


Oligonuclear Manganese Complexes with Multiple Redox Properties for High-Contrast Electrochromism
 and
and 
Abstract
1. Introduction
2. Results and Discussion
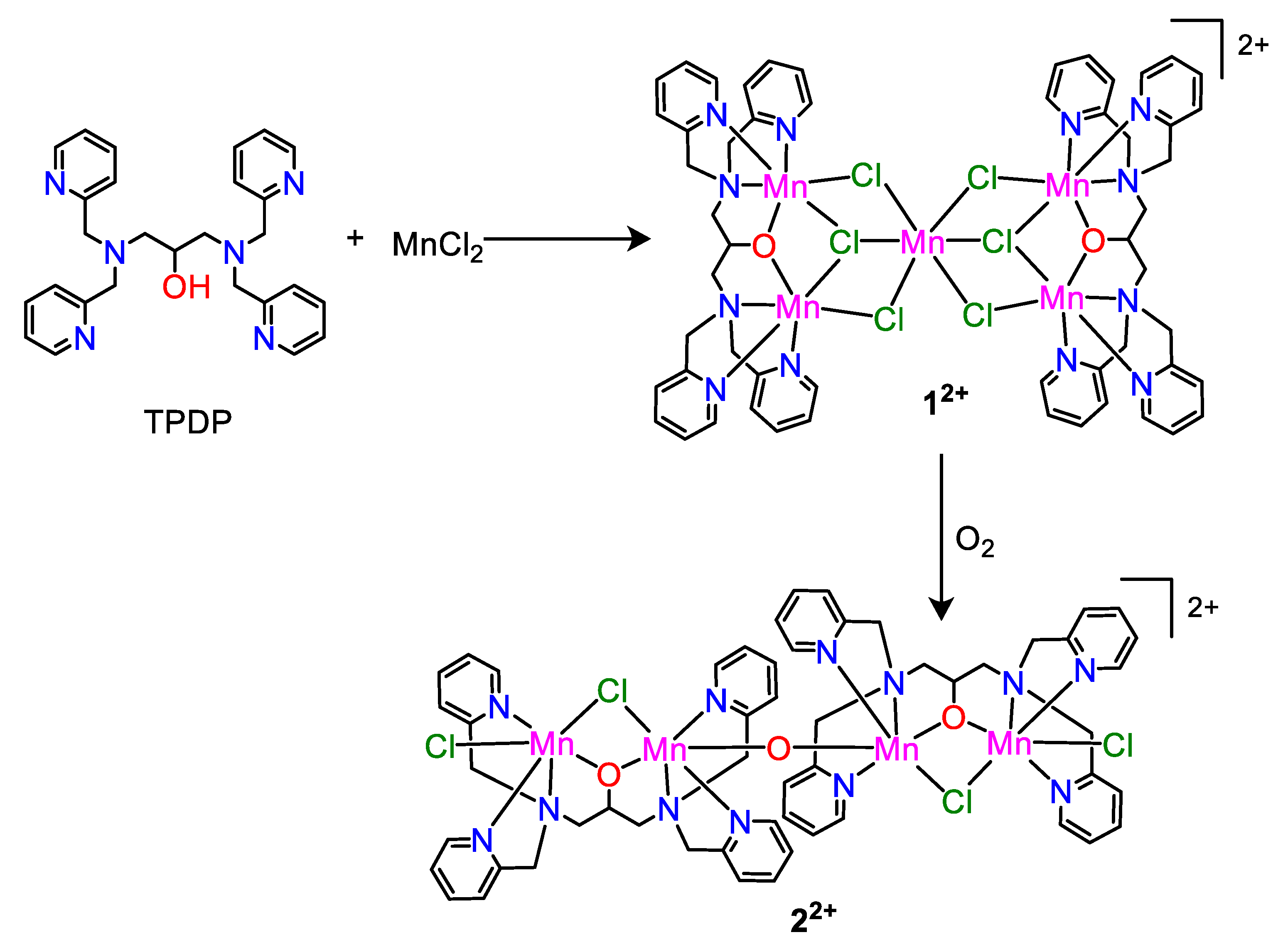
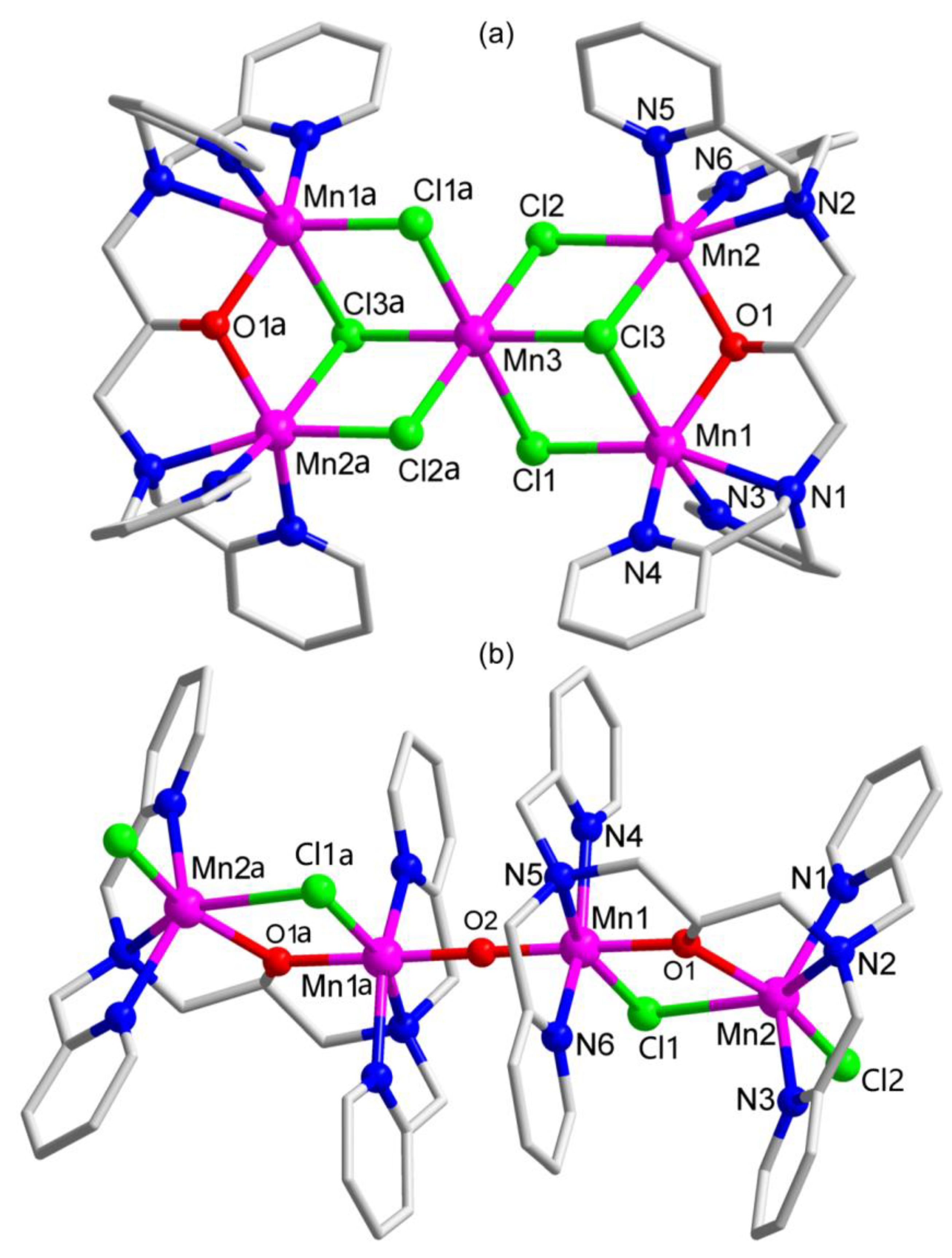
2.1. Synthesis and Crystal Structure
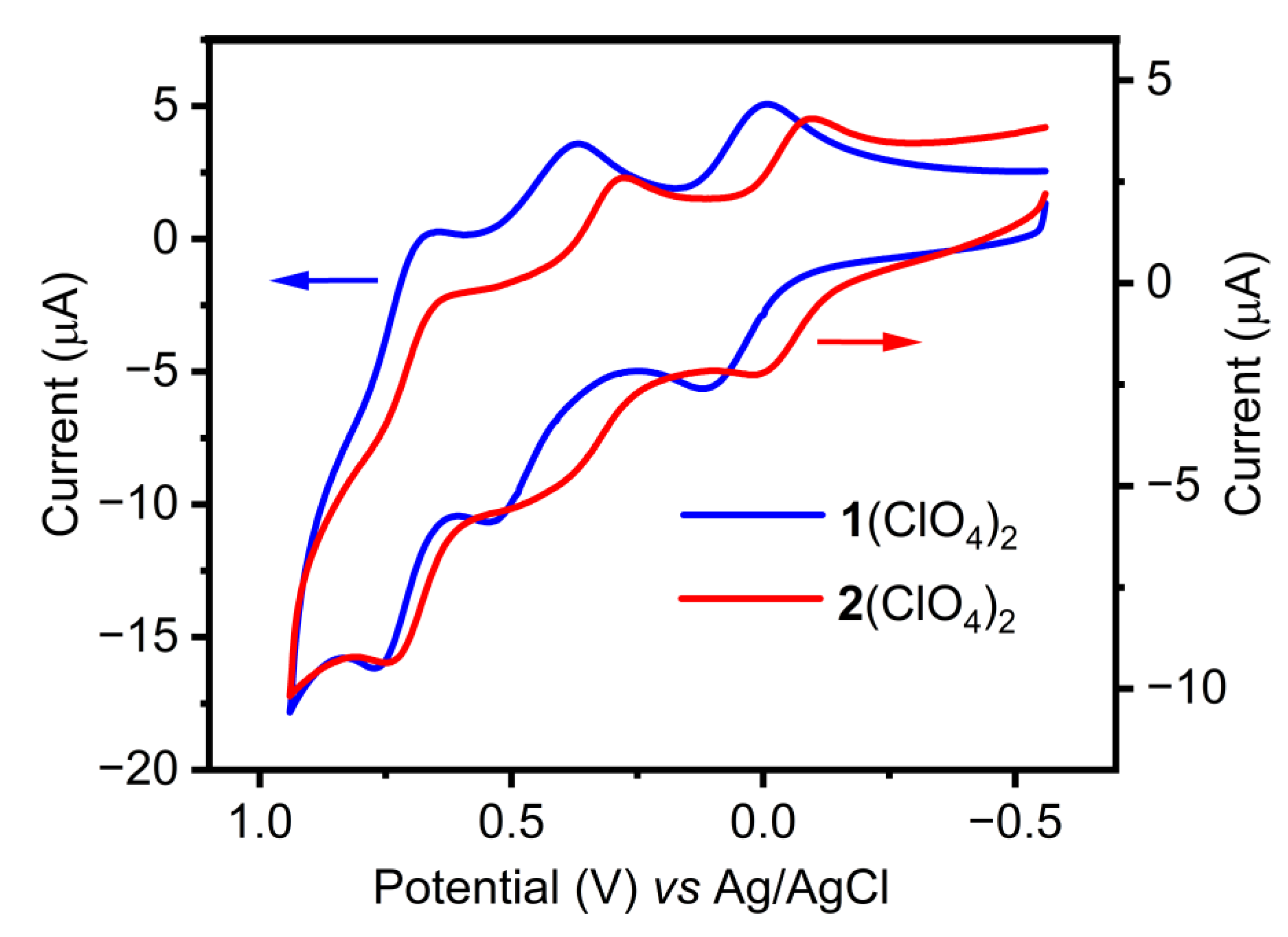
2.2. Electrochemical Characterization
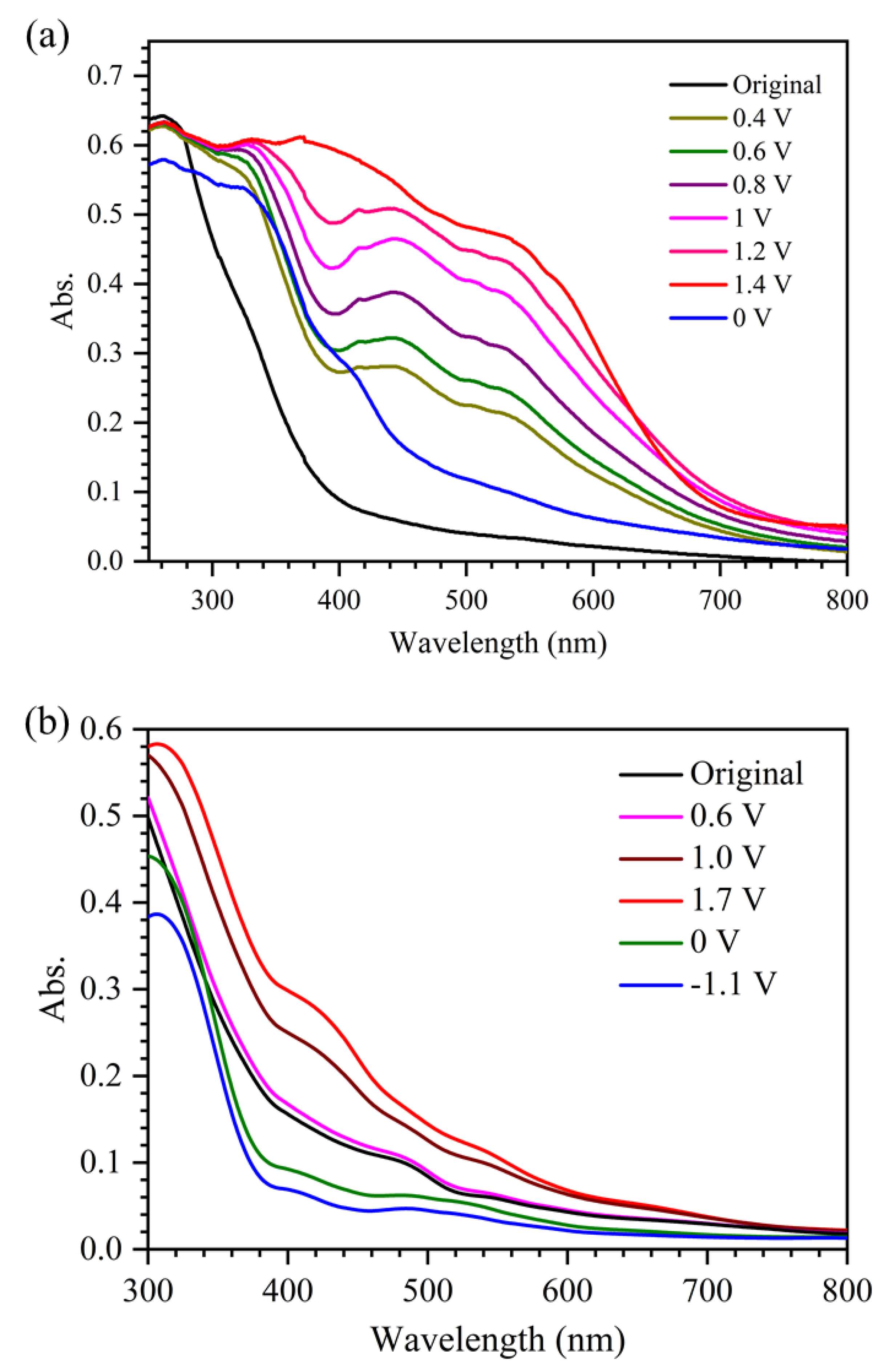
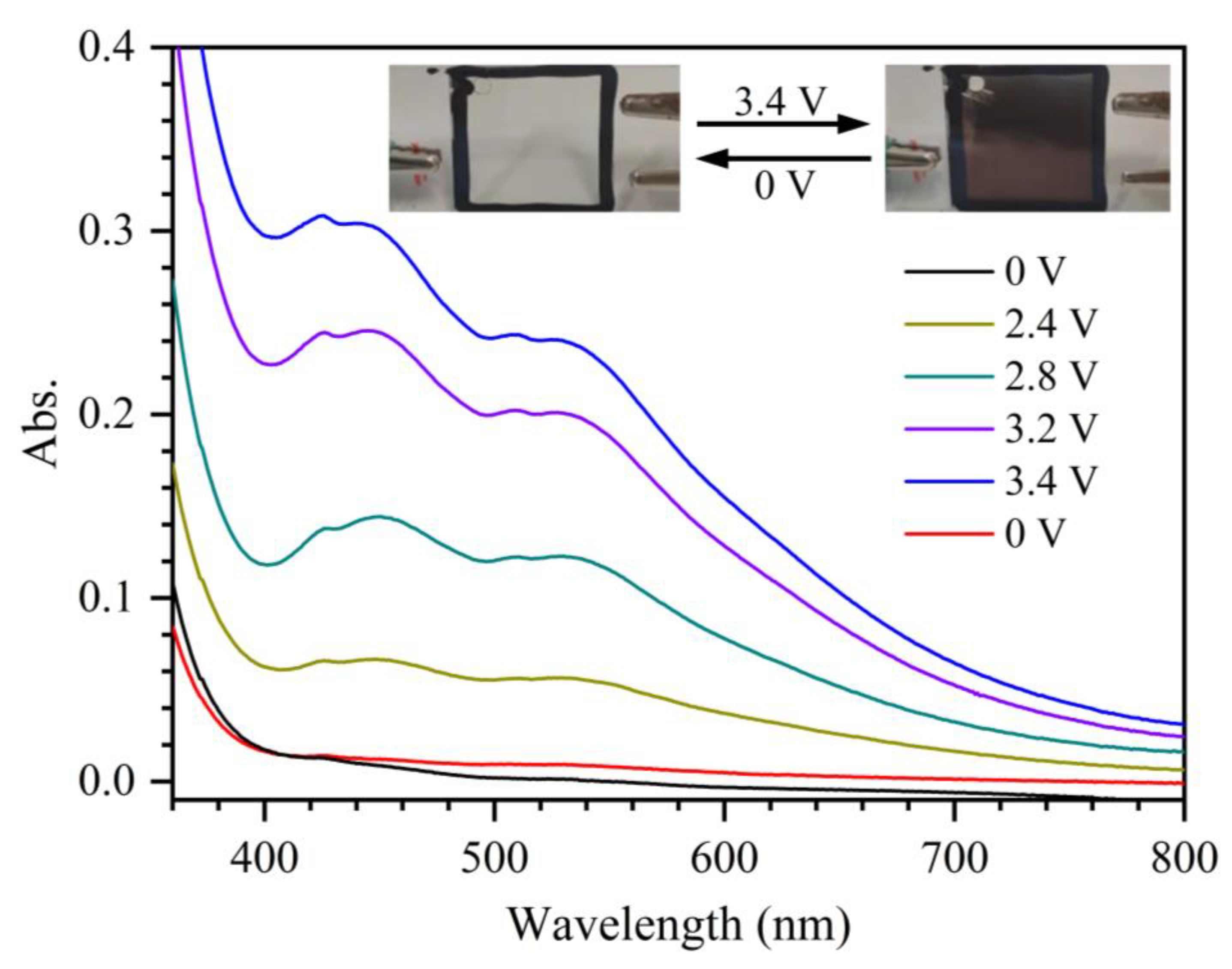
2.3. Spectroelectrochemical Study
2.4. Electrochromic Device
3. Materials and Methods
3.1. General Methods
3.2. Synthesis of 1(ClO4)2
3.3. Synthesis of 2(ClO4)2
3.4. X-Ray Crystallography
3.5. Fabrication of Electrochromic Devices
3.6. Characterization of Electrochromic Devices
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| TPDP | 1,3-bis(bis(2-pyridinylmethyl)amino)-2-propanol |
| LMCT | Ligand-to-metal charge transfer |
| ECD | Electrochromic device |
| ITO | Indium tin oxide |
| CV | Cyclic voltammetry |
| MLCT | Metal-to-ligand charge transfer |
| IVCT | Intervalence charge transfer |
References
- Felder, P.S.; Keller, S.; Gasser, G. Polymetallic Complexes for Applications as Photosensitisers in Anticancer Photodynamic Therapy. Adv. Ther. 2020, 3, 1900139. [Google Scholar] [CrossRef]
- Li, X.; Zhao, X.; Wang, W.; Shi, Z.; Zhang, Y.; Tian, Q.; Yao, Y.; He, C.; Duan, C. Biomedical applications of multinuclear Pt(II)/Ru(II)/Ir(III) metallo-supramolecular assemblies for intensive cancer therapy. Coord. Chem. Rev. 2023, 495, 215366. [Google Scholar] [CrossRef]
- Wang, K.; Gao, E. Recent Advances in Multinuclear Complexes as Potential Anticancer and DNA Binding Agents. Anti-Cancer Agents Med. Chem. 2014, 14, 147–169. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-Z.; Tian, C.-B.; Sun, Q.-F. Coordination-Directed Self-Assembly of Functional Polynuclear Lanthanide Supramolecular Architectures. Chem. Rev. 2022, 122, 6374–6458. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, Y.; Kitagawa, Y. Thermo-sensitive luminescence of lanthanide complexes, clusters, coordination polymers and metal-organic frameworks with organic photosensitizers. J. Mater. Chem. C 2019, 7, 7494–7511. [Google Scholar] [CrossRef]
- Zhang, S.; Zhao, L. Macrocycle-Encircled Polynuclear Metal Clusters: Controllable Synthesis, Reactivity Studies, and Applications. Acc. Chem. Res. 2018, 51, 2535–2545. [Google Scholar] [CrossRef]
- Tang, J.; Zhao, L. Polynuclear organometallic clusters: Synthesis, structure, and reactivity studies. Chem. Commun. 2020, 56, 1915–1925. [Google Scholar] [CrossRef]
- Hartinger, C.G.; Phillips, A.D.; Nazarov, A.A. Polynuclear Ruthenium, Osmium and Gold Complexes. The Quest for Innovative Anticancer Chemotherapeutics. Curr. Top. Med. Chem. 2011, 11, 2688–2702. [Google Scholar] [CrossRef]
- Kostakis, G.E.; Perlepes, S.P.; Blatov, V.A.; Proserpio, D.M.; Powell, A.K. High-nuclearity cobalt coordination clusters: Synthetic, topological and magnetic aspects. Coord. Chem. Rev. 2012, 256, 1246–1278. [Google Scholar] [CrossRef]
- Nesterov, D.S.; Nesterova, O.V.; Pombeiro, A.J.L. Homo- and heterometallic polynuclear transition metal catalysts for alkane C-H bonds oxidative functionalization: Recent advances. Coord. Chem. Rev. 2018, 355, 199–222. [Google Scholar] [CrossRef]
- Mondal, I.; Chattopadhyay, S. Development of multi-metallic complexes using metal-salen complexes as building blocks. J. Coord. Chem. 2019, 72, 3183–3209. [Google Scholar] [CrossRef]
- Horiuchi, S.; Umakoshi, K. Recent advances in pyrazolato-bridged homo- and heterometallic polynuclear platinum and palladium complexes. Coord. Chem. Rev. 2023, 476, 214924. [Google Scholar] [CrossRef]
- Li, K.; Del Rosal, I.; Zhao, Y.; Maron, L.; Zhu, C. Planar Tetranuclear Uranium Hydride Cluster Supported by ansa-Bis(cyclopentadienyl) Ligands. Angew. Chem. Int. Ed. 2024, 63, e202405494. [Google Scholar] [CrossRef]
- McEvoy, J.P.; Brudvig, G.W. Water-splitting chemistry of photosystem II. Chem. Rev. 2006, 106, 4455–4483. [Google Scholar] [CrossRef]
- Kanady, J.S.; Tsui, E.Y.; Day, M.W.; Agapie, T. A Synthetic Model of the Mn3Ca Subsite of the Oxygen-Evolving Complex in Photosystem II. Science 2011, 333, 733–736. [Google Scholar] [CrossRef]
- Kanady, J.S.; Lin, P.H.; Carsch, K.M.; Nielsen, R.J.; Takase, M.K.; Goddard, W.A., 3rd; Agapie, T. Toward models for the full oxygen-evolving complex of photosystem II by ligand coordination to lower the symmetry of the Mn3CaO4 cubane: Demonstration that electronic effects facilitate binding of a fifth metal. J. Am. Chem. Soc. 2014, 136, 14373–14376. [Google Scholar] [CrossRef]
- Kärkäs, M.D.; Johnston, E.V.; Verho, O.; Åkermark, B. Artificial Photosynthesis: From Nanosecond Electron Transfer to Catalytic Water Oxidation. Acc. Chem. Res. 2014, 47, 100–111. [Google Scholar] [CrossRef]
- Richmond, C.J.; Miras, H.N.; de la Oliva, A.R.; Zang, H.; Sans, V.; Paramonov, L.; Makatsoris, C.; Inglis, R.; Brechin, E.K.; Long, D.L.; et al. A flow-system array for the discovery and scale up of inorganic clusters. Nat. Chem. 2012, 4, 1037–1043. [Google Scholar] [CrossRef]
- Cirera, J.; Jiang, Y.; Qin, L.; Zheng, Y.-Z.; Li, G.; Wu, G.; Ruiz, E. Ferromagnetism in polynuclear systems based on non- linear Mn2IIMnIII building blocks. Inorg. Chem. Front. 2016, 3, 1272–1279. [Google Scholar] [CrossRef]
- Li Manni, G. Modeling magnetic interactions in high-valent trinuclear [Mn3(IV)O4]4+ complexes through highly compressed multi-configurational wave functions. PCCP 2021, 23, 19766–19780. [Google Scholar] [CrossRef]
- Cañada-Vilalta, C.; Streib, W.E.; Huffman, J.C.; O’Brien, T.A.; Davidson, E.R.; Christou, G. Polynuclear manganese complexes with the dicarboxylate ligand m-phenylenedipropionate: A hexanuclear mixed-valence (3MnIII, 3MnIV) complex. Inorg. Chem. 2004, 43, 101–115. [Google Scholar] [CrossRef] [PubMed]
- Christou, G.; Gatteschi, D.; Hendrickson, D.N.; Sessoli, R. Single-Molecule Magnets. MRS Bull. 2011, 25, 66–71. [Google Scholar] [CrossRef]
- Ozcan, S.; Kobak, R.Z.; Budak, O.; Koca, A.; Bayir, Z.A. Synthesis, Electrochemistry, Spectroelectrochemistry, and Electrochromism of Metallophthalocyanines Substituted with Four (2,4,5-trimethylphenyl)ethynyl Groups. Electroanalysis 2022, 34, 1610–1620. [Google Scholar] [CrossRef]
- Fukuda, Y.; Hirota, M.; Kon-No, M.; Nakao, A.; Umezawa, K. New chromotropic behavior of manganese complexes with 1,4,7-triazacyclononane-N,N′,N”-tricarboxylates. Inorg. Chim. Acta 2002, 339, 322–326. [Google Scholar] [CrossRef]
- Silver, J.; Lukes, P.; Hey, P.; Ahmet, M.T. Electrochromism in the Transition-Metal Phthalocyanines. 2. Structural-Changes in the Properties of [Cr(Pc)] and [Mn(Pc)] Films. J. Mater. Chem. 1992, 2, 841–847. [Google Scholar] [CrossRef]
- Heras Ojea, M.J.; Hay, M.A.; Cioncoloni, G.; Craig, G.A.; Wilson, C.; Shiga, T.; Oshio, H.; Symes, M.D.; Murrie, M. Ligand-directed synthesis of Mn twisted bow-ties. Dalton Trans. 2017, 46, 11201–11207. [Google Scholar] [CrossRef]
- Mikata, Y.; Wakamatsu, M.; So, H.; Abe, Y.; Mikuriya, M.; Fukui, K.; Yano, S. N,N,N’,N’-Tetrakis(2-quinolylmethyl)-2-hydroxy-1,3-propanediamine (Htqhpn) as a supporting ligand for a low-valent (μ-O)2 tetranuclear manganese core. Inorg. Chem. 2005, 44, 7268–7270. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Mok, H.J.; Staples, R.J.; Armstrong, W.H. Shape-Shifting Tetranuclear Oxo-Bridged Manganese Cluster: Relevance to Photosystem II Water Oxidase Active Site. J. Am. Chem. Soc. 2004, 126, 9202–9204. [Google Scholar] [CrossRef]
- Chan, M.K.; Armstrong, W.H. A Novel Tetranuclear Manganese Complex That Displays Multiple High-Potential Redox Processes—Synthesis, Structure, and Properties of ([Mn2(TPHPN)(O2CCH3)(H2O)]2O)(ClO4)4-2CH3OH. J. Am. Chem. Soc. 1989, 111, 9121–9122. [Google Scholar] [CrossRef]
- Bansal, D.; Mondal, A.; Lakshminarasimhan, N.; Gupta, R. Oxo-bridged trinuclear and tetranuclear manganese complexes supported with nitrogen donor ligands: Syntheses, structures and properties. Dalton Trans. 2019, 48, 7918–7927. [Google Scholar] [CrossRef]
- Huang, T.; Du, P.; Cheng, X.; Lin, Y.M. Manganese Complexes with Consecutive Mn(IV) --> Mn(III) Excitation for Versatile Photoredox Catalysis. J. Am. Chem. Soc. 2024, 146, 24515–24525. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Hoffmann, P.; Steinmetzer, J.; Askes, S.H.C.; Kupfer, S.; Görls, H.; Gräfe, S.; Neugebauer, U.; Gandra, U.R.; Schiller, A. Visible light-activated biocompatible photo-CORM for CO-release with colorimetric and fluorometric dual turn-on response. Polyhedron 2019, 172, 175–181. [Google Scholar] [CrossRef]
- Gandra, U.R.; Jana, B.; Hammer, P.; Mohideen, M.I.H.; Neugebauer, U.; Schiller, A. Lysosome targeted visible light-induced photo-CORM for simultaneous CO-release and singlet oxygen generation. Chem. Commun. 2024, 60, 2098–2101. [Google Scholar] [CrossRef] [PubMed]
- Elshaarawy, R.F.M.; Lan, Y.; Janiak, C. Oligonuclear homo- and mixed-valence manganese complexes based on thiophene- or aryl-carboxylate ligation: Synthesis, characterization and magnetic studies. Inorg. Chim. Acta 2013, 401, 85–94. [Google Scholar] [CrossRef]
- Ali, B.; Iqbal, M.A. Coordination Complexes of Manganese and Their Biomedical Applications. ChemistrySelect 2017, 2, 1586–1604. [Google Scholar] [CrossRef]
- Trehoux, A.; Roux, Y.; Guillot, R.; Mahy, J.-P.; Avenier, F. Catalytic oxidation of dibenzothiophene and thioanisole by a diiron(III) complex and hydrogen peroxide. J. Mol. Catal. A Chem. 2015, 396, 40–46. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision A.03. Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Lee, C.T.; Yang, W.T.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Casida, M.E.; Jamorski, C.; Casida, K.C.; Salahub, D.R. Molecular excitation energies to high-lying bound states from time-dependent density-functional response theory: Characterization and correction of the time-dependent local density approximation ionization threshold. J. Chem.Phys. 1998, 108, 4439–4449. [Google Scholar] [CrossRef]
- Stratmann, R.E.; Scuseria, G.E.; Frisch, M.J. An efficient implementation of time-dependent density-functional theory for the calculation of excitation energies of large molecules. J. Chem Phys. 1998, 109, 8218–8224. [Google Scholar] [CrossRef]
- Barone, V.; Cossi, M.; Tomasi, J. A new definition of cavities for the computation of solvation free energies by the polarizable continuum model. J. Chem. Phys. 1997, 107, 3210–3221. [Google Scholar] [CrossRef]
- Cossi, M.; Scalmani, G.; Rega, N.; Barone, V. New developments in the polarizable continuum model for quantum mechanical and classical calculations on molecules in solution. J. Chem. Phys. 2002, 117, 43–54. [Google Scholar] [CrossRef]
- Andrae, D.; Häussermann, U.; Dolg, M.; Stoll, H.; Preuss, H. Energy-adjustedab initio pseudopotentials for the second and third row transition elements. Theor. Chim. Acta 1990, 77, 123–141. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| E1/2(A) | E1/2(B) | E1/2(C) | |
|---|---|---|---|
| 1(ClO4)2 | 0.058 ([Mn2II,II-MnII-Mn2II,II]/ [Mn2II,II-MnIII-Mn2II,II]) | 0.45 ([Mn2II,II-MnIII-Mn2II,II]/ [Mn2III,II-MnIII-Mn2III,II]) | 0.72 ([Mn2III,II-MnIII-Mn2III,II]/ [Mn2III,III-MnIII-Mn2III,III]) |
| 2(ClO4)2 | −0.040 [MnIIMnII−O−MnIIMnII]/ [MnIIMnIII−O−MnIIMnII] | 0.33 [MnIIMnIII−O−MnIIMnII]/ [MnIIMnIII−O−MnIIIMnII] | 0.69 [MnIIMnIII−O−MnIIIMnII]/ [MnIIIMnIII−O−MnIIIMnIII] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, Y.-T.; Deng, H.-T.; Zhang, L.-Y.; Li, M.-D.; Dai, F.-R.; Chen, Z.-N. Oligonuclear Manganese Complexes with Multiple Redox Properties for High-Contrast Electrochromism. Molecules 2025, 30, 2054. https://doi.org/10.3390/molecules30092054
Wu Y-T, Deng H-T, Zhang L-Y, Li M-D, Dai F-R, Chen Z-N. Oligonuclear Manganese Complexes with Multiple Redox Properties for High-Contrast Electrochromism. Molecules. 2025; 30(9):2054. https://doi.org/10.3390/molecules30092054
Chicago/Turabian StyleWu, Yi-Ting, Hao-Tian Deng, Li-Yi Zhang, Meng-Die Li, Feng-Rong Dai, and Zhong-Ning Chen. 2025. "Oligonuclear Manganese Complexes with Multiple Redox Properties for High-Contrast Electrochromism" Molecules 30, no. 9: 2054. https://doi.org/10.3390/molecules30092054
APA StyleWu, Y.-T., Deng, H.-T., Zhang, L.-Y., Li, M.-D., Dai, F.-R., & Chen, Z.-N. (2025). Oligonuclear Manganese Complexes with Multiple Redox Properties for High-Contrast Electrochromism. Molecules, 30(9), 2054. https://doi.org/10.3390/molecules30092054