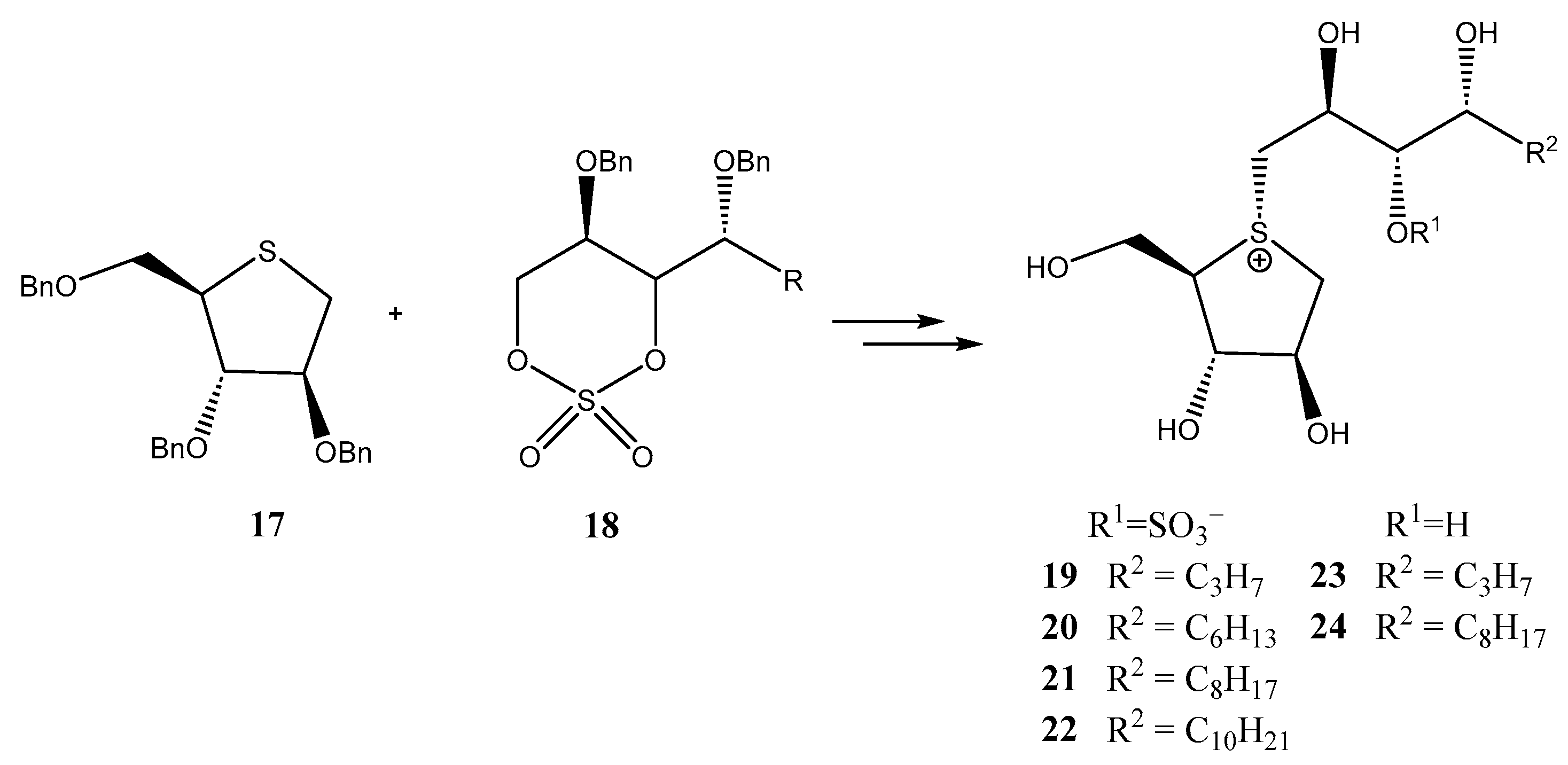
- Takashima, K.; Sakano, M.; Kinouchi, E.; Nakamura, S.; Marumoto, S.; Ishikawa, F.; Ninomiya, K.; Nakanishi, I.; Morikawa, T.; Tanabe, G. Elongation of the side chain by linear alkyl groups increases the potency of salacinol, a potent α-glucosidase inhibitor from the Ayurvedic traditional medicine “Salacia”, against human intestinal maltase. Bioorg. Med. Chem. Lett. 2021, 33, 127751. [Google Scholar] [CrossRef] [PubMed]
- Takashima, K.; Nakamura, S.; Nagayama, M.; Marumoto, S.; Ishikawa, F.; Xie, W.; Nakanishi, I.; Muraoka, O.; Morikawa, T.; Tanabe, G. Role of the thiosugar ring in the inhibitory activity of salacinol, a potent natural α-glucosidase inhibitor. RSC Adv. 2024, 14, 4471–4481. [Google Scholar] [CrossRef] [PubMed]
- Fowler, P.A.; Haines, A.H.; Taylor, R.J.K.; Chrystal, E.J.T.; Gravestock, M.B. Synthesis and biological activity of acyclic analogues of nojirimycin. J. Chem. Soc. Perkin Trans. 1 1994, 1, 2229–2235. [Google Scholar] [CrossRef]
- Ishikawa, F.; Jinno, K.; Kinouchi, E.; Ninomiya, K.; Marumoto, S.; Xie, W.; Muraoka, O.; Morikawa, T.; Tanabe, G. Diastereoselective synthesis of salacinol-type α-glucosidase inhibitors. J. Org. Chem. 2018, 83, 185–193. [Google Scholar] [CrossRef]
- Crawford, C.J.; Seeberger, P.H. Advances in glycoside and oligosaccharide synthesis. Chem. Soc. Rev. 2023, 52, 7773–7801. [Google Scholar] [CrossRef]
- Bennai, N.; Chabrier, A.; Fatthalla, M.I.; Tran, C.; Yen-Pon, E.; Belkadi, M.; Alami, M.; Grimaud, L.; Messaoudi, S. Reversing reactivity: Stereoselective desulfurative 1,2-trans-O-glycosylation of anomeric thiosugars with carboxylic acids under copper or cobalt catalysis. J. Org. Chem. 2020, 85, 8893–8909. [Google Scholar] [CrossRef]
- Banisalman, K.A.F.; Polykandritou, A.; Barnieh, F.M.; Morais, G.R.; Falconer, R.A. Chemoselective solution- and solid-phase synthesis of disulfide linked glycopeptides. J. Org. Chem. 2022, 87, 14026–14036. [Google Scholar] [CrossRef]
- Neralkar, M.; Tian, L.; Redman, R.L.; Krauss, I.J. Synthesis of mannosidase-stable Man3 and Man4 glycans containing S-linked Manα1→2Man termini. Org. Lett. 2021, 23, 3053–3057. [Google Scholar] [CrossRef]
- Morrone-Pozzuto, P.; Uhrig, M.L.; Agusti, R. Synthesis of oligosaccharides containing the S-Galp(α1 → 3)Galp unit, glycomimetic of the epitope recognized by lytic antibodies. J. Org. Chem. 2022, 87, 13455–13468. [Google Scholar] [CrossRef]
- Bege, M.; Singh, V.; Sharma, N.; Debreczeni, N.; Bereczki, I.; Poonam; Herczegh, P.; Rathi, B.; Singh, S.; Borbás, A. In vitro and in vivo antiplasmodial evaluation of sugar-modified nucleoside analogues. Sci. Rep. 2023, 13, 12228. [Google Scholar] [CrossRef]
- Azzam, R.A.; Gad, N.M.; Elgemeie, G.H. Novel thiophene thioglycosides substituted with the benzothiazole moiety: Synthesis, characterization, antiviral and anticancer evaluations, and NS3/4A and USP7 enzyme inhibitions. ACS Omega 2022, 7, 35656–35667. [Google Scholar] [CrossRef]
- de la Luz Quintana, I.; Paul, A.; Chowdhury, A.; Moulton, K.D.; Kulkarni, S.S.; Dube, D.H. Thioglycosides act as metabolic inhibitors of bacterial glycan biosynthesis. ACS Infect. Dis. 2023, 9, 2025–2035. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-S.; Gao, X.; del Solar, V.; Yu, X.; Antonopoulos, A.; Friedman, A.E.; Matich, E.K.; Atilla-Gokcumen, G.E.; Nasirikenari, M.; Lau, J.T.; et al. Thioglycosides are efficient metabolic decoys of glycosylation that reduce selectin dependent leukocyte adhesion. Cell Chem. Biol. 2018, 25, 1519–1532. [Google Scholar] [CrossRef]
- He, P.; Zhang, X.; Xia, K.; Green, D.E.; Baytas, S.; Xu, Y.; Pham, T.; Liu, J.; Zhang, F.; Almond, A.; et al. Chemoenzymatic synthesis of sulfur-linked sugar polymers as heparanase inhibitors. Nat. Commun. 2022, 13, 7438. [Google Scholar] [CrossRef]
- Burrini, N.; Pâris, A.; Collet, G.; Lafite, P.; Daniellou, R. Biocatalytic synthesis of coumarin S-glycosides: Towards non-cytotoxic probes for biomedical imaging and sensing. Molecules 2024, 29, 1322. [Google Scholar] [CrossRef]
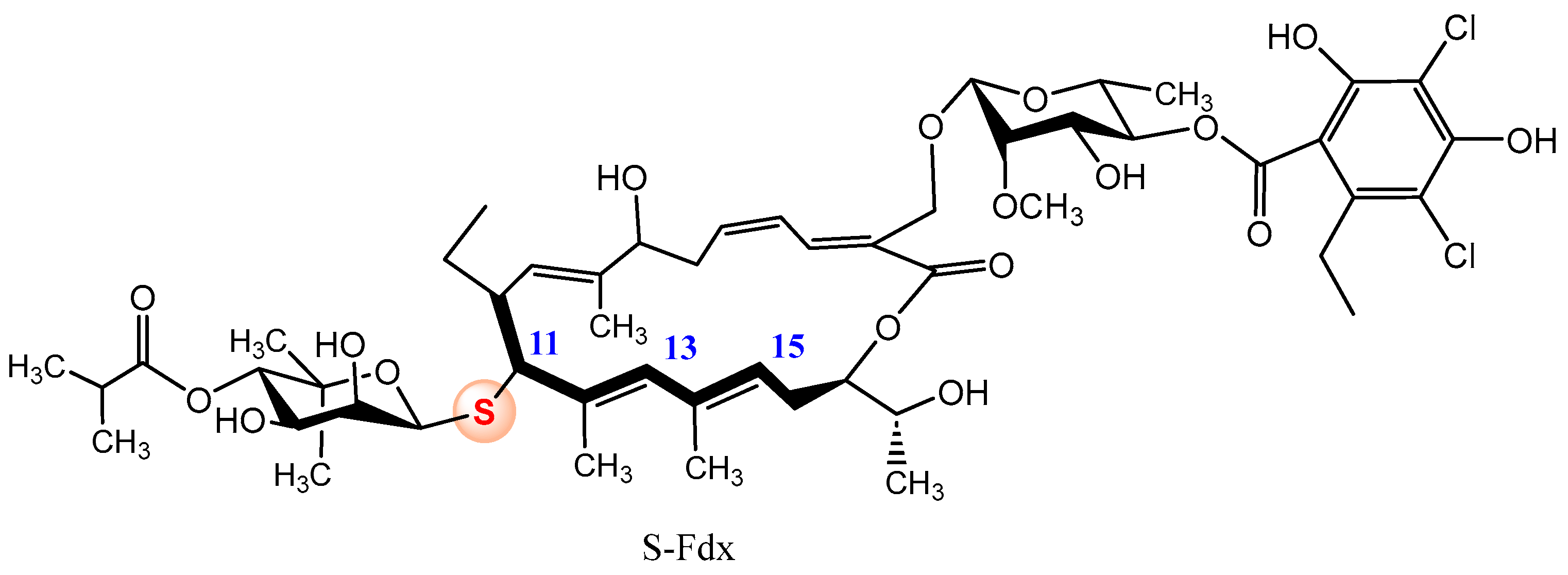
- Ferrara, I.; Chesnokov, G.A.; Dittmann, S.; Blacque, O.; Sievers, S.; Gademann, K. Formal single atom editing of the glycosylated natural product fidaxomicin improves acid stability and retains antibiotic activity. JACS Au 2024, 4, 2267–2280. [Google Scholar] [CrossRef]
- Chen, J.-S.; Lo, T.-C.; Hsieh, Y.-C.; Chen, C.-H.; Lin, M.; Lin, H.-Y.; Hung, M.-W.; Wu, H.-R.; Luo, S.-Y. Utilizing reusable catalyst phosphotungstic acid for the synthesis of thioglycoside from per-O-acetyl saccharides with microwave assisted and de-O-acetylation with methanol. ACS Omega 2023, 8, 8885–8893. [Google Scholar] [CrossRef]
- Feng, G.-J.; Luo, T.; Guo, Y.-F.; Liu, C.-Y.; Dong, H. Concise synthesis of 1-thioalkyl glycoside donors by reaction of per-O-acetylated sugars with sodium alkanethiolates under solvent-free conditions. J. Org. Chem. 2022, 87, 3638–3646. [Google Scholar] [CrossRef]
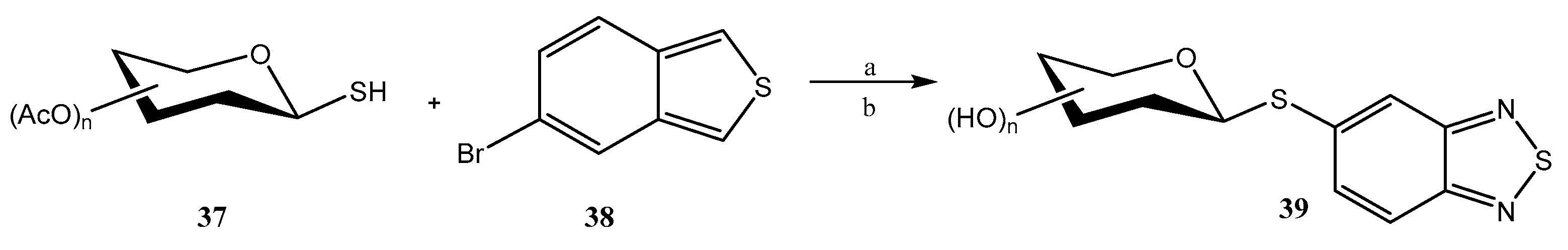
- Santos, B.F.; Silva, B.A.L.; Domingues, N.L.C. Pd-catalyzed functionalization of benzo-2,1,3-thiadiazole at the C-5-position using 1-thiosugars. New, J. Chem. 2022, 46, 19785–19789. [Google Scholar] [CrossRef]
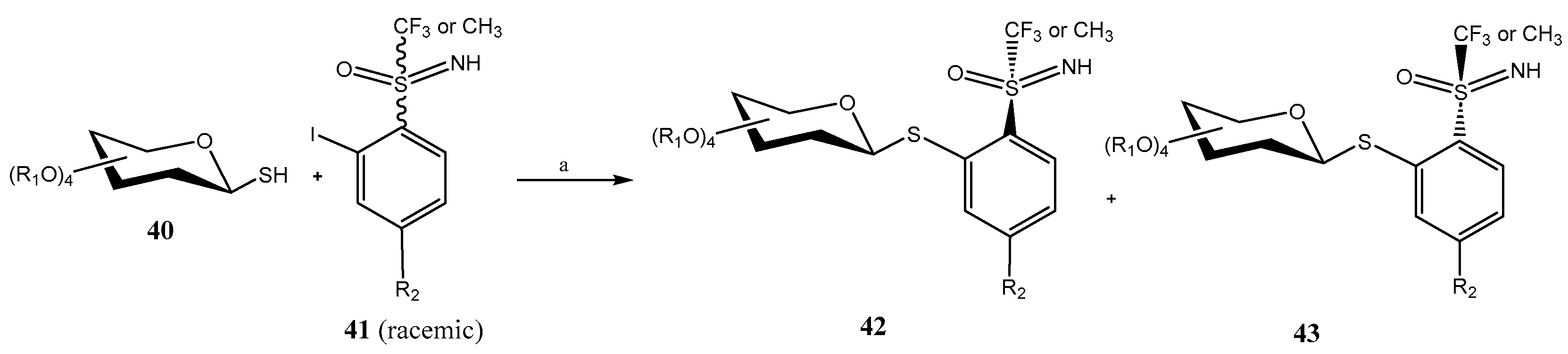
- Bennai, N.; Ibrahim, N.; Marrot, J.; Belkadi, M.; Alami, M.; Magnier, E.; Anselmi, E.; Messaoudi, S. Synthesis of S-trifluoromethyl S-arylsulfoximine thioglycosides through Pd-catalyzed Migita cross-coupling. Eur. J. Org. Chem. 2020, 2020, 4972–4981. [Google Scholar] [CrossRef]
- Wu, Y.; Zhou, W.-J.; Yao, L.; Niu, Y.; Zhao, H.; Peng, C.; Han, B.; Huang, W.; Zhan, G. Organocatalytic atroposelective synthesis of naphthoquinone thioglycosides from aryl-naphthoquinones and thiosugars. Chem. Commun. 2023, 59, 7279–7282. [Google Scholar] [CrossRef] [PubMed]
- Dubey, A.; Mandal, P.K. An Efficient one-pot protocol for direct access to diarylmethyl thioglycosides with para-quinone methides via S-glycosyl isothiouronium salts. Synlett 2020, 31, 1713–1719. [Google Scholar] [CrossRef]
- Venkatesh, R.; Tiwari, V.; Kandasamy, J. Copper(I)-catalyzed Sandmeyer-type S-arylation of 1-thiosugars with aryldiazonium salts under mild conditions. J. Org. Chem. 2022, 87, 11414–11432. [Google Scholar] [CrossRef] [PubMed]
- Feng, G.-J.; Guo, Y.-F.; Tang, Y.; Li, M.; Jia, Y.; Li, Z.; Wang, S.; Liu, H.; Wu, Y.; Dong, H. Design, synthesis, and biological evaluation of thioglucoside analogues of gliflozin as potent new gliflozin drugs. J. Med. Chem. 2023, 66, 12536–12543. [Google Scholar] [CrossRef]
- Feng, G.-J.; Wang, S.-S.; Lv, J.; Luo, T.; Wu, Y.; Dong, H. Improved synthesis of 1-glycosyl thioacetates and its application in the synthesis of thioglucoside gliflozin analogues. Eur. J. Org. Chem. 2021, 2021, 2940–2949. [Google Scholar] [CrossRef]
- Li, S.; Wang, Y.; Zhong, L.; Wang, S.; Liu, Z.; Dai, Y.; He, Y.; Feng, Z. Boron-promoted umpolung reaction of sulfonyl chlorides for the stereospecific synthesis of thioglycosides via reductive deoxygenation coupling reactions. Org. Lett. 2022, 24, 2463–2468. [Google Scholar] [CrossRef]
- Malapit, C.A.; Prater, M.B.; Cabrera-Pardo, J.R.; Li, M.; Pham, T.D.; McFadden, T.P.; Blank, S.; Minteer, S.D. Advances on the merger of electrochemistry and transition metal catalysis for organic synthesis. Chem. Rev. 2022, 122, 3180–3218. [Google Scholar] [CrossRef]
- Li, F.; Liu, H.; Xing, W.; Zhang, Q.; Wang, L. Electrochemical nickel-catalyzed cross-coupling of glycosyl thiols with preactivated phenols and ketones. Org. Biomol. Chem. 2024, 22, 3597–3601. [Google Scholar] [CrossRef]
- Zhu, M.; Alami, M.; Messaoudi, S. Electrochemical nickel-catalyzed Migita cross-coupling of 1-thiosugars with aryl, alkenyl and alkynyl bromides. Chem. Commun. 2020, 56, 4464–4467. [Google Scholar] [CrossRef]
- Wang, R.-Q.; Jiang, Q.-H.; Wang, H.-X.; Zhang, X.-W.; Yan, N. Electrochemically mediated S-glycosylation of 1-thiosugars with xanthene derivatives. Org. Lett. 2023, 25, 4252–4257. [Google Scholar] [CrossRef]
- Zhu, M.; Ghouilem, J.; Messaoudi, S. Visible-light-mediated Stadler−Ziegler arylation of thiosugars with anilines. ACS Org. Inorg. Au. 2022, 2, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Liang, Q.; Shi, L.; Wen, J.; Liu, X.; Hong, X.; Zha, X.; Ji, F. Site-selective S-arylation of 1-thiosugars with aryl thianthrenium salts through copper(I)-mediated, photoredox catalyzed reactions. Adv. Synth. Catal. 2024, 36, 2344–2351. [Google Scholar] [CrossRef]
- Zhang, L.; He, S.; Hou, J.; Ye, M.; Chen, J.; Lv, G.; Huang, T.; Yang, Z.; Wu, Y. Visible-light-mediated synthesis of non-anomeric S-aryl glycosides via a photoactive electron-donor–acceptor complex. Chem. Commun. 2023, 59, 13759–13762. [Google Scholar] [CrossRef]
- Belmonte-Reche, E.; Benassi, A.; Peñalver, P.; Cucchiarini, A.; Guédin, A.; Mergny, J.L.; Rosu, F.; Gabelica, V.; Freccero, M.; Doria, F.; et al. Thiosugar naphthalene diimide conjugates: G-quadruplex ligands with antiparasitic and anticancer activity. Eur. J. Med. Chem. 2022, 232, 114183. [Google Scholar] [CrossRef]
- Ceccherini, V.; Giorgi, E.; Mannelli, M.; Cirri, D.; Gamberi, T.; Gabbiani, C.; Pratesi, A. Synthesis, chemical characterization, and biological evaluation of hydrophilic gold(I) and silver(I) N-heterocyclic carbenes as potential anticancer agents. Inorg. Chem. 2024, 63, 16949–16963. [Google Scholar] [CrossRef]
- Ryzhakov, D.; Beillard, A.; Le Bideau, F.; Al-Shuaeeb, R.A.A.; Alami, M.; Bantreil, X.; Bonnemoy, A.; Gautier, A.; Lamaty, F.; Messaoudi, S. Azoliums and Ag(I)-N-heterocyclic carbene thioglycosides: Synthesis, reactivity and bioactivity. Eur. J. Org. Chem. 2022, 2022, e202101499. [Google Scholar] [CrossRef]
- Cristófalo, A.E.; Cagnoni, A.J.; Uhrig, M.L. Synthesis of N-acetylglucosamine and N-acetylallosamine resorcinarene-based multivalent β-thio-glycoclusters: Unexpected affinity of N-acetylallosamine ligands towards wheat germ agglutinin. Org. Biomol. Chem. 2020, 18, 6853–6865. [Google Scholar] [CrossRef]
- Hopkinson, M.N.; Richter, C.; Schedler, M.; Glorius, F. An Overview of N-heterocyclic carbenes. Nature 2014, 510, 485–496. [Google Scholar] [CrossRef]
- Shit, P.; Kundu, M.; Misra, A.K. Straightforward stereoselective synthesis of 1-thio-β-d-mannosides and 1-thio-β -l-rhamnosides. Tetrahedron Lett. 2023, 118, 154391. [Google Scholar] [CrossRef]
- Csávás, M.; Eszenyi, D.; Mező, E.; Lázár, L.; Debreczeni, N.; Tóth, M.; Somsák, L.; Borbás, A. Stereoselective synthesis of carbon-sulfur-bridged glycomimetics by photoinitiated thiol-ene coupling reactions. Int. J. Mol. Sci. 2020, 21, 573. [Google Scholar] [CrossRef]
- Reintjens, N.R.M.; Witte, M.D.; Minnaard, A.J. Site-selective introduction of thiols in unprotected glycosides. Org. Biomol. Chem. 2023, 21, 5098–5103. [Google Scholar] [CrossRef] [PubMed]
- Kern, M.K.; Pohl, N.L.B. Automated solution-phase synthesis of S-glycosides for the production of oligomannopyranoside derivatives. Org. Lett. 2020, 22, 4156–4159. [Google Scholar] [CrossRef] [PubMed]
- Meena; Sam, M.; Pierce, K.; Szostak, J.W.; McLaughlin, L.W. 2′,3′-Dideoxy-3′-thionucleoside triphosphates: Syntheses and polymerase substrate activities. Org. Lett. 2007, 9, 1161–1163. [Google Scholar] [CrossRef]
- Zargar, I.A.; Sakander, N.; Mukherjee, D. 2-Ketophenyl assisted biomimetic synthesis of 3-thio substituted glycals at room temperature. Eur. J. Org. Chem. 2023, 26, e202300780. [Google Scholar] [CrossRef]
- Liu, Y.; Jiao, Y.; Luo, H.; Huang, N.; Lai, M.; Zou, K.; Yao, H. Catalyst-controlled regiodivergent synthesis of 1- and 3-thiosugars with high stereoselectivity and chemoselectivity. ACS Catal. 2021, 11, 5287–5293. [Google Scholar] [CrossRef]
- Qin, K.; Zhang, H.; Zhao, Z.; Chen, X. Protein S-glyco-modification through an elimination–addition mechanism. J. Am. Chem. Soc. 2020, 142, 9382–9388. [Google Scholar] [CrossRef]
- Shit, P.; Kundu, M.; Misra, A.K. Expeditious preparation of 1,6-anhydro-1-thio-β-d-hexopyranose derivatives. Carbohydr. Res. 2023, 525, 108765. [Google Scholar] [CrossRef]
- Kundu, M.; Misra, A.K. Direct synthesis of unsymmetrical glycosyl disulfides from glycosyl bromides. Eur. J. Org. Chem. 2021, 2021, 3759–3767. [Google Scholar] [CrossRef]
- Anghinoni, J.M.; Birmann, P.T.; da Rocha, M.J.; Gomes, C.S.; Davies, M.J.; Brüning, C.A.; Savegnago, L.; Lenardão, E.J. Recent advances in the synthesis and antioxidant activity of low molecular mass organoselenium molecules. Molecules 2023, 28, 7349. [Google Scholar] [CrossRef]
- Jain, V.K.; Priyadarsini, K.I. Selenium compounds as promising antiviral agents. New J. Chem. 2024, 48, 6534–6552. [Google Scholar] [CrossRef]
- Ibáñez-Escribano, A.; Fonseca-Berzal, C.; Martínez-Montiel, M.; Álvarez-Márquez, M.; Gómez-Núñez, M.; Lacueva-Arnedo, M.; Espinosa-Buitrago, T.; Martín-Pérez, T.; Escario, J.A.; Merino-Montiel, P.; et al. Thio- and selenosemicarbazones as antiprotozoal agents against Trypanosoma cruzi and Trichomonas vaginalis. J. Enzyme Inhib. Med. Chem. 2022, 37, 781–791. [Google Scholar] [CrossRef] [PubMed]
- Pyka, P.; Garbo, S.; Fioravanti, R.; Jacob, C.; Hittinger, M.; Handzlik, J.; Zwergel, C.; Battistelli, C. Selenium-containing compounds: A new hope for innovative treatments in Alzheimer’s disease and Parkinson’s disease. Drug Discov. Today 2024, 29, 104062. [Google Scholar] [CrossRef] [PubMed]
- Velueta-Viveros, M.; Martínez-Bailén, M.; Puerta, A.; Romero-Hernández, L.L.; Křen, V.; Merino-Montiel, P.; Montiel-Smith, S.; Fernandes, M.X.; Moreno-Vargas, A.J.; Padrón, J.M.; et al. Carbohydrate-derived bicyclic selenazolines as new dual inhibitors (cholinesterases/OGA) against Alzheimer’s disease. Bioorg. Chem. 2022, 127, 105983. [Google Scholar] [CrossRef] [PubMed]
- Roldán-Peña, J.M.; Alejandre-Ramos, D.; López, Ó.; Maya, I.; Lagunes, I.; Padrón, J.M.; Peña-Altamira, L.E.; Bartolini, M.; Monti, B.; Bolognesi, M.L.; et al. New tacrine dimers with antioxidant linkers as dual drugs: Anti-Alzheimer’s and antiproliferative agents. Eur. J. Med. Chem. 2017, 138, 761–773. [Google Scholar] [CrossRef]
- Roldán-Peña, J.M.; Puerta, A.; Dinić, J.; Stojanov, S.J.; González-Bakker, A.; Hicke, F.J.; Mishra, A.; Piyasaengthong, A.; Maya, I.; Walton, J.W.; et al. Biotinylated selenocyanates: Potent and selective cytostatic agents. Bioorg. Chem. 2023, 133, 106410. [Google Scholar] [CrossRef]
- Begines, P.; Martos, S.; Lagunes, I.; Maya, I.; Padrón, J.M.; López, Ó.; Fernández-Bolaños, J.G. Chemoselective preparation of new families of phenolic-organoselenium hybrids-a biological assessment. Molecules 2022, 27, 1315. [Google Scholar] [CrossRef]
- Ramos-Inza, S.; Plano, D.; Sanmartín, C. Metal-based compounds containing selenium: An appealing approach towards novel therapeutic drugs with anticancer and antimicrobial effects. Eur. J. Med. Chem. 2022, 244, 114834. [Google Scholar] [CrossRef]
- Begines, P.; Sevilla-Horrillo, L.; Puerta, A.; Puckett, R.; Bayort, S.; Lagunes, I.; Maya, I.; Padrón, J.M.; López, Ó.; Fernández-Bolaños, J.G. Masked phenolic-selenium conjugates: Potent and selective antiproliferative agents overcoming P-gp resistance. Pharmaceuticals 2020, 13, 358. [Google Scholar] [CrossRef]
- Lagunes, I.; Begines, P.; Silva, A.; Galán, A.R.; Puerta, A.; Fernandes, M.X.; Maya, I.; Fernández-Bolaños, J.G.; López, Ó.; Padrón, J.M. Selenocoumarins as new multitarget antiproliferative agents: Synthesis, biological evaluation and in silico calculations. Eur. J. Med. Chem. 2019, 179, 493–501. [Google Scholar] [CrossRef]
- Blumberg, K.; Fuccello, A.; van Es, T. Selenium derivatives of l-arabinose, d-ribose, and d-xylose. Carbohydr. Res. 1977, 59, 351–362. [Google Scholar] [CrossRef]
- Lucas, M.A.; Nguyen, O.T.K.; Schiesser, C.H.; Zheng, S.-L. Preparation of 5-selenopentopyranose sugars from pentose starting materials by samarium(II) iodide or (phenylseleno)formate mediated ring closures. Tetrahedron 2000, 56, 3995–4000. [Google Scholar] [CrossRef]
- Schiesser, C.H. The quest for selenocycles: From an ESR spectrum to a commercial product. J. Chem. Res. 2022, 46. [Google Scholar] [CrossRef]
- Liu, H.; Pinto, M.B. Synthesis of zwitterionic selenonium and sulfonium sulfates from d-mannose as potential glycosidase inhibitors. Can. J. Chem. 2006, 84, 497–505. [Google Scholar] [CrossRef]
- Schiesser, C.H.; Storkey, C.; Davies, M.J. Seleno-compounds and therapeutic uses thereof. U.S. Patent 2015/0191446 A1, 24 July 2014. [Google Scholar]
- Merino-Montiel, P.; López, Ó.; Fernández-Bolaños, J.G. l-Isofucoselenofagomine and derivatives: Dual activities as antioxidants and as glycosidase inhibitors. Tetrahedron 2012, 68, 3591–3595. [Google Scholar] [CrossRef]
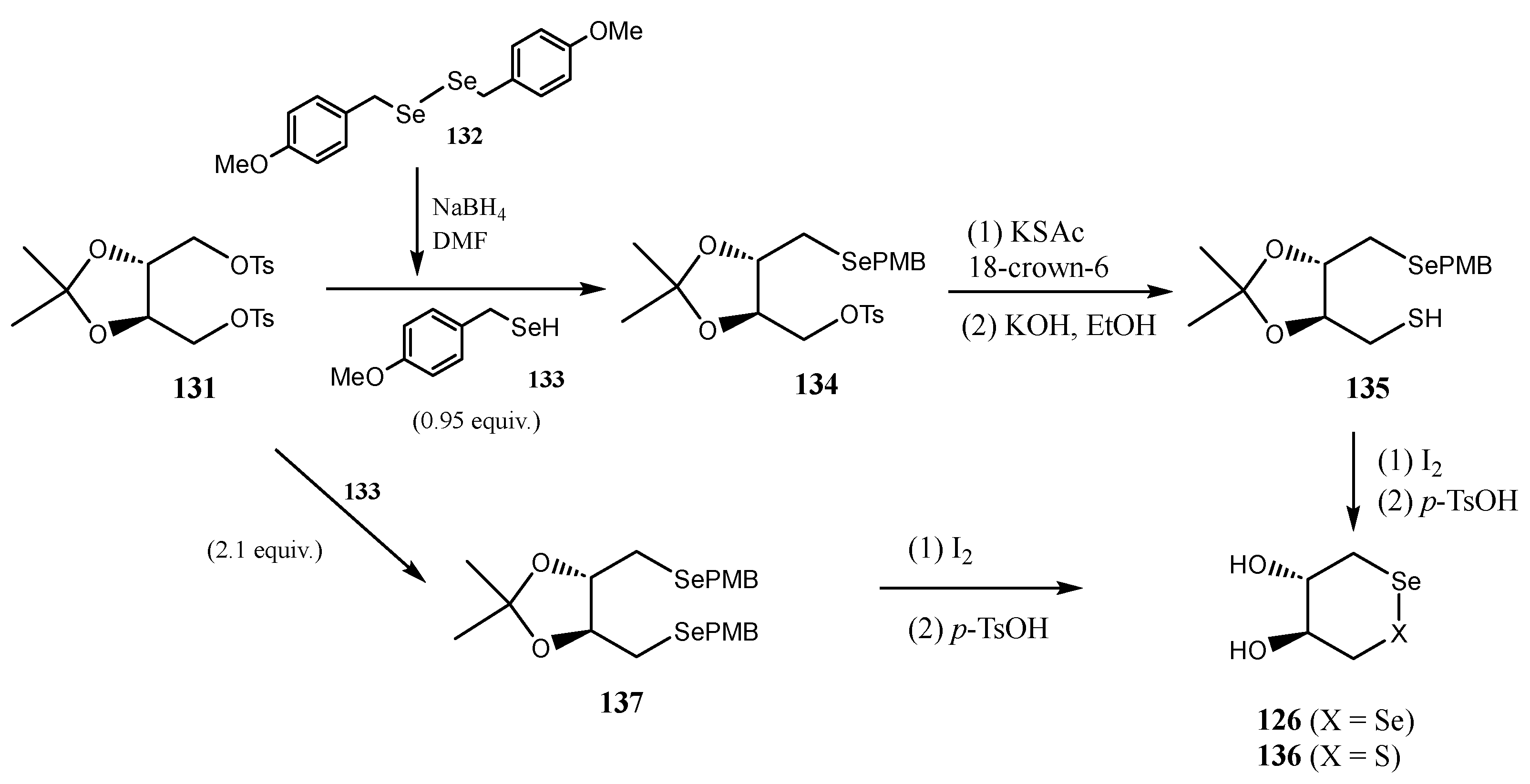
- Davies, M.J.; Schiesser, C.H. 1,4-Anhydro-4-seleno-d-talitol (SeTal): A remarkable selenium-containing therapeutic molecule. New J. Chem. 2019, 43, 9759–9765. [Google Scholar] [CrossRef]
- Zacharias, T.; Flouda, K.; Jepps, T.A.; Gammelgaard, B.; Schiesser, C.H.; Davies, M.J. Effects of a novel selenium substituted-sugar (1,4-anhydro-4-seleno-d-talitol, SeTal) on human coronary artery cell lines and mouse aortic rings. Biochem. Pharmacol. 2020, 173, 113631. [Google Scholar] [CrossRef]
- Voss, G.T.; de Oliveira, R.L.; Davies, M.J.; Domingues, W.B.; Campos, V.F.; Soares, M.P.; Luchese, C.; Schiesser, C.H.; Wilhelm, E.A. Suppressive effect of 1,4-anhydro-4-seleno-d-talitol (SeTal) on atopic dermatitis-like skin lesions in mice through regulation of inflammatory mediators. J. Trace Elem. Med. Biol. 2021, 67, 126795. [Google Scholar] [CrossRef]
- Voss, G.T.; Davies, M.J.; Schiesser, C.H.; de Oliveira, R.L.; Nornberg, A.B.; Soares, V.R.; Barcellos, A.M.; Luchese, C.; Fajardo, A.R.; Wilhelm, E.A. Treating atopic-dermatitis-like skin lesions in mice with gelatin-alginate films containing 1,4-anhydro-4-seleno-d-talitol (SeTal). Int. J. Pharm. 2023, 642, 123174. [Google Scholar] [CrossRef]
- di Vito, R.; Acito, M.; Fatigoni, C.; Schiesser, C.H.; Davies, M.J.; Mangiavacchi, F.; Villarini, M.; Santi, C.; Moretti, M. Genotoxicity assessment of 1,4-anhydro-4-seleno-d-talitol (SeTal) in human liver HepG2 and HepaRG cells. Toxicology 2023, 499, 153663. [Google Scholar] [CrossRef]
- Iwaoka, M.; Takahashi, T.; Tomoda, S. Syntheses and structural characterization of water-soluble selenium reagents for the redox control of protein disulfide bonds. Heteroat. Chem. 2001, 12, 293–299. [Google Scholar] [CrossRef]
- Arai, K.; Kumakura, F.; Takahira, M.; Sekiyama, N.; Kuroda, N.; Suzuki, T.; Iwaoka, M. Effects of ring size and polar functional groups on the glutathione peroxidase-like antioxidant activity of water-soluble cyclic selenides. J. Org. Chem. 2015, 80, 5633–5642. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.G.; Kumar, P.; Phadnis, P.; Iwaoka, M.; Priyadarsini, K.I. Free radical induced selenoxide formation in isomeric organoselenium compounds: The effect of chemical structures on antioxidant activity. New J. Chem. 2019, 43, 13357–13362. [Google Scholar] [CrossRef]
- Arai, K.; Tashiro, A.; Osaka, Y.; Iwaoka, M. Glutathione peroxidase-like activity of amino-substituted water-soluble cyclic selenides: A shift of the major catalytic cycle in methanol. Molecules 2017, 22, 354. [Google Scholar] [CrossRef] [PubMed]
- Kunwar, A.; Verma, P.; Bhilwade, H.N.; Iwaoka, M.; Priyadarsini, K.I. Dihydroxyselenolane (DHS) supplementation improves survivalfollowing whole-body irradiation (WBI) by suppressing tissue-specificinflammatory responses. Mutat. Res. 2016, 807, 33–46. [Google Scholar] [CrossRef]
- Verma, P.; Kunwar, A.; Arai, K.; Iwaoka, M.; Priyadarsini, K.I. Mechanism of radioprotection by dihydroxy-1-selenolane (DHS): Effect of fatty acid conjugation and role of glutathione peroxidase (GPx). Biochimie 2018, 144, 122–133. [Google Scholar] [CrossRef]
- Iwaoka, M.; Sano, N.; Lin, Y.-Y.; Katakura, A.; Noguchi, M.; Takahashi, K.; Kumakura, F.; Arai, K.; Singh, B.G.; Kunwar, A.; et al. Fatty acid conjugates of water-soluble (±)-trans-selenolane-3,4-diol: Effects of alkyl chain length on the antioxidant capacity. ChemBioChem 2015, 16, 1226–1234. [Google Scholar] [CrossRef]
- Arai, K.; Sato, Y.; Nakajima, I.; Saito, M.; Sasaki, M.; Kanamori, A.; Iwaoka, M. Glutathione peroxidase-like functions of 1,2-diselenane-4,5-diol and its amphiphilic derivatives: Switchable catalytic cycles depending on peroxide substrates. Bioorg. Med. Chem. 2021, 29, 115866. [Google Scholar] [CrossRef]
- Arai, K.; Ueno, H.; Asano, Y.; Chakrabarty, G.; Shimodaira, S.; Mugesh, G.; Iwaoka, M. Protein folding in the presence of water-soluble cyclic diselenides with novel oxidoreductase and isomerase activities. ChemBioChem 2018, 19, 207–211. [Google Scholar] [CrossRef]
- Iwaoka, M.; Katakura, A.; Mishima, J.; Ishihara, Y.; Kunwar, A.; Priyadarsini, K.I. Mimicking the lipid peroxidation inhibitory activity of phospholipid hydroperoxide glutathione peroxidase (GPx4) by using fatty acid conjugates of a water-soluble selenolane. Molecules 2015, 20, 12364–12375. [Google Scholar] [CrossRef]
- Chakrabarty, G.; NaveenKumar, S.K.; Kumar, S.; Mugesh, G. Modulation of redox signaling and thiol homeostasis in red blood cells by peroxiredoxin mimetics. ACS Chem. Biol. 2020, 15, 2673–2682. [Google Scholar] [CrossRef]
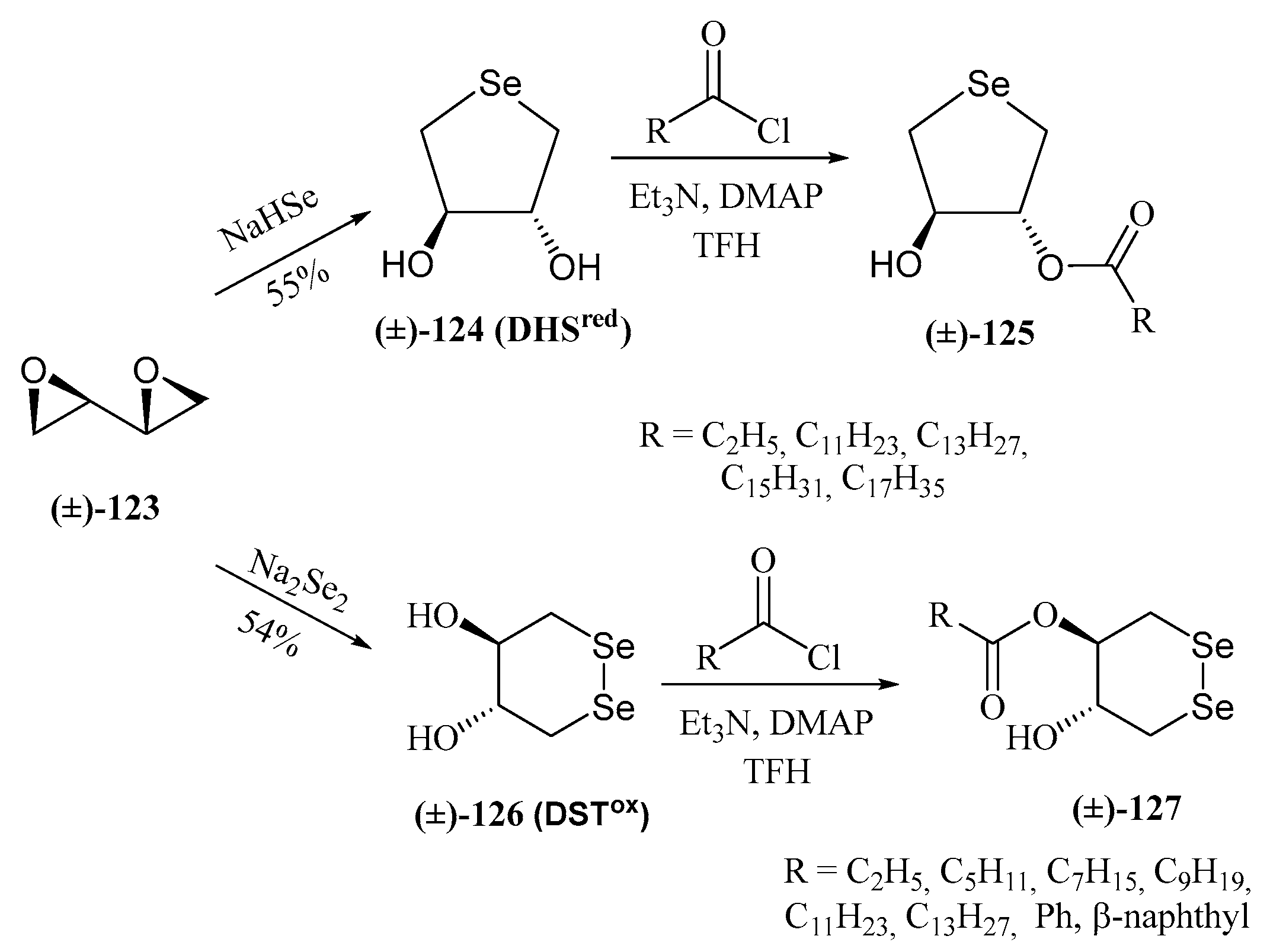
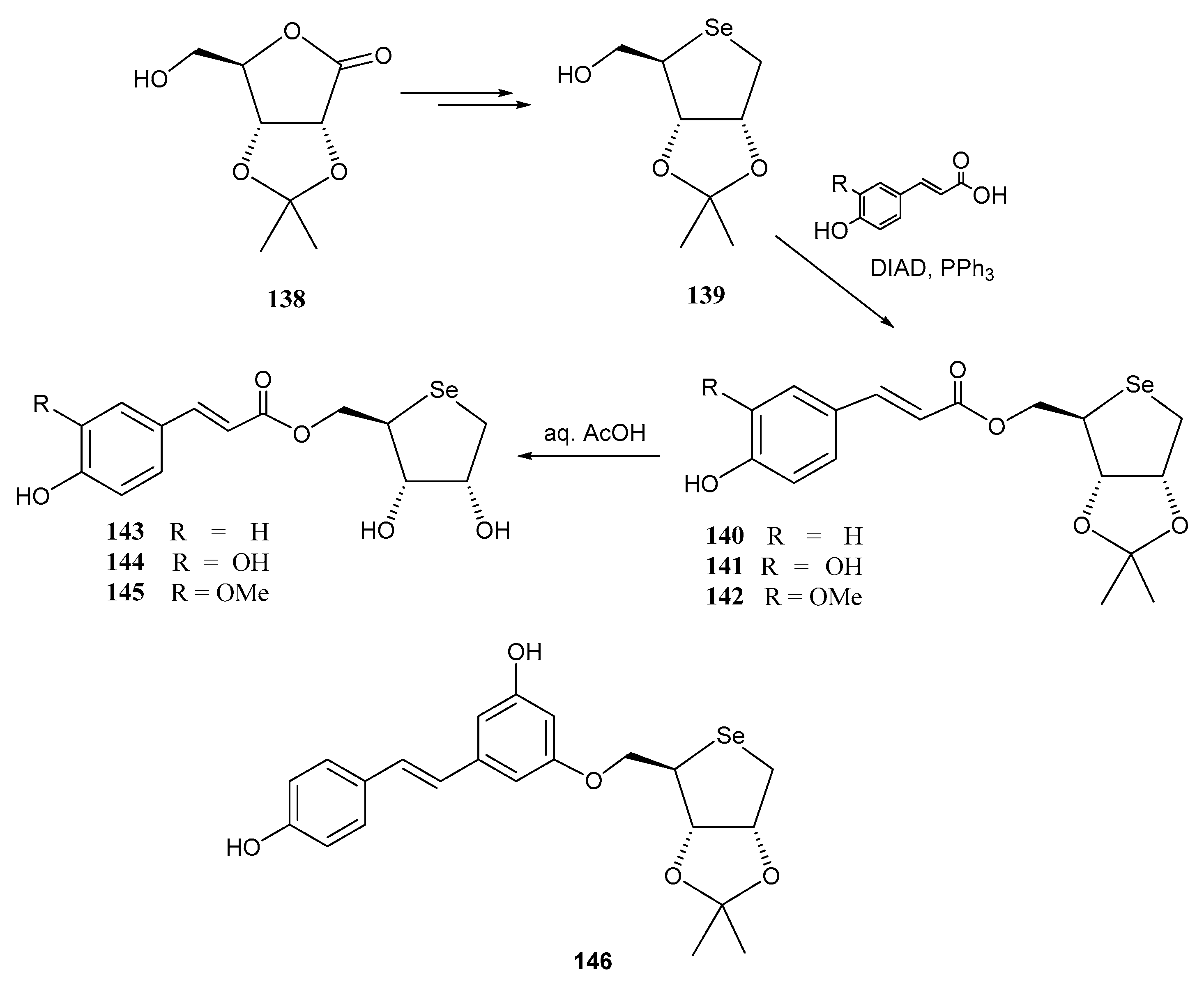
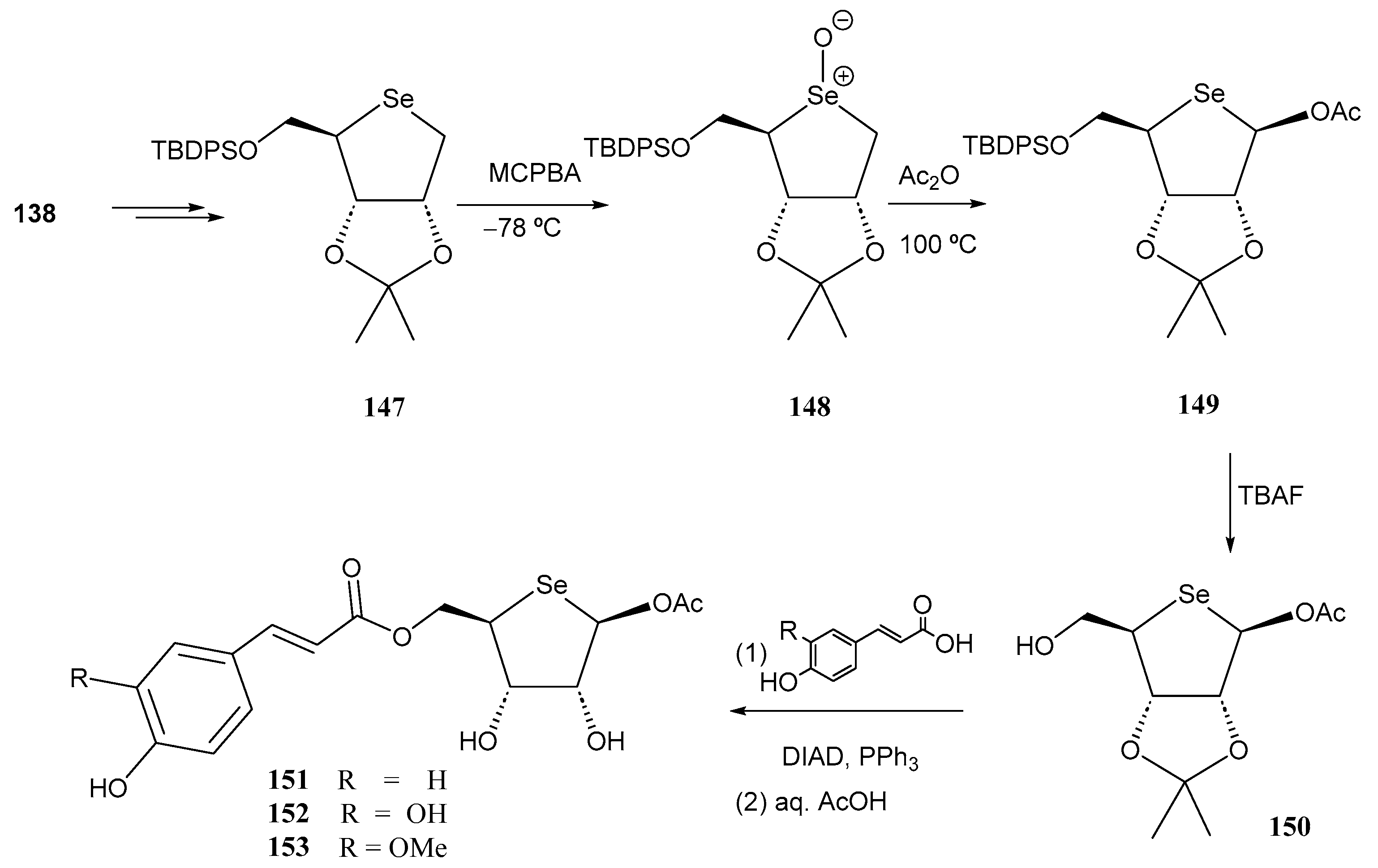
- Cimmino, G.; De Nisco, M.; Piccolella, S.; Gravina, C.; Pedatella, S.; Pacifico, S. Innovative cosmeceutical ingredients: Harnessing selenosugar-linked hydroxycinnamic acids for antioxidant and wound-healing properties. Antioxidants 2024, 13, 744. [Google Scholar] [CrossRef] [PubMed]
- Serpico, L.; De Nisco, M.; Cermola, F.; Manfra, M.; Pedatella, S. Stereoselective synthesis of selenium-containing glycoconjugates via the Mitsunobu reaction. Molecules 2021, 26, 2541. [Google Scholar] [CrossRef] [PubMed]
- Serpico, L.; Iacono, S.D.; De Stefano, L.; De Martino, S.; Battisti, M.; Dardano, P.; Pedatella, S.; De Nisco, M. pH-sensitive release of antioxidant Se-glycoconjugates through a flexible polymeric patch. Eur. Polym. J. 2022, 178, 111486. [Google Scholar] [CrossRef]
- Zhang, L.-X.; Li, C.-X.; Kakar, M.U.; Khan, M.S.; Wu, P.-F.; Amir, R.M.; Dai, D.-F.; Naveed, M.; Li, Q.-Y.; Saeed, M.; et al. Resveratrol (RV): A pharmacological review and call for further research. Biomed. Pharmacother. 2021, 143, 112164. [Google Scholar] [CrossRef]
- Cimmino, G.; De Nisco, M.; Alonso, C.; Gravina, C.; Piscopo, V.; Lemos, R.; Coderch, L.; Piccolella, S.; Pacifico, S.; Pedatella, S. Novel synthesized seleno-glycoconjugates as cosmeceutical ingredients: Antioxidant activity and in vitro skin permeation. Eur. J. Med. Chem. Rep. 2024, 12, 100240. [Google Scholar] [CrossRef]
- Iwaoka, M.; Hiyoshi, Y.; Arai, S.; Ito, T. Synthesis of 4-selenothreofuranose derivatives via Pummerer-type reactions of trans-3,4-dioxygenated tetrahydroselenophenes mediated by a selenonium intermediate. ACS Omega 2021, 6, 17621–17634. [Google Scholar] [CrossRef]
- Lee, H.; Jarhad, D.B.; Yu, J.; Lee, C.; Jeong, L.S. Asymmetric synthesis of 2-C-methyl-4-selenonucleosides as anti-hepatitis C virus agents. J. Org. Chem. 2019, 84, 14414–14426. [Google Scholar] [CrossRef]
- Jeong, L.S.; Tosh, D.K.; Kim, H.O.; Wang, T.; Hou, X.; Yun, H.S.; Kwon, Y.; Lee, S.K.; Choi, J.; Zhao, L.X. First synthesis of 4-selenonucleosides showing unusual southern conformation. Org. Lett. 2008, 10, 209–212. [Google Scholar] [CrossRef]
- Okano, Y.; Saito-Tarashima, N.; Kurosawa, M.; Iwabu, A.; Ota, M.; Watanabe, T.; Kato, F.; Hishiki, T.; Fujimuro, M.; Minakawa, N. Synthesis and biological evaluation of novel imidazole nucleosides as potential anti-dengue virus agents. Bioorg. Med. Chem. 2019, 27, 2181–2186. [Google Scholar] [CrossRef]
- Sahu, P.K.; Kim, G.; Yu, J.; Ahn, J.Y.; Song, J.; Choi, Y.; Jin, X.; Kim, J.-H.; Lee, S.K.; Park, S.; et al. Stereoselective synthesis of 4-selenonucleosides via seleno-Michael reaction as potent antiviral agents. Org. Lett. 2014, 16, 5796–5799. [Google Scholar] [CrossRef]
- Yu, J.; Kim, J.-H.; Lee, H.W.; Alexander, V.; Ahn, H.-C.; Choi, W.J.; Choi, J.; Jeong, L.S. New RNA purine building blocks, 4′-selenopurine nucleosides: First synthesis and unusual mixture of sugar puckerings. Chem. Eur. J. 2013, 19, 5528–5532. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Kim, J.W.; Chandra, G.; Saito–Tarashima, N.; Nogi, Y.; Ota, M.; Minakawa, N.; Jeong, L.S. Synthesis of oligonucleotides containing 5-homo-4-selenouridine derivative and its increased resistance against nuclease. Bioorg. Med. Chem. Lett. 2023, 83, 129172. [Google Scholar] [CrossRef] [PubMed]
- Ota, M.; Takahashi, H.; Nogi, Y.; Kagotani, Y.; Saito-Tarashima, N.; Kondo, J.; Minakawa, N. Synthesis and properties of fully-modified 4-selenoRNA, an endonuclease-resistant RNA analog. Bioorg. Med. Chem. 2022, 76, 117093. [Google Scholar] [CrossRef]
- Kim, J.-H.; Yu, J.; Alexander, V.; Choi, J.H.; Song, J.; Lee, H.W.; Kim, H.O.; Choi, J.; Lee, S.K.; Jeong, L.S. Structure-activity relationships of 2′-modified-4′-selenoarabinofuranosyl-pyrimidines as anticancer agents. Eur. J. Med. Chem. 2014, 83, 208–225. [Google Scholar] [CrossRef]
- Byun, W.S.; Jin, M.; Yu, J.; Kim, W.K.; Song, J.; Chung, H.-J.; Jeong, L.S.; Lee, S.K. A novel selenonucleoside suppresses tumor growth by targeting Skp2 degradation in paclitaxel-resistant prostate cancer. Biochem. Pharmacol. 2018, 158, 84–94. [Google Scholar] [CrossRef]
- Chinetti, G.; Fruchart, J.-C.; Staels, B. Peroxisome proliferator-activated receptors (PPARs): Nuclear receptors at the crossroads between lipid metabolism and inflammation. Inflamm. Res. 2000, 49, 497–505. [Google Scholar] [CrossRef]
- An, S.; Yu, J.; Choi, H.; Ko, H.; Ahn, S.; Shin, J.C.; Pyo, J.J.; Jeong, L.S.; Noh, M. Selenium bioisosteric replacement of adenosine derivatives promoting adiponectin secretion increases the binding affinity to peroxisome proliferator-activated receptor δ. Bioorg. Med. Chem. 2020, 28, 115226. [Google Scholar] [CrossRef]
- Choi, H.; An, S.; Hyun, Y.E.; Noh, M.; Jeong, L.S. Design, synthesis and biological evaluation of truncated 1′-homologated 4′-selenonucleosides as PPAR γ/δ dual modulators. Bioorg. Chem. 2025, 154, 108042. [Google Scholar] [CrossRef]
- Yu, J.; Zhao, L.X.; Park, J.; Lee, H.W.; Sahu, P.K.; Cui, M.; Moss, S.M.; Hammes, E.; Warnick, E.; Gao, Z.-G.; et al. N6-Substituted 5-N-methylcarbamoyl-4-selenoadenosines as potent and selective A3 adenosine receptor agonists with unusual sugar puckering and nucleobase orientation. J. Med. Chem. 2017, 60, 3422–3437. [Google Scholar] [CrossRef]
- Kim, M.; Choi, H.; Nayak, A.; Tripathi, S.K.; Aswar, V.R.; Gaikwad, V.B.; Jacobson, K.A.; Jeong, L.S. Structure−activity relationship of truncated 4-selenonucleosides: A3 adenosine receptor activity and binding selectivity. ACS Med. Chem. Lett. 2024, 15, 1620–1626. [Google Scholar] [CrossRef]
- Choi, H.; Jacobson, K.A.; Yu, J.; Jeong, L.S. Design and synthesis of 2,6-disubstituted-4′-selenoadenosine-5′-N,N-dimethyluronamide derivatives as human A3 adenosine receptor antagonists. Pharmaceuticals 2021, 14, 363. [Google Scholar] [CrossRef] [PubMed]
- Raics, M.; Timári, I.; Diercks, T.; Szilágyi, L.; Gabius, H.-J.; Kövér, K.E. Selenoglycosides as lectin ligands: 77Se-edited CPMG-HSQMBC NMR spectroscopy to monitor biomedically relevant interactions. ChemBioChem 2019, 20, 1688–1692. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Hayashi, C.; Komura, N.; Tamai, R.; Uzawa, J.; Ogawa, J.; Tanaka, H.-N.; Imamura, A.; Ishida, H.; Kiso, M.; et al. Synthesis and glycan−protein interaction studies of Se-sialosides by 77Se NMR. Org. Lett. 2019, 21, 6393–6396. [Google Scholar] [CrossRef] [PubMed]
- Diercks, T.; Medrano, F.J.; FitzGerald, F.G.; Beckwith, D.; Pedersen, M.J.; Reihill, M.; Ludwig, A.-K.; Romero, A.; Oscarson, S.; Cudic, M.; et al. Galectin–glycan interactions: Guidelines for monitoring by 77Se NMR spectroscopy, and solvent (H2O/D2O) impact on binding. Chem. Eur. J. 2021, 27, 316–325. [Google Scholar] [CrossRef]
- Raics, M.; Balogh, A.K.; Kishor, C.; Timári, I.; Medrano, F.J.; Romero, A.; Go, R.M.; Blanchard, H.; Szilágyi, L.; Kövér, K.E.; et al. Investigation of the molecular details of the interactions of selenoglycosides and human galectin-3. Int. J. Mol. Sci. 2022, 23, 2494. [Google Scholar] [CrossRef]
- Dibello, E.; Oddone, N.; Franco, J.; Illyés, T.-Z.; Medeiros, A.; Kiss, A.; Hőgye, F.; Kövér, K.E.; Szilágyi, L.; Comini, M.A. Selenosugars targeting the infective stage of Trypanosoma brucei with high selectivity. Int. J. Parasitol. Drugs Drug Resist. 2024, 24, 100529. [Google Scholar] [CrossRef]
- McDonagh, A.W.; Mahon, M.F.; Murphy, P.V. Lewis acid induced anomerization of Se-glycosides. Application to synthesis of α-Se-GalCer. Org. Lett. 2016, 18, 552–555. [Google Scholar] [CrossRef]
- Zhu, M.; Alami, M.; Messaoudi, S. Room-temperature Pd-catalyzed synthesis of 1-(hetero)aryl selenoglycosides. Org. Lett. 2020, 22, 6584–6589. [Google Scholar] [CrossRef]
- Ding, Y.-N.; Huang, Y.-C.; Shi, W.-Y.; Zheng, N.; Wang, C.-T.; Chen, X.; An, Y.; Zhang, Z.; Liang, Y.-M. Modular synthesis of aryl thio/selenoglycosides via the Catellani strategy. Org. Lett. 2021, 23, 5641–5646. [Google Scholar] [CrossRef]
- Catellani, M.; Frignani, F.; Rangoni, A. A complex catalytic cycle leading to a regioselective synthesis of o,o′-disubstituted vinylarenes. Angew. Chem. Int. Ed. Engl. 1997, 36, 119–122. [Google Scholar] [CrossRef]
- Azeem, Z.; Mandal, P.K. Atom-economic synthesis of unsymmetrical gem-diarylmethylthio/seleno glycosides via base mediated C(O)−S/Se bond cleavage and acyl transfer approach of glycosylthio/selenoacetates. J. Org. Chem. 2023, 88, 1695–1712. [Google Scholar] [CrossRef] [PubMed]
- Shit, P.; Sahaji, S.; Misra, A.K. Synthesis of selenoglycosides and selenium linked disaccharides using reductive cleavage of diselenides. Carbohydr. Res. 2022, 516, 108554. [Google Scholar] [CrossRef] [PubMed]
- Romanò, C.; Bengtsson, D.; Infantino, A.S.; Oscarson, S. Synthesis of fluoro- and seleno-containing d-lactose and d-galactose analogues. Org. Biomol. Chem. 2023, 21, 2545–2555. [Google Scholar] [CrossRef] [PubMed]
- Manna, T.; Misra, A.K. Glycosyl selenoacetates: Versatile building blocks for the preparation of stereoselective selenoglycosides and selenium linked disaccharides. Org. Biomol. Chem. 2019, 17, 8902–8912. [Google Scholar] [CrossRef]
- Angeli, A.; Ferraroni, M.; Granchi, C.; Minutolo, F.; Chen, X.; Shriwas, P.; Russo, E.; Leo, A.; Selleri, S.; Carta, F.; et al. First-in-class dual targeting compounds for the management of seizures in glucose transporter type 1 deficiency syndrome. J. Med. Chem. 2023, 66, 10010–10026. [Google Scholar] [CrossRef]
- Ma, T.; Li, C.; Liang, H.; Wang, Z.; Yu, L.; Xue, W. InBr3-catalyzed synthesis of aryl 1,2-trans-thio(seleno)glycosides. Synlett 2017, 28, 2311–2314. [Google Scholar] [CrossRef]
- Fomitskaya, P.A.; Argunov, D.A.; Tsvetkov, Y.E.; Lalov, A.V.; Ustyuzhanina, N.E.; Nifantiev, N.E. Further investigation of the 2-azido-phenylselenylation of glycals. Eur. J. Org. Chem. 2021, 2021, 5897–5904. [Google Scholar] [CrossRef]
- Guberman, M.; Pieber, B.; Seeberger, P.H. Safe and scalable continuous flow azidophenylselenylation of galactal to prepare galactosamine building blocks. Org. Process Res. Dev. 2019, 23, 2764–2770. [Google Scholar] [CrossRef]
- Zhu, F.; O’Neill, S.; Rodriguez, J.; Walczak, M.A. Stereoretentive reactions at the anomeric position: Synthesis of selenoglycosides. Angew. Chem. Int. Ed. 2018, 57, 7091–7095. [Google Scholar] [CrossRef]
- Miller, E.M.; Walczak, M.A. Light-mediated cross-coupling of anomeric trifluoroborates. Org. Lett. 2021, 23, 4289–4293. [Google Scholar] [CrossRef]
- Compañón, I.; Guerreiro, A.; Mangini, V.; Castro-López, J.; Escudero-Casao, M.; Avenoza, A.; Busto, J.H.; Castillón, S.; Jiménez-Barbero, J.; Asensio, J.L.; et al. Structure-based design of potent tumor-associated antigens: Modulation of peptide presentation by single-atom O/S or O/Se substitutions at the glycosidic linkage. J. Am. Chem. Soc. 2019, 141, 4063–4072. [Google Scholar] [CrossRef] [PubMed]
- Kazakova, E.D.; Yashunsky, D.V.; Krylov, V.B.; Bouchara, J.-P.; Cornet, M.; Valsecchi, I.; Fontaine, T.; Latgé, J.-P.; Nifantiev, N.E. Biotinylated oligo-α-(1→4)-d-galactosamines and their N-acetylated derivatives: α-stereoselective synthesis and immunology application. J. Am. Chem. Soc. 2020, 142, 1175–1179. [Google Scholar] [CrossRef] [PubMed]
- Tokatly, A.I.; Vinnitsky, D.Z.; Kamneva, A.A.; Yashunsky, D.V.; Tsvetkov, Y.E.; Nifantiev, N.E. Glycosylation with derivatives of phenyl 2-azido-2-deoxy-1-seleno-α-d-gluco- and -α-d-mannopyranosides. Russ. Chem. Bull. 2023, 72, 785–792. [Google Scholar] [CrossRef]
- Li, D.; Wang, J.; Wang, X.; Qiao, Z.; Wang, L.; Wang, P.; Song, N.; Li, M. β-Glycosylations with 2-deoxy-2-(2,4-dinitrobenzenesulfonyl)-amino-glucosyl/galactosyl selenoglycosides: Assembly of partially N-acetylated β-(1→ 6)-oligoglucosaminosides. J. Org. Chem. 2023, 88, 9004–9025. [Google Scholar] [CrossRef]
- Li, X.; Hou, Y.; Zhao, J.; Li, J.; Wang, S.; Fang, J. Combination of chemotherapy and oxidative stress to enhance cancer cell apoptosis. Chem. Sci. 2020, 11, 3215–3222. [Google Scholar] [CrossRef]
- Manna, T.; Misra, A.K. On-water synthesis of glycosyl selenocyanate derivatives and their application in the metal free organocatalytic preparation of nonglycosidic selenium linked pseudodisaccharide derivatives. RSC Adv. 2021, 11, 10902–10911. [Google Scholar] [CrossRef]
- Manna, T.; Rana, A.; Misra, A.K. Synthesis of unsymmetrical glycosyl diselenides by the treatment of symmetrical diselenides with glycosyl selenocyanates. Tetrahedron 2021, 95, 132358. [Google Scholar] [CrossRef]
- Rana, A.; Kundu, M.; Misra, A.K. Convenient synthesis of mixed S-Se-linked pseudodisaccharides by sulfur and selenium exchange. Tetrahedron 2022, 115, 132804. [Google Scholar] [CrossRef]
- Wang, L.; Cao, W.; Xu, H. Tellurium-containing polymers: Towards biomaterials and optoelectronic materials. ChemNanoMat 2016, 2, 479–488. [Google Scholar] [CrossRef]
- Comasseto, J.V.; Barrientos-Astigarraga, R.E. Add a little tellurium to your synthetic plans! Aldrichimica Acta 2000, 33, 66–78. [Google Scholar]
- Irfan, M.; Rehman, R.; Razali, M.R.; Ur-Rehman, S.; Ur-Rehman, A.; Iqbal, M.A. Organotellurium compounds: An overview of synthetic methodologies. Rev. Inorg. Chem. 2020, 40, 193–232. [Google Scholar] [CrossRef]
- Jain, S.; Satpute, S.S.; Jha, R.K.; Patel, M.S.; Kumar, S. Bidentate ligand driven intramolecularly Te…O bonded organotellurium cations from synthesis, stability to catalysis. Chem. Eur. J. 2024, 30, e202303089. [Google Scholar] [CrossRef] [PubMed]
- Purohit, S.; Oswal, P.; Bahuguna, A.; Tyagi, A.; Bhatt, N.; Kumar, A. Catalytic system having an organotellurium ligand on graphene oxide: Immobilization of Pd(0) nanoparticles and application in heterogeneous catalysis of cross-coupling reactions. RSC Adv. 2024, 14, 27092–27109. [Google Scholar] [CrossRef]
- Comasseto, J.V.; Gariani, R.A. Biotransformations on organic selenides and tellurides: Synthetic applications. Tetrahedron 2009, 65, 8447–8459. [Google Scholar] [CrossRef]
- Zhao, H.; Xu, X.; Zhang, S.; Sun, J.; He, W.; Zhang, L.; Cheng, Z. NIR Photocontrolled organotellurium-mediated reversible-deactivation radical polymerization by activation of C–Te bonds with organotellurium radicals. Macromolecules 2024, 57, 1182–1194. [Google Scholar] [CrossRef]
- Takagi, K.; Hayashi, S.; Sakakibara, N. Controlled cationic polymerization with organotellurium catalysts utilizing redox-mediated chalcogen bonding interaction. Macromolecules 2024, 57, 3358–13367. [Google Scholar] [CrossRef]
- Jiang, Y.; Kibune, M.; Tosaka, M.; Yamago, S. Practical synthesis of dendritic hyperbranched polyacrylates and their topological block polymers by organotellurium-mediated emulsion polymerization in water. Angew. Chem. Int. Ed. 2023, 62, e202306916. [Google Scholar] [CrossRef]
- Bhardwaj, A.; Dubey, A.; Tufail, A.; Tufail, N.; Jangra, K.; Garg, S. Experimental and computational insights into organotellurium (IV) complexes as potent PfNDH2 inhibitors: Synthesis, characterization, and antimalarial evaluation. Appl. Organometal. Chem. 2025, 39, e7778. [Google Scholar] [CrossRef]
- Angeli, A.; Pinteala, M.; Maier, S.S.; Toti, A.; Mannelli, L.D.C.; Ghelardini, C.; Selleri, S.; Carta, F.; Supuran, C.T. Tellurides bearing benzensulfonamide as carbonic anhydrase inhibitors with potent antitumor activity. Bioorg. Med. Chem. Lett. 2021, 45, 128147. [Google Scholar] [CrossRef]
- Wollenhaupt, S.G.N.; Soares, A.T.; Salgueiro, W.G.; Noremberg, S.; Reis, G.; Viana, C.; Gubert, P.; Soares, F.A.; Affeldt, R.F.; Lüdtke, D.S.; et al. Seleno- and telluro-xylofuranosides attenuate Mn-induced toxicity in C. elegans via the DAF-16/FOXO pathway. Food Chem. Toxicol. 2014, 64, 192–199. [Google Scholar] [CrossRef]
- Jiao, A.; Yang, N.; Wang, J.; Xu, X.; Jin, Z. Cyclodextrin-derived chalcogenides as glutathione peroxidase mimics and their protection of mitochondria against oxidative damage. J. Incl. Phenom. Macrocycl. Chem. 2013, 75, 155–163. [Google Scholar] [CrossRef]
- Nguyen, O.T.K.; Schiesser, C.H. Preparation of 5-telluropentopyranose sugars from common pentose starting materials. Tetrahedron Lett. 2002, 43, 3799–3800. [Google Scholar] [CrossRef]
- Tanini, D.; Ricci, L.; Capperucci, A. Rongalite-promoted on water synthesis of functionalised tellurides and ditellurides. Adv. Synth. Catal. 2020, 362, 1323–1332. [Google Scholar] [CrossRef]
- Kashyap, C.; Mazumder, L.J.; Rohman, S.S.; Ullah, S.S.; Guha, A.K. Re-visiting the antioxidant activity of Se- and Te-carbohydrates: A theoretical study. ChemistrySelect 2019, 4, 1470–1475. [Google Scholar] [CrossRef]
- Yamago, S.; Kokubo, K.; Masuda, S.; Yoshida, J.-i. Practical synthesis of telluroglycosides. Synlett 1996, 1996, 929–930. [Google Scholar] [CrossRef]
- Tsegay, S.; Williams, R.J.; Williams, S.J. Synthesis of glycosyl fluorides from thio-, seleno-, and telluroglycosides and glycosyl sulfoxides using aminodifluorosulfinium tetrafluoroborates. Carbohydr. Res. 2012, 357, 16–22. [Google Scholar] [CrossRef]
- Nagatomo, M.; Kamimura, D.; Matsui, Y.; Masuda, K.; Inoue, M. Et3B-mediated two- and three-component coupling reactions via radical decarbonylation of α-alkoxyacyl tellurides: Single-step construction of densely oxygenated carboskeletons. Chem. Sci. 2015, 6, 2765–2769. [Google Scholar] [CrossRef]
- Fukuda, T.; Nagatomo, M.; Inoue, M. Total synthesis of diospyrodin and its three diastereomers. Org. Lett. 2020, 22, 6468–6472. [Google Scholar] [CrossRef]
- Nagatomo, M.; Zhang, K.; Fujino, H.; Inoue, M. Et3B/Et2AlCl/O2-mediated radical coupling reaction between α-alkoxyacyl tellurides and 2-hydroxybenzaldehyde derivatives. Chem. Asian J. 2020, 15, 3820–3824. [Google Scholar] [CrossRef]
- Fujino, H.; Nagatomo, M.; Paudel, A.; Panthee, S.; Hamamoto, H.; Sekimizu, K.; Inoue, M. Unified total synthesis of polyoxins J, L, and fluorinated analogues on the basis of decarbonylative radical coupling reactions. Angew. Chem. Int. Ed. 2017, 56, 11865–11869. [Google Scholar] [CrossRef]
- Nagatomo, M.; Fujimoto, Y.; Masuda, K.; Inoue, M. Construction of a 6/5/9-membered tricyclic structure of cladiellins via radical-polar crossover reaction. J. Antibiot. 2019, 72, 486–489. [Google Scholar] [CrossRef] [PubMed]
- Kuwana, D.; Ovadia, B.; Kamimura, D.; Nagatomo, M.; Inoue, M. Installation of O-heterocycles to N-heteroarenes via an Et3B/O2-mediated radical reaction of α-alkoxy and α-alkoxyacyl tellurides. Asian J. Org. Chem. 2019, 8, 1088–1091. [Google Scholar] [CrossRef]
- Masuda, K.; Nagatomo, M.; Inoue, M. Direct assembly of multiply oxygenated carbon chains by decarbonylative radical–radical coupling reactions. Nat. Chem. 2017, 9, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Kawamata, T.; Yamaguchi, A.; Nagatomo, M.; Inoue, M. Convergent total synthesis of asimicin via decarbonylative radical dimerization. Chem. Eur. J. 2018, 24, 18907–18912. [Google Scholar] [CrossRef]










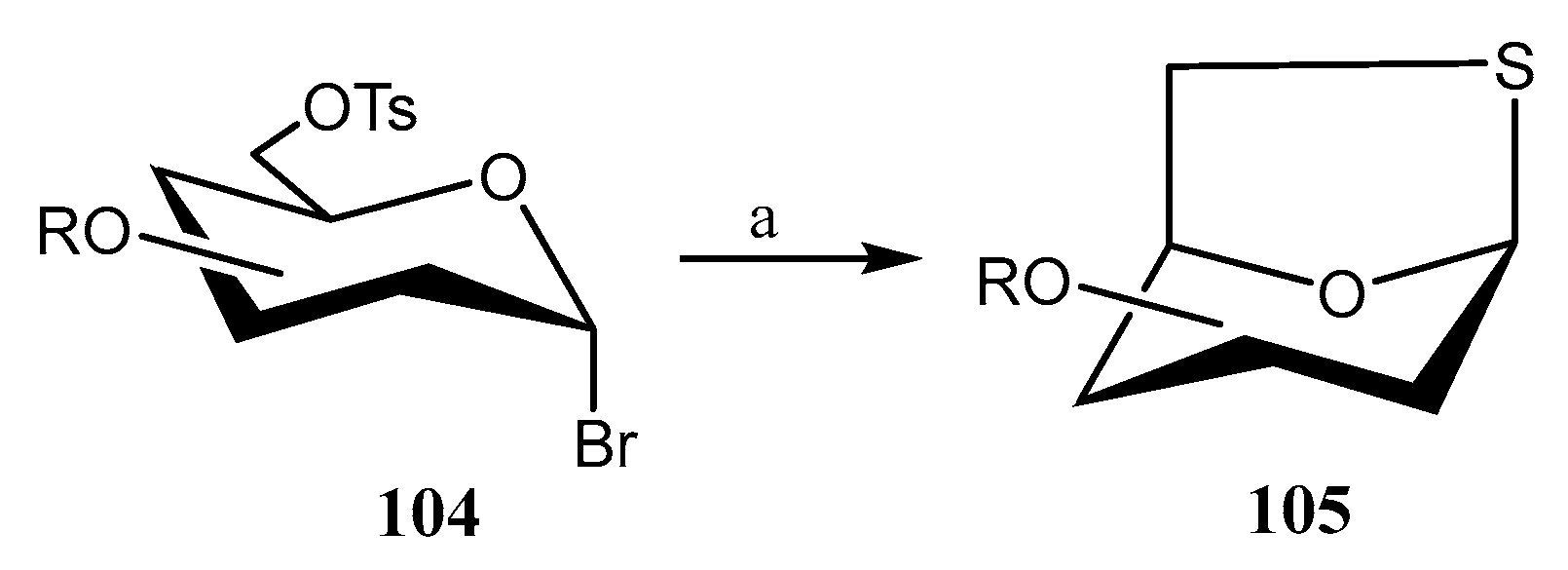
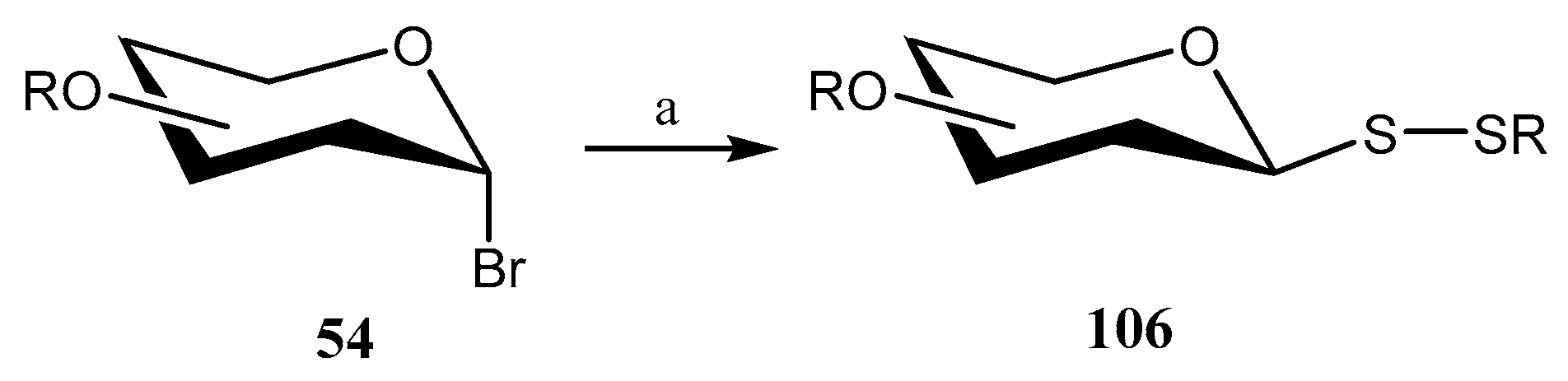
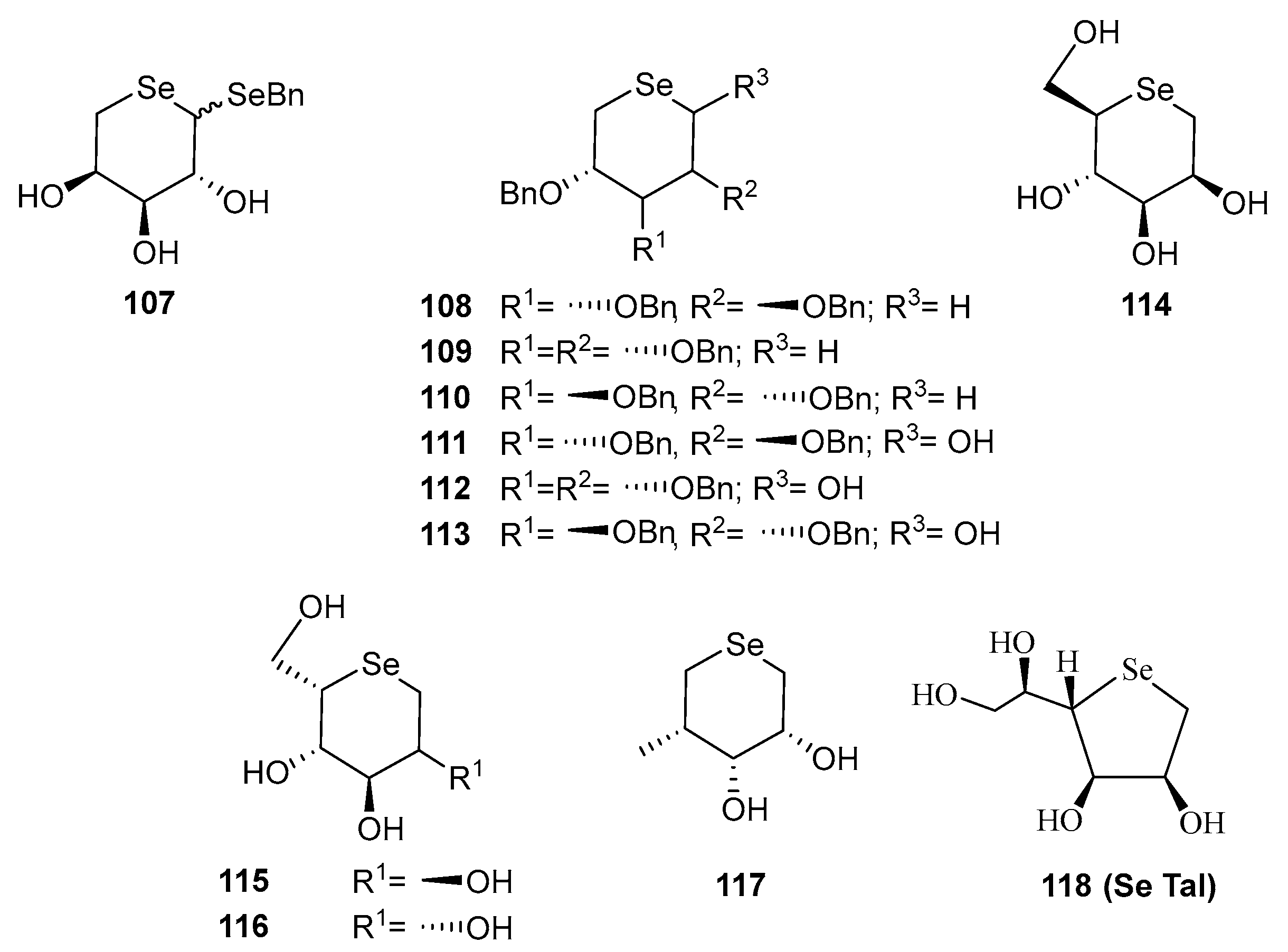
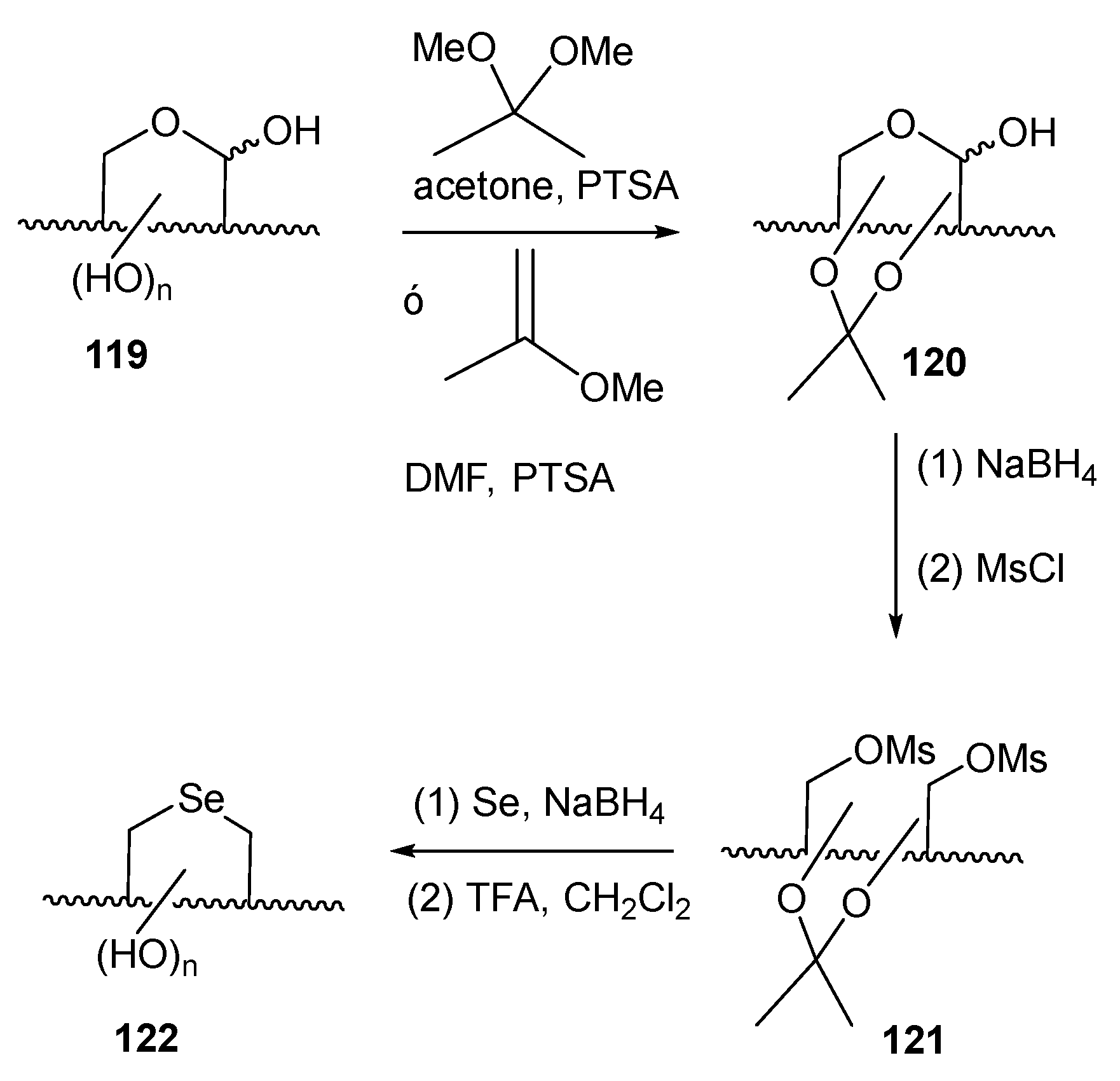













Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martínez-Pascual, R.; Valera-Zaragoza, M.; Fernández-Bolaños, J.G.; López, Ó. Exploring the Chemistry and Applications of Thio-, Seleno-, and Tellurosugars. Molecules 2025, 30, 2053. https://doi.org/10.3390/molecules30092053
Martínez-Pascual R, Valera-Zaragoza M, Fernández-Bolaños JG, López Ó. Exploring the Chemistry and Applications of Thio-, Seleno-, and Tellurosugars. Molecules. 2025; 30(9):2053. https://doi.org/10.3390/molecules30092053
Chicago/Turabian StyleMartínez-Pascual, Roxana, Mario Valera-Zaragoza, José G. Fernández-Bolaños, and Óscar López. 2025. "Exploring the Chemistry and Applications of Thio-, Seleno-, and Tellurosugars" Molecules 30, no. 9: 2053. https://doi.org/10.3390/molecules30092053
APA StyleMartínez-Pascual, R., Valera-Zaragoza, M., Fernández-Bolaños, J. G., & López, Ó. (2025). Exploring the Chemistry and Applications of Thio-, Seleno-, and Tellurosugars. Molecules, 30(9), 2053. https://doi.org/10.3390/molecules30092053