Abstract
Phaeosphaeride A and its analogues have been extensively explored for their potential pharmacological applications, particularly in the development of anticancer agents. In this study, the synthesis of structurally modified phaeosphaeride analogues is reported. The structures of the synthesized analogues bearing the tetrahydro- and hexahydro-2H-furo[3,2-b]pyran-2-one and hexahydropyrano[3,2-b]pyrrol-2(1H)-one moieties were assessed and the new compounds were evaluated for their antiproliferative activity against two cancer cell lines. Despite successful synthesis and structural modification, the majority of the phaeosphaeride analogues exhibited limited bioactivity. Structure-activity relationship studies suggested that specific modifications did not enhance anticancer potency. The hydroxy groups and the alkyl moiety in cyclic or non-cyclic phaeosphaeride analogues contribute to the activity, as shown by the activity of compounds 24 and 25. The presence of double bonds and oxygen or nitrogen heteroatoms in furopyranones or pyranopyrrolones 9, 28, 29 and 33a, does not significantly impact cytotoxic activity. These findings highlight the challenges in optimizing phaeosphaerides for anticancer applications and provide insights into future structural modifications to improve their therapeutic potential. Moreover, our studies open a synthetic route for the development of new phaeosphaeride analogues.
1. Introduction
Natural products have been a rich source of inspiration for drug discovery since ancient times [1]. Nowadays, numerous studies demonstrate the efficacy of natural products, their derivatives and synthetic analogues in targeting biological pathways and treating or preventing human diseases such as cancer, infections, cardiovascular diseases and others [2,3]. Natural product drug discovery is often a complicated process due to the structural complexity of natural products, requiring multistep, expensive, and time-consuming synthesis. In response to current drug discovery needs, natural product synthesis and the development of natural product-based chemical libraries remain at the forefront of pharmaceutical research [4,5,6].
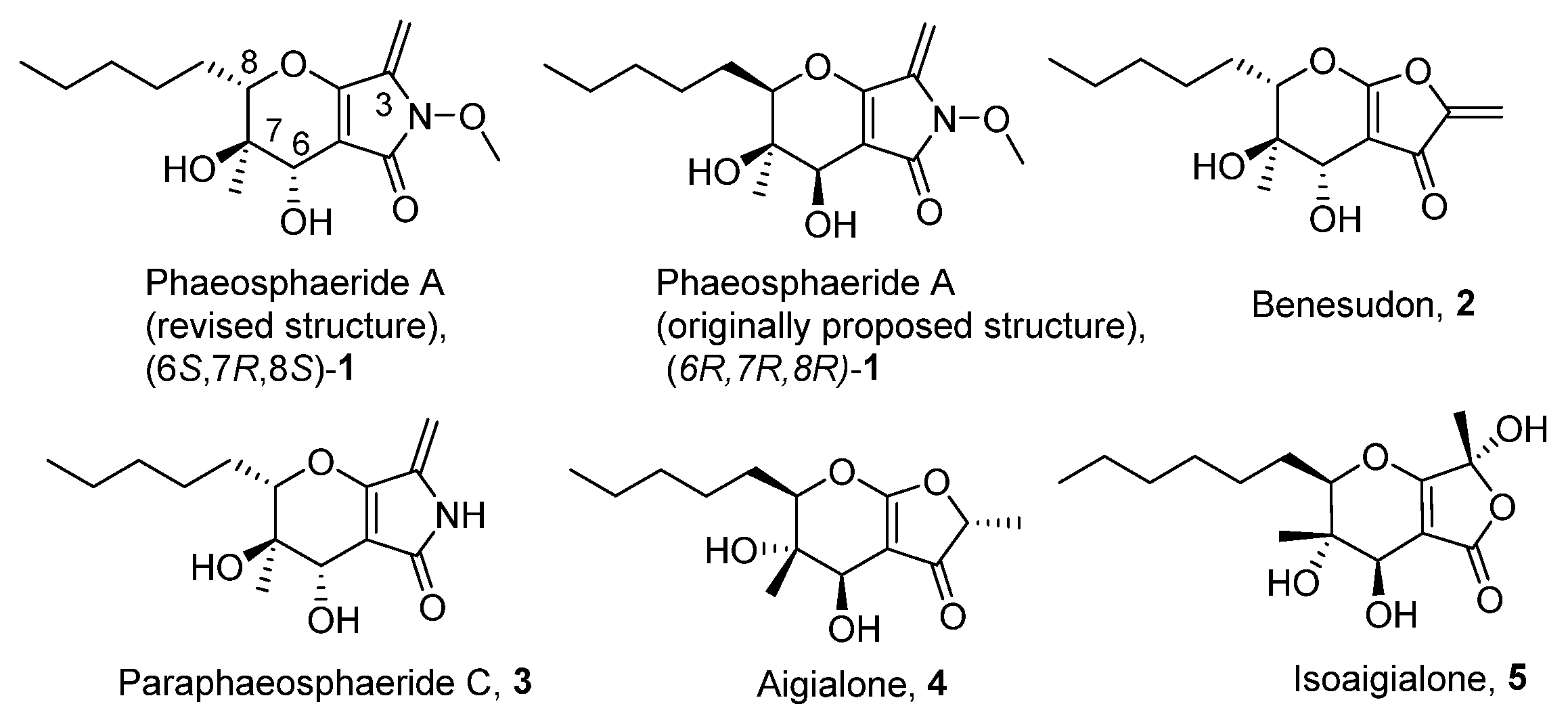
In 2006 phaeosphaerides A and B were isolated by an endophytic fungus from the genus Phaeosphaeria [7]. Phaeosphaeride A was found to inhibit the Signal Transducer and Activator of Transcription 3 (STAT3) signalling pathway and display promising anticancer activities. In the following years, phaeosphaerides have attracted the attention of our group and Kobayashi’s as synthetic targets [8,9,10]. Initial efforts were focused to structural elucidation and determination of the mode of action of phaeosphaerides. Subsequent biological studies and SAR investigation by Berestetskiy et al. pointed out the herbicidal potential of phaeosphaeride A [11], while Abzianidze et al. studied the anticancer activities of synthetic phaeosphaeride derivatives [12,13]. Since phaeosphaerides [14], paraphaeosphaerides 3 [15], aigialone 4 [16], isoaigialone 5 [15] and related compounds [17] are biologically active natural products, we aimed to synthesize hydrofuropyrans and hydropyranopyrrolones as synthetic analogues of phaeosphaerides and study their antiproliferative properties (Figure 1).
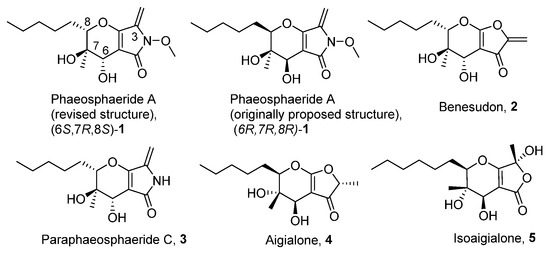
Figure 1.
Phaeosphaerides and other structural related natural products.
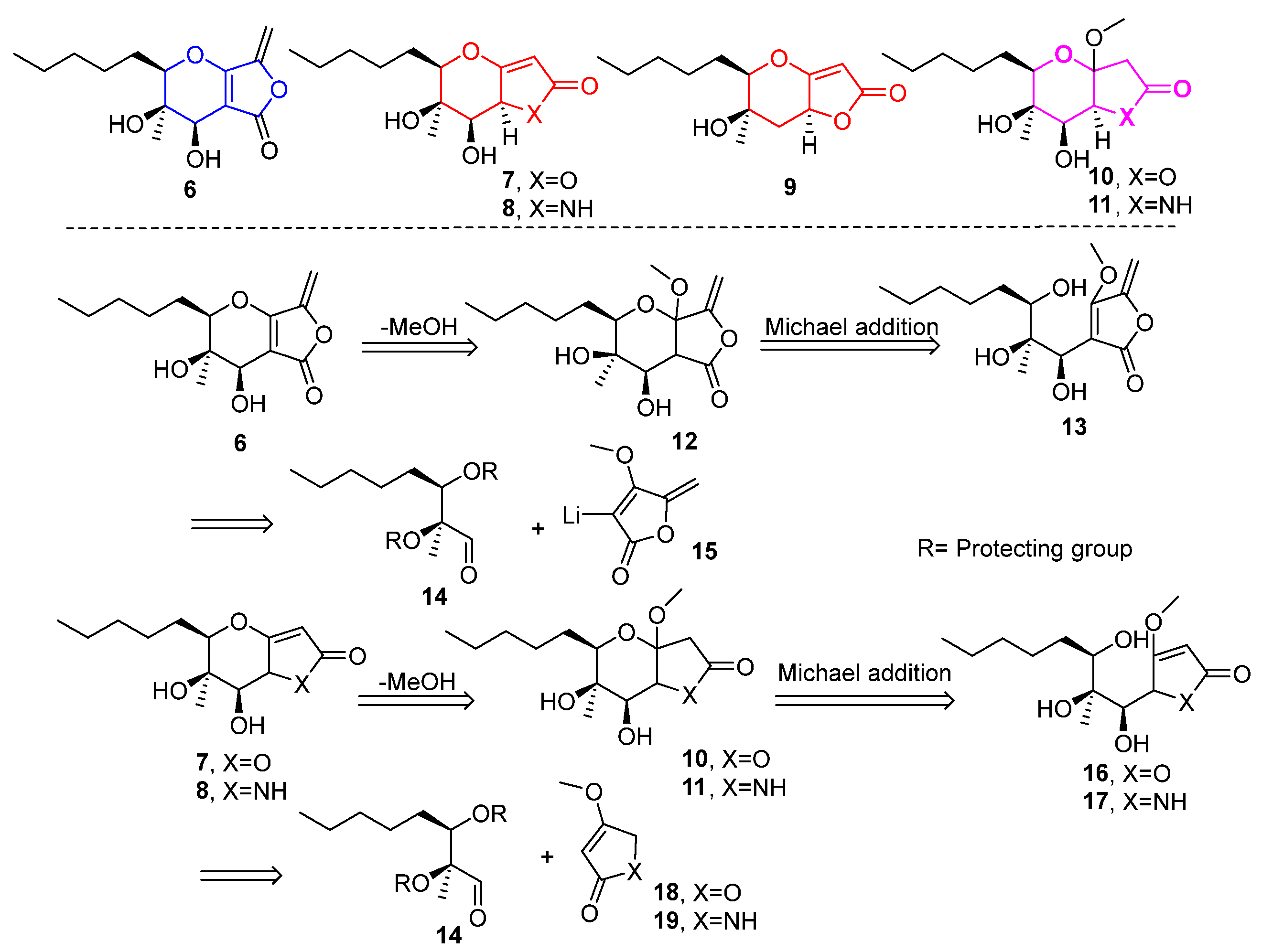
Our first plan included efforts to synthesize 6, containing the tetrahydro-5H-furo[3,4-b]pyran-5-one scaffold, which is naturally occurring. The same protocol could then be applied to the synthesis of tetrahydro- and hexahydro-2H-furo[3,2-b]pyran-2-one 7, 9, 10 and tetrahydro- and hexahydropyrano[3,2-b]pyrrol-2(1H)-ones 8, 11. The synthetic route would be similar to that initially reported by our group during the total synthesis of the proposed structure of phaeosphaeride A, (6R,7R,8R)-1, with significant differences after the aldehyde synthesis stage [9]. Herein, key synthetic steps were designed to include the addition of lithiated methyl tetronate 15 to 14, the vinylogous aldol reaction of 18, 19 to 14, deprotection of the hydroxy protecting groups and intramolecular oxy-Michael reactions for the formation of the 3,4-dihydro-2H-pyran (Figure 2).
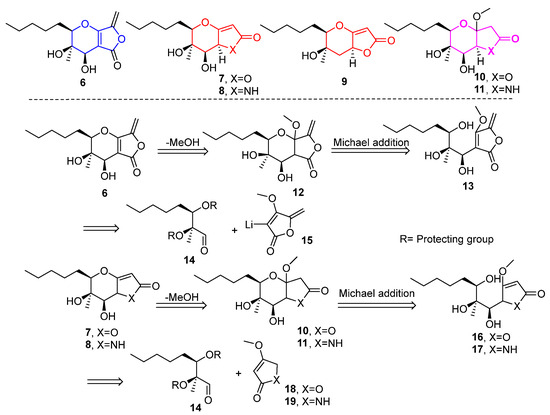
Figure 2.
Retrosynthetic disconnections for phaeosphaeride analogues. Lactone 6 and analogues 7–11 were selected as synthetic targets in this work.
2. Results
2.1. Synthesis of Phaeosphaeride Analogues
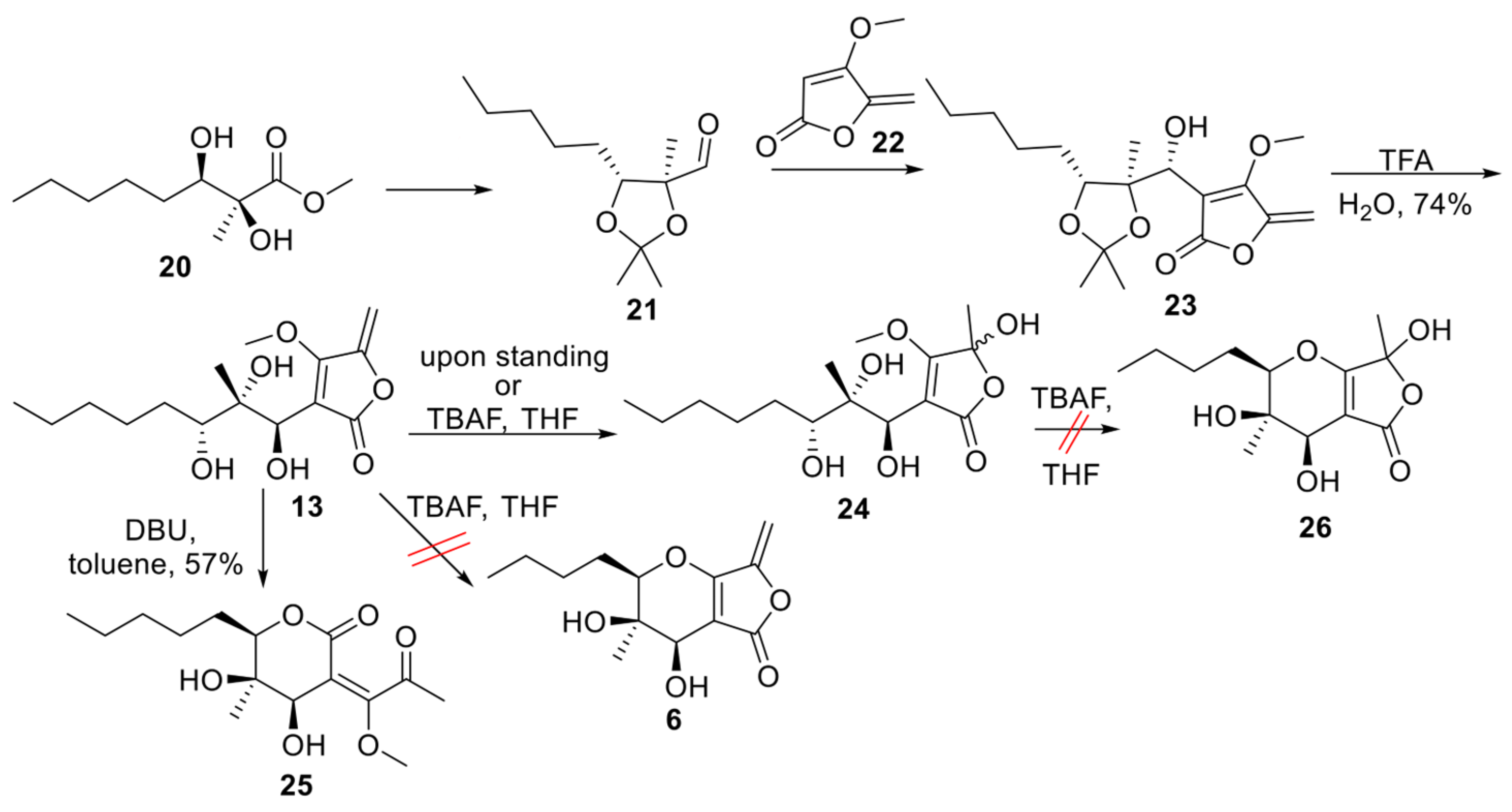
Aldehyde 21 (Scheme 1) represents a versatile chiral intermediate to access phaeosphaeride analogues. According to our previous work, the synthesis of compound 21 involved the addition of vinyl lithium reagent of 22 to the acetonide-protected aldehyde 21 following the acetonide group deprotection using TFA. Although compound 13 was stable enough to be purified by column chromatography, upon standing, it was hydrated to compound 24. Therefore, 13 was advanced to the next step immediately after its purification. Subsequently, intramolecular oxy-Michael addition using various reagents and conditions was attempted. Initially, 1.5 equivalent of TBAF (1.6M) in THF was used at 60 °C following the previously described procedure for the Michael reaction in phaeosphaeride derivatives synthesis (Table 1). However, the hydration of the exocyclic double bond took place and the formation of isomer 24 (20% yield) was again observed together with a complicated reaction mixture containing retro aldol reaction products. In a second attempt, a different base DBU was selected instead of TBAF. The reaction was carried out using 1.5 equivalents of DBU in toluene at rt and stirring overnight. In this case, the six-membered lactone 25 formation took place and the desired product 6 could not be detected. On the other hand, the cyclisation of 13 in acidic conditions using p-TSA resulted in complicated mixtures. Our experiments indicate that the intramolecular Michael reaction is challenging to perform, and tetronate 13 preferably produces lactone 25.

Table 1.
Conditions for the cyclisation of tetronate 13.
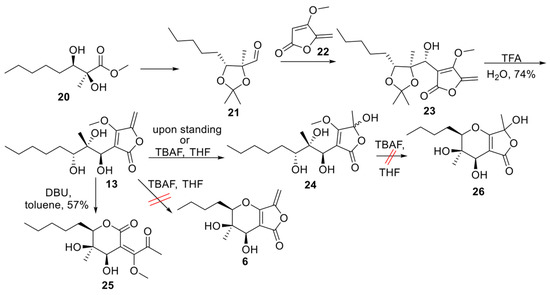
Scheme 1.
Cyclisation of tetronate 13 [18].
Scheme 1.
Cyclisation of tetronate 13 [18].
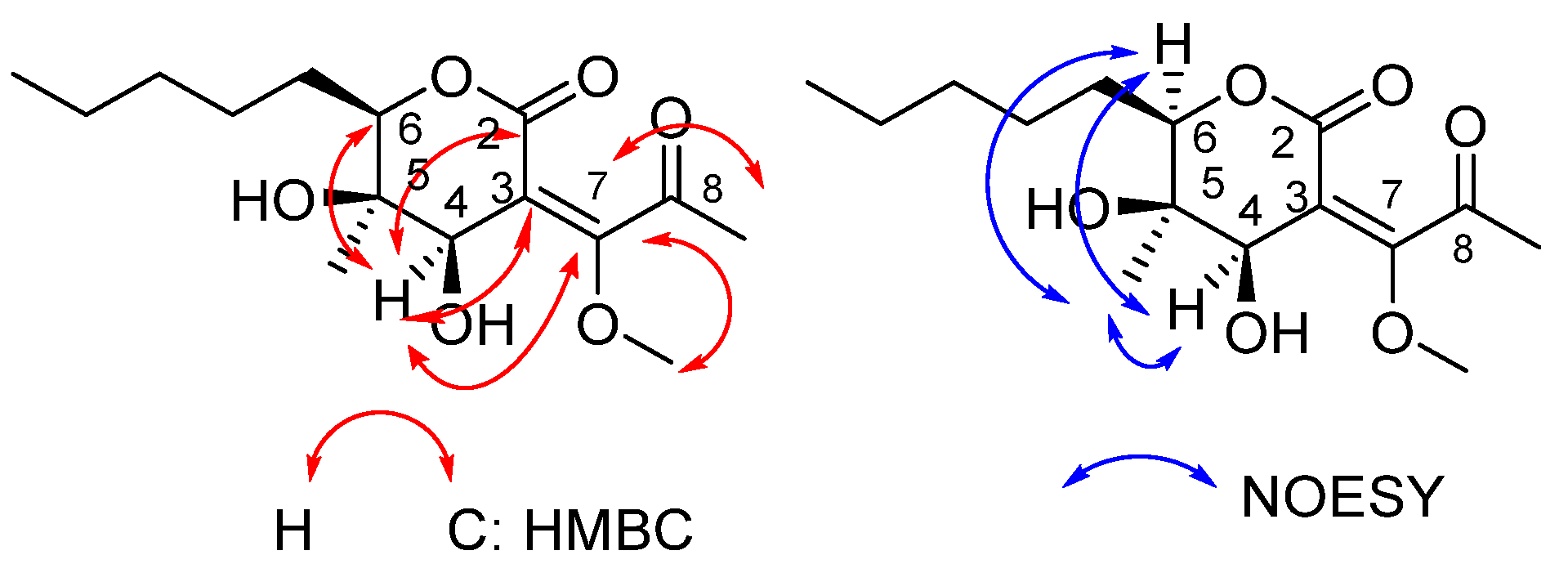
The two-dimensional HMBC and NOESY correlations, as well as the final structure assigned to product 25, are shown in Figure 3. The two-dimensional spectra demonstrate the characteristic correlation between the C-7 of the double bond and H atoms of the methoxy group. HMBC correlations of H-4 /C-2 and C-7 indicated the formation of the tetrahydro-2H-pyran-2-one core. The chemical shift in the ketone carbonyl carbon at 198.3 ppm is highly diagnostic of the structure of 25. The assigned structure was further supported by detailed analysis of the 1D and 2D NMR data (Table 2).
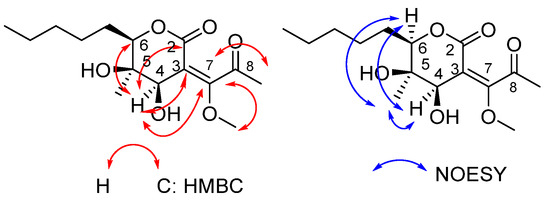
Figure 3.
Key HMBC and NOESY and correlations of compound 25.
Table 2.
1H and 13C NMR Data for 25, at 500 and 125 MHz.
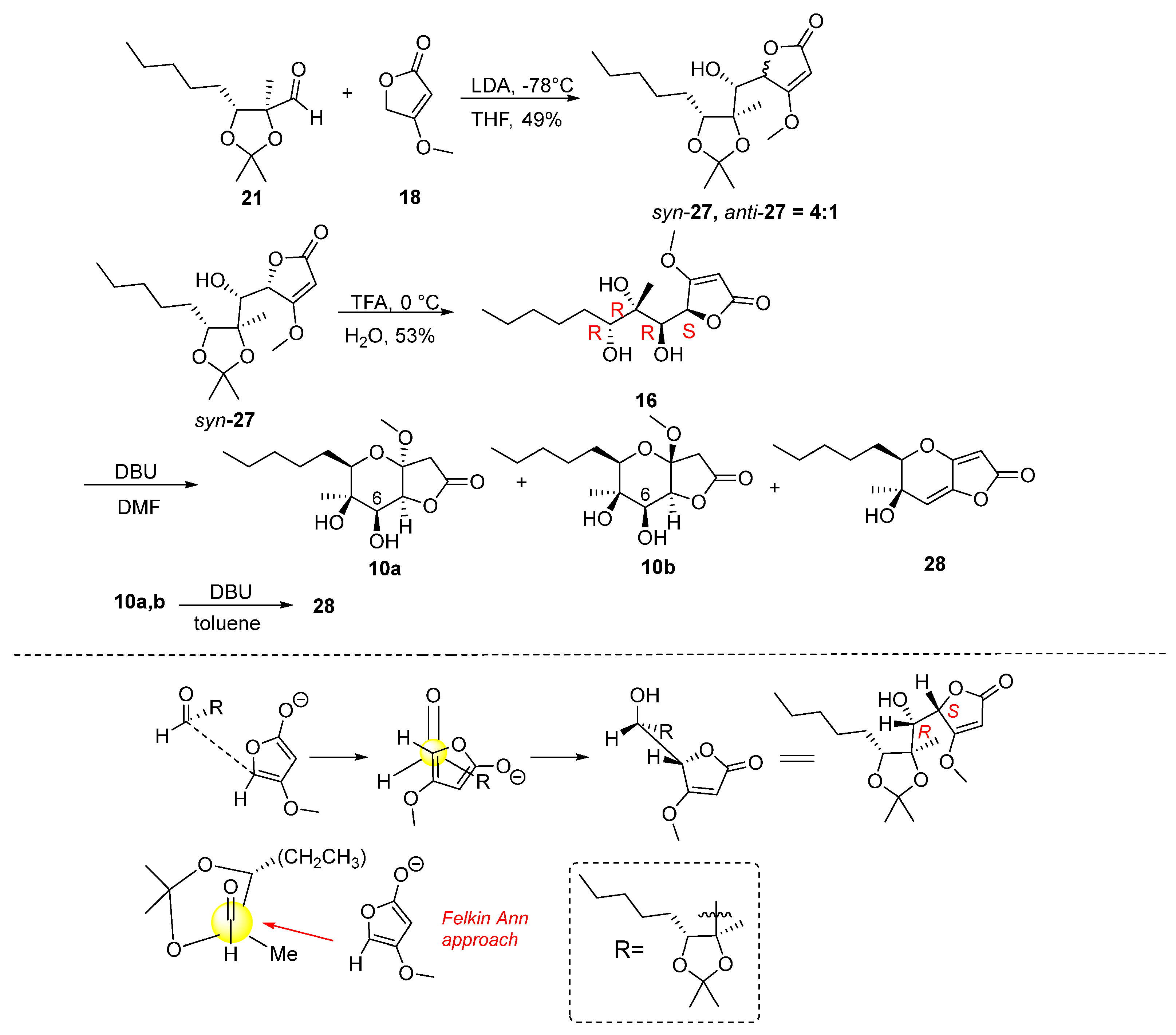
Attention was then turned to the synthesis of analogues 7 and 9 (Figure 2). Specifically, the synthesis started via a reaction between aldehyde 21 and the lithium derivative of 4-methoxyfuran-2(5H)-one 18. The reaction was carried out using LDA in THF solvent at −78 °C for two hours (Scheme 2), which resulted in the formation of butenolides 27 in 49% yield and in a ratio of syn:anti = 4:1. We were able to isolate the major isomer syn-27, the stereochemistry of which was determined at a later stage. The addition of 4-methoxyfuran-2(5H)-one to 21 furnishes mainly the Felkin–Anh product syn-27 as shown in Scheme 2. However, it should not be excluded that butanolide syn-27 is the thermodynamic stable product resulting from the epimerization of the butenolide stereogenic center under basic conditions as previously reported by Karak et al. [19].
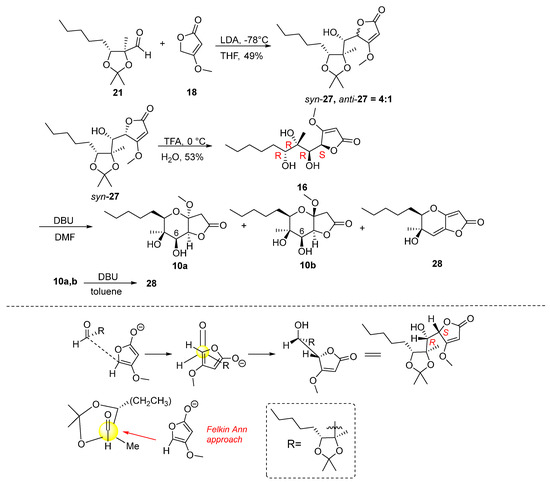
Scheme 2.
Synthesis of γ-butyrolactones 10a,b and butenolide 28.
Having syn-27 in hand, the synthetic course proceeded to the deprotection step using TFA (Scheme 2). Butenolide 16 was obtained in 53% yield and was then stirred under basic conditions to form the tetrahydropyran ring (Table 3). The reaction was carried out with 1.5 equiv. of DBU in DMF solvent at 60 °C for 72 h, which yielded a mixture of two cyclization product isomers 10a,b in a 4:1 ratio and in 69% yield. Their complete separation was not possible by column chromatography in various solvents. However, we were able to isolate and fully characterize a small amount of the minor isomer 10b, with a purity of 70% relative to the major one 10a. Along with the mixture of isomers, the reaction also resulted in the formation of product 28 in 39%. Compound 28 was formed from MeOH elimination and dehydration of alcohols 10a,b.
Table 3.
Cyclisation of 16 in DMF.
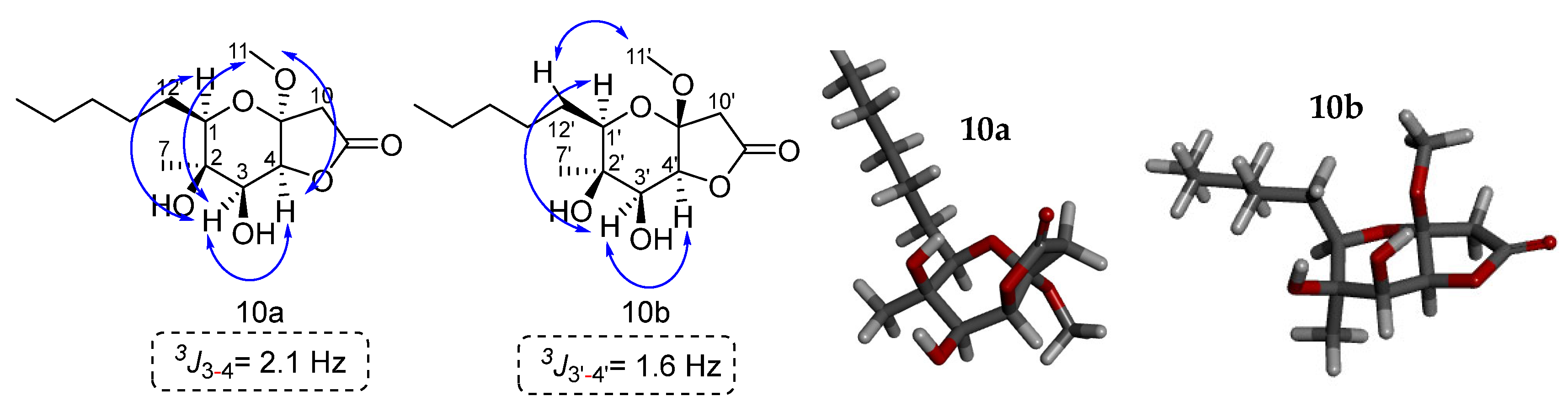
Our next efforts were focused on the structure determination of 10b. It was possible to isolate also a small amount of minor isomer 10b to assign its stereochemistry. The sample had a purity of 70% of 10b and 30% of 10a. The two-dimensional NOESY spectrum shows key correlations between the C-1′ hydrogen and the C-3′ hydrogen. Additionally, a correlation is observed between the C-3′ and C-4′ hydrogens, suggesting the syn arrangement between them. The coupling constant 3J3′-4′ = 1.6 Hz confirms this stereochemistry. For 10b the methoxy group shows a correlation of its hydrogens with the C-12′ hydrogens. For 10a the stereochemistry for the methoxy group could be determined by the association of its hydrogens with the C-3 and C-4 hydrogens. Another sample that was enriched with 10a (10a:10b = 4:1) was used for the 2D NMR experiments. NOESY correlations for 10a indicated a syn relationship between H-1, H-3, CH3O and H-4 on the dihydropyran ring. This is verified by the coupling constant 3J3-4 of 2.1 Hz between H-3 and H-4. Based on these results we can conclude that the relative stereochemistry of the two isomers is this shown in Figure 4.
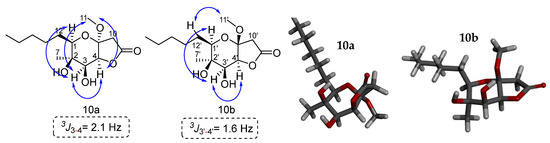
Figure 4.
Key NOESY correlations of lactones 10a and 10b. The modelling of compounds 10a and 10b was performed with the MM2 method molecular energy calculations using Chem3D 15.0 software. BIOVIA Discovery Studio was used for the visualization of the 3D model.
To improve the cyclization reaction of 16, it was carried out under the same conditions changing the time from 72 to 24 h (Table 2). It was observed that in a shorter reaction time, the mixture of isomers was formed in a higher yield of 74%, while 28 was isolated in a lower yield (12%). At the same time, an attempt was made to form compound 28 without obtaining the mixture of isomers. Thus, compounds 10a,b reacted with three equivalents of DBU in DMF with at 80 °C for 48 h. From the reaction both the mixture of isomers 10a,b and compound 28 were obtained in 46% yield and 47% yield, respectively. In conclusion, the formation of 28 is favoured with an increase in base equivalents, temperature and reaction time. In another attempt, a mixture of 10a,b reacted with three equivalents of DBU in DMF at 80 °C overnight. The reaction led to 28 in 23%. When changing the solvent from DMF to toluene the reaction, proceeded slowly. Thus, using 1.5 equivalents of DBU at 80 °C for 48 h with unchanged starting material, 10a,b was received, together with a small amount of 28 (6%).
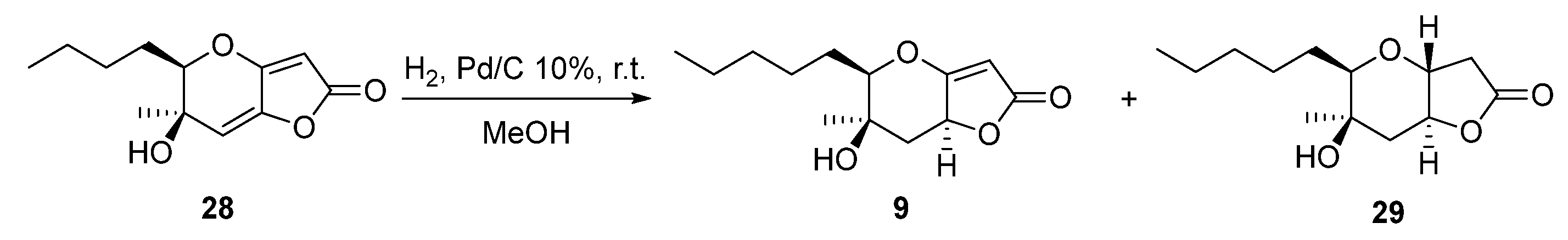
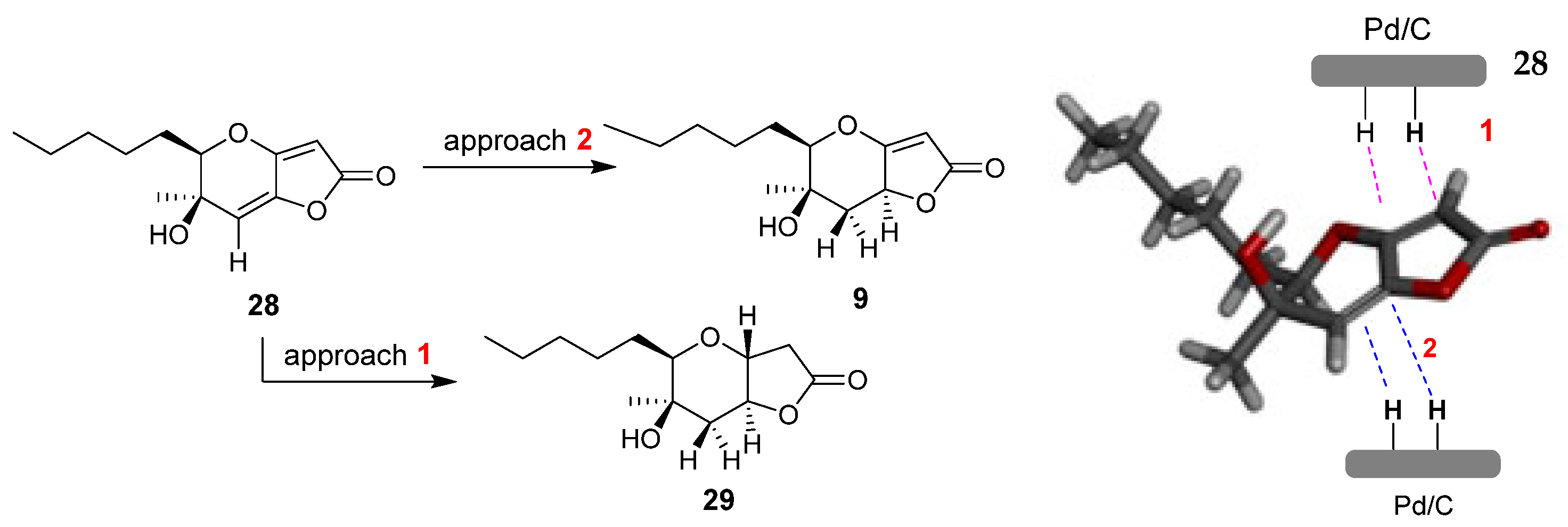
Another interesting phaeosphaeride analogue was obtained by reacting compound 28 under reductive catalytic hydrogenation conditions (Scheme 3). Specifically, the reaction was carried out in the presence of 10% Pd/C catalyst in MeOH solvent at room temperature for 30 min under an H2 atmosphere. The 1H NMR spectrum data indicated that the reduction took place at the double bond of the six membered ring forming compound 9 in 79% yield. The fully hydrogenated product 29 was also isolated in 18% yield.
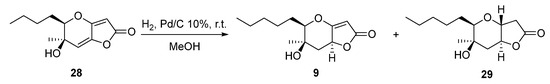
Scheme 3.
Catalytic hydrogenation reaction of compound 28.
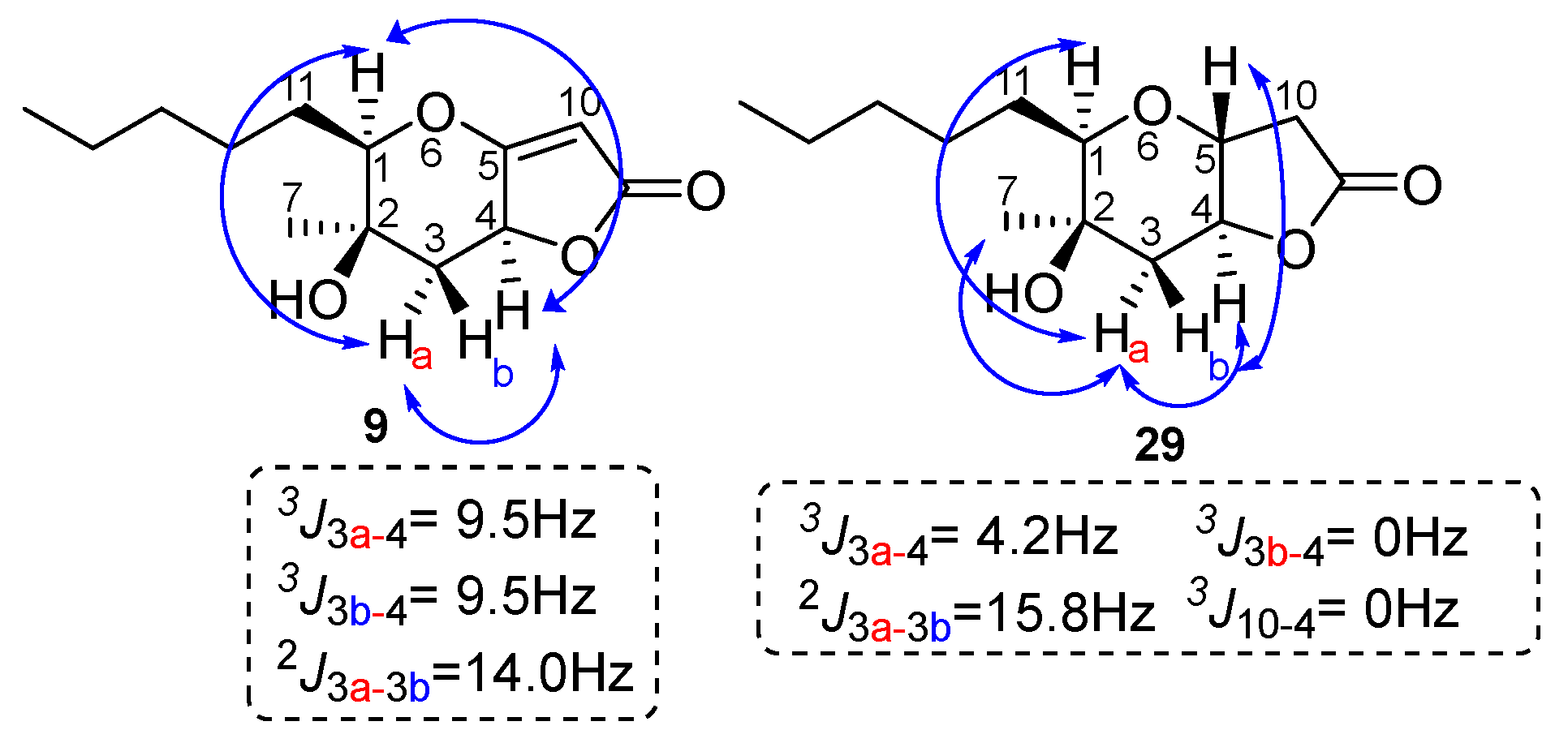
To assign the stereochemistry of derivative 9, the two-dimensional NOESY spectrum was recorded. As shown in Figure 5, there is a correlation between the C-4 hydrogen and the hydrogen of C-1 hydrogen. Thus, we conclude that the stereochemistry of compound 9 is that shown in Figure 5 below. This stereochemistry is considered more likely because the catalyst approaches 28 from side 2, due to the smaller hindrance of the methyl group compared to the hydroxy group on the opposite side (Figure 6). Compound 29 is produced when the catalyst approaches 28 from side 1, with the double bond of the five-membered unsaturated lactone first being reduced, or possibly being produced by the subsequent reduction of 9. For compound 29, the NOEs on H-5 with H-3b and H-3a with H-1, H-7 and H-4 defined the H-5, pentyl and OH group on the same side of the hexahydro-2H-furo[3,2-b]pyran-2-one, 28.
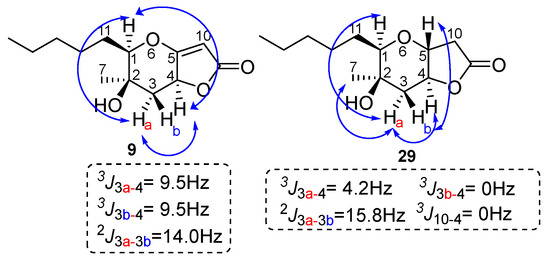
Figure 5.
Relative stereochemistry for the furo[3,2-b]pyran-2-one system of 9 and 29. Key NOE interactions are indicated by arrows.
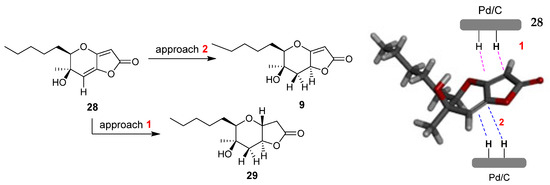
Figure 6.
Catalytic hydrogenation of 28. The modelling of compound 28 was performed with the MM2 method molecular energy calculations using Chem3D 15.0 software. BIOVIA Discovery Studio was used for the visualization of the 3D model.
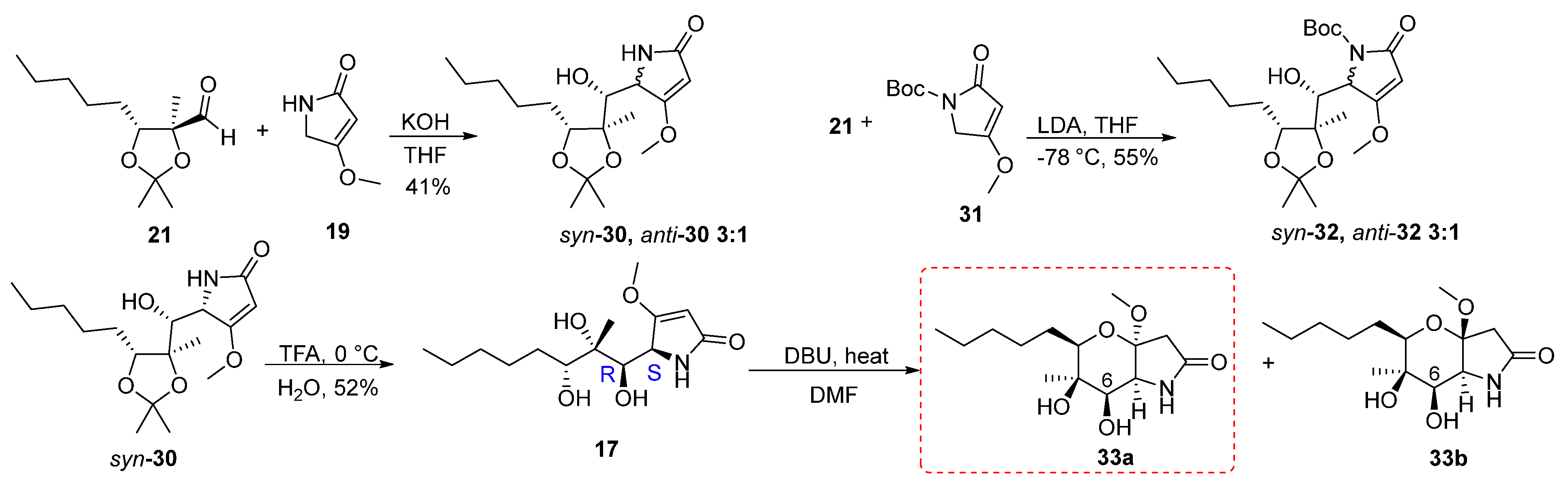
The synthetic route followed to obtain the tetrahydropyrano[3,2-b]pyrrol-2(1H)-one 11 started from the vinylogous aldol reaction of aldehyde 21 with 19 (Scheme 4). First, the reaction was carried out using 1.6 equivalents of 4-methoxy-3-pyrrolin-2-one 19, by adding an aqueous solution of KOH base (4M) in THF solvent at room temperature overnight. Formation of the anion of compound 19 and the addition to the aldehyde was carried out at 60 °C, after which the reaction was brought to room temperature. The reaction gave a mixture of adducts 30 in a ratio of syn:anti = 3:1 in a yield of 41%. The determination of the stereochemistry of the products was attempted at a later stage after the cyclization and formation of the pyran ring. The separation of the two isomers was troublesome due to the excess of 19, as the products and starting material had similar Rf values when developed by thin layer chromatography. To optimize the reaction, one equivalent of 19 was used to facilitate the isolation process of the products. Furthermore, to control the formation of isomers, the addition of aldehyde 21 to the anion of compound 19 was carried out at 0 °C, while the reaction was then allowed to reach room temperature. But even in this case, two isomers were formed in a ratio of 4:1 and 13% yield, without the starting materials being completely consumed. When 21 reacted with the Boc protected pyrrolin-2-one 31 at −78 °C with LDA as a base in THF the vinylogous aldol reaction yielded adducts 32 in 55% yield in syn:anti = 3:1 ratio.
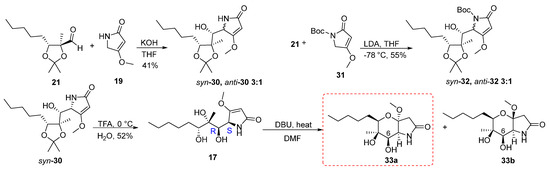
Scheme 4.
Synthesis of the pyrano[3,2-b]pyrrol-2(1H)-one 33a.
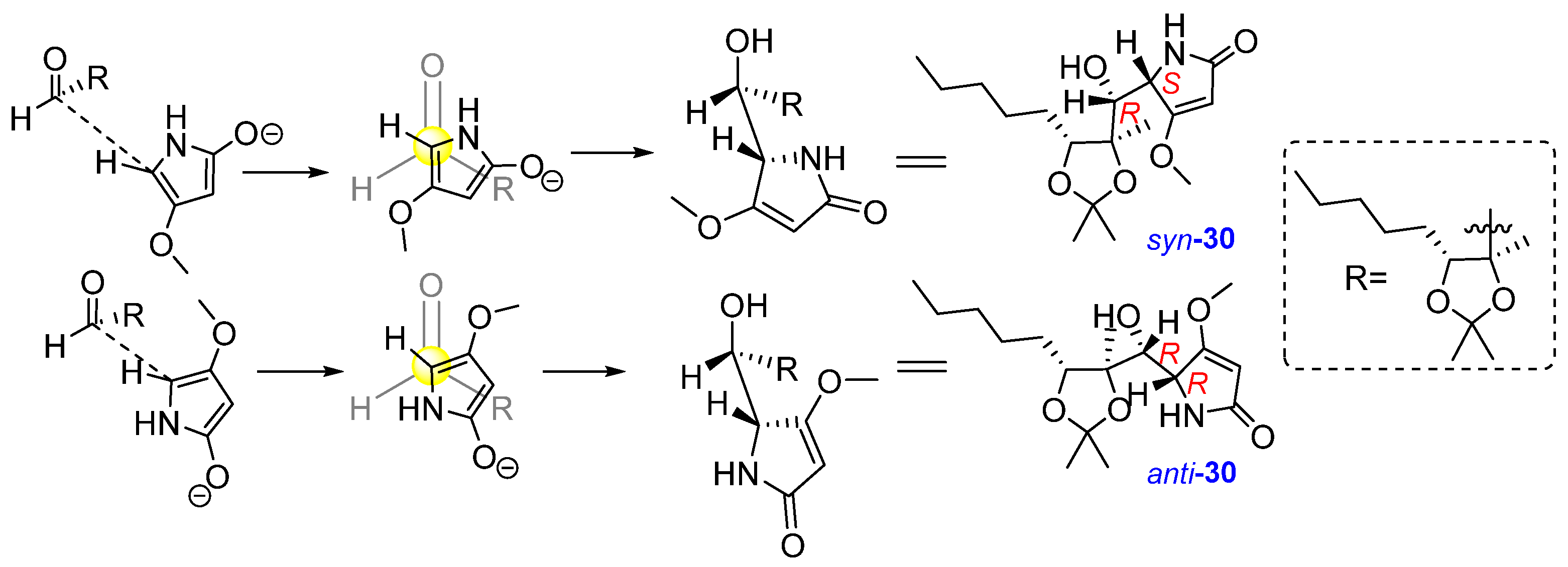
The addition of the 4-methoxy-1,5-dihydro-2H-pyrrol-2-one 19 to aldehyde 21 could result from the following vinylogous aldol reaction, shown in Figure 7. Syn-anti isomerization on the asymmetric centre of dihydro-2-pyrrolidone could also occur.
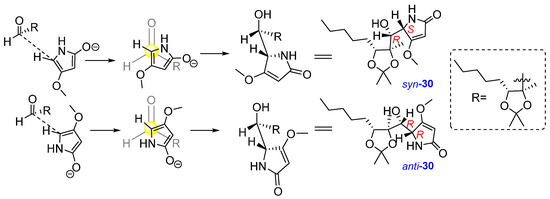
Figure 7.
Formation of adducts syn-30 and anti-30.
Having obtained the mixture of the two isomers 30 from the vinylogous aldol reaction, we proceeded to the next step of deprotection. During this reaction, syn-30 was deprotected using TFA to give 17 in 50% yield. Then, 17 reacted with 1.5 equiv. of TBAF in THF solvent at 60 °C for 1 h; however, a complex mixture of compounds was received. In a second attempt, the reaction was carried out under the same conditions and left stirring overnight (Table 4). From the 1H spectrum a complex mixture of compounds was observed. In addition, the DBU base was used to achieve cyclization. The reaction was initially carried out using 1.5 equivalents of DBU in toluene solvent at room temperature overnight. However, we did not observe any change in the starting material. Thus, the reaction was carried out again under the same conditions but with heating at 60 °C, which led to the formation of two isomers of forms 33a,b, in a 62% yield and in a 10:1 ratio. We were able to isolate and characterize only 33a.
Table 4.
Cyclisation of 33a in DMF.
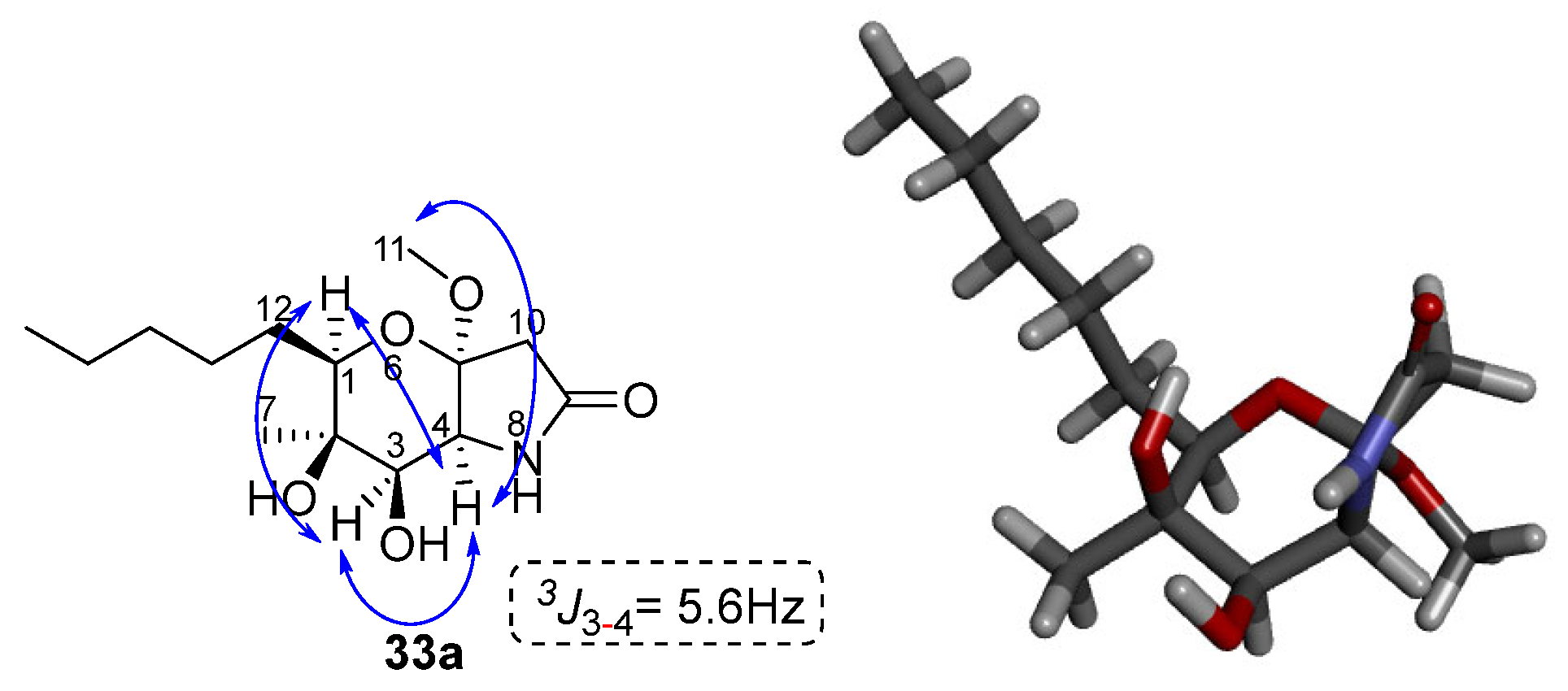
The stereochemistry of compound 33a was determined based on the two-dimensional NOESY spectrum data. Specifically, as shown in Figure 8, correlation of the hydrogens of methyl 7 and N-H with the hydrogen of C-3 is observed. In addition, there is a correlation between the C-1 hydrogen with the C-3 and C-4 hydrogens. Finally, the C-4 hydrogen is correlated with the 11 methoxy group. Based on these observations, the stereochemistry of the cyclized product 33a can be assigned as shown in Figure 8 below.
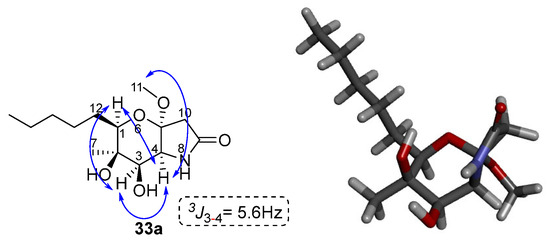
Figure 8.
Key NOESY and correlations of compound 33a. The modelling of compound 33a was performed with the MM2 method molecular energy calculations using Chem3D 15.0 software. BIOVIA Discovery Studio was used for the visualization of the 3D model.
2.2. Cell Viability Studies
In the final phase of our study, we aimed to determine the anticancer potential of newly synthesized phaeosphaeride derivatives. We utilized two distinct cancer cell lines representing different malignancies: HeLa cells derived from cervical cancer and MOLT-4 cells from acute lymphoblastic leukemia. The cytotoxic activity of selected compounds was evaluated using the MTT metabolic assay. Cells were exposed to various concentrations of the compounds (ranging from 5 to 100 μM) for a duration of 48 h. The results, expressed as EC50 values (the concentration causing 50% loss of cell viability compared to non-treated cells), are presented in Table 5.
Table 5.
Half maximal effective concentration (EC50 Values, μΜ) of selected compounds against HeLa and MOLT-4 cancer cell lines. Results are presented as mean ± SE of three independent experiments.
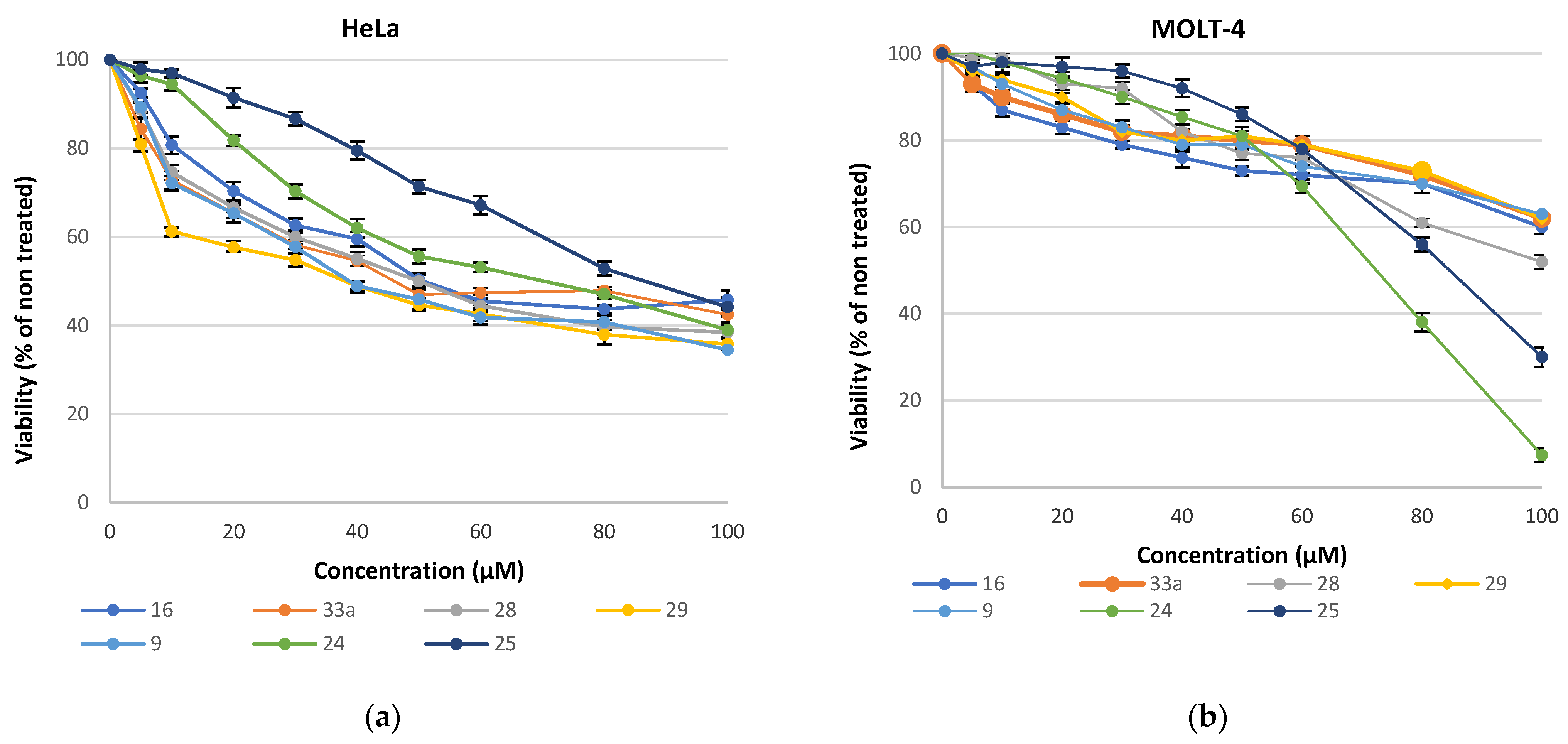
Analysis of the cytotoxic effects revealed that MOLT-4 cells (Figure 9b) exhibited greater resistance (EC50 > 70 μM) compared to HeLa cells (Figure 9a), except for 25, which had a similar limited effect on both cell lines. Treatment of HeLa cells with the compounds showed a notably higher impact with the most potent compounds exhibiting EC50 values in the 40 μM range.
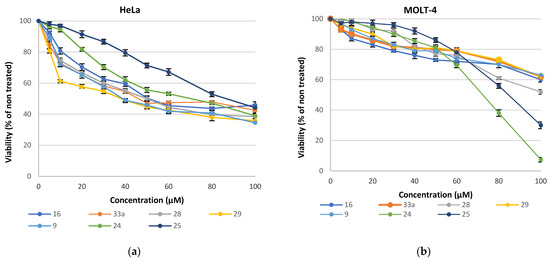
Figure 9.
Growth inhibition of (a) HeLa cells; (b) Molt-4 cells grown in the presence of different concentrations of the synthesized compounds for 48 h. Values are means of three independent experiments, with the standard errors of the mean represented by vertical bars.
3. Discussion
Inspired by the structures of natural phaeosphaerides and paraphaesphaerides a series of analogues bearing the tetrahydro- and hexahydro-2H-furo[3,2-b]pyran-2-one and hexahydropyrano[3,2-b]pyrrol-2(1H)-one moieties were synthesized. Key synthetic steps included the vinylogous aldol reactions to aldehyde 21, deprotection of the acetonide protecting group and intramolecular oxy-Michael reactions for the formation of the 3,4-dihydro-2H-pyran ring. The synthetic approach proved challenging in terms of diasteroselectivity and stability of synthetic intermediates; however, it has produced divergent sp3-enriched structures. Despite successful synthesis and structural modification, the majority of the phaeosphaeride analogues exhibited limited bioactivity against 2 cancer cells, HeLa and MOLT-4. The structure–activity relationship analysis suggested that the new modifications on the phaeosphaeride bicyclic system did not enhance anticancer potency and the α,β-unsaturated carbonyl functionality is important for activity. Phaeosphaerides appear to act through mixed mechanisms of action involving inhibition of the IL-6-activated STAT3 signalling pathway [18], oxidative stress and the modulation of JNK, ERK1/2, and p38 signalling pathways, according to Hirayama and Abzianidze [20,21]. As recently reported by Hirayama et al. phaeosphaeride A potently inhibits the proliferation of HeLa cells with IC50 value of 8.8 μΜ [18]. These studies indicate that the presence of exomethylene group at the C-3 position of phaeosphaeride is important for activity, suggesting that phaeosphaerides act as Michael acceptors in cancer cells. Our findings further support the importance of exomethylene group for activity, but also its high reactivity with nucleophiles such as water (demonstrated by the formation of 24). In addition, the hydroxy groups and the alkyl moiety in cyclic or non-cyclic phaeosphaeride analogues contribute to the activity, as shown by the antiproliferative activity of 24 and 25. The presence of double bonds and oxygen or nitrogen heteroatoms in furopyranones or pyranopyrrolones 9, 28, 29 and 33a does not significantly impact cytotoxic activity. In HeLa cells, the synthesized compounds demonstrate moderate activity (EC50 values in the 40 μM range), and weaker activity against MOLT-4 cells. The most active compounds against MOLT-4 are the α,β-unsaturated carbonyl compounds 2-furanone 24 and lactone 25.
4. Materials and Methods
General Experimental Details
Reactions were carried out under an atmosphere of Argon unless otherwise specified. Commercial starting materials were purchased from Merck (Darmstadt, Germany) or Alfa Aesar (Ward Hill, MA, USA) and used without further purification. Reactions were monitored by TLC using silica plates 60-F264 and UV light as a visualizing agent or aqueous ceric sulphate/phosphomolybdic acid, ethanolic p-anisaldehyde solution, potassium permanganate solution and heat as developing agents. 1H and 13C NMR spectra were recorded at 500 and 126 MHz (Agilent) with tetramethylsilane as an internal standard. Chemical shifts are given in δ values (ppm) from internal reference peaks (TMS 1H 0.00; CDCl3 1H 7.26, 13C 77.16, (CD3)2SO 1H 2.50, 13C 39.52, (CD3)2CO 1H 2.05, 13C 29.84, 206.26). LC-MS analysis was performed on a LC-20AD Shimadzu connected to Shimadzu LCMS-2010EV (Shimadzu Kyoto, Japan) equipped with C18 analytical column (Supelco discovery C18, 5 μm 250 × 4.6 mm). HRMS experiments were carried out on a QExactive Plus Mass Spectrometer (Agilent, Santa Clara, CA, USA); flow rate 0.5 mL / min, 90% CAN + 0.1% HCOOH; spray voltage = 3 kV; capillary temperature = 300 °C. Melting points (mp) are uncorrected. Optical rotation was measured on a Kruss P-3000 polarimeter with a sodium lamp at the solvent, temperature, and concentration indicated for each compound. ESI-MS analysis and NMR spectra of all synthesized compounds are reported in the Supplementary Materials.
4-methoxy-5-methylene-3-((1S,2R,3S)-1,2,3-trihydroxy-2-methyloctyl)furan-2(5H)-one, 13
To a round bottom flask containing 23 [8] (19.3 mg, 0.057 mmol), a solution of TFA, in water (0.5 mL, 6.521 mmol, 1:1 v/v), was added. The reaction was stirred for 2 h at 0 °C, quenched with saturated aqueous NaHCO3 and extracted with DCM. The combined organic layers were washed with brine, dried over Na2SO4, and concentrated under reduced pressure. The residue was purified by column chromatography (gradient elution PS–EA 2:1 to 1:2) to afford 13 (74%) as orange oil. 13: 1H NMR (500 MHz, CDCl3) δ 5.16 (d, J = 3.0 Hz, 1H), 5.16 (d, J = 3.0 Hz, 1H), 4.95 (s, 1H), 4.21 (s, 4H), 3.63 (dd, J = 9.7, 2.7 Hz, 1H), 3,10 (br, 1H), 2.7 (br, 1H), 1.67–1.54 (m, 3H), 1.51–1.40 (m, 2H), 1.37–1.27 (m, 3H), 1.19 (s, 3H), 0.88 (t, J = 7.0 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 171.0, 163.7, 149.3, 105.3, 94.8, 77.1, 74.9, 69.8, 60.5, 31.8, 30.9, 25.9, 22.6, 18.9, 14.0; ESI-MS, positive mode: m/z calcd mass for C15H24O6 [M+Na]+ = 323.1471, was found to be 322.95.
5-hydroxy-4-methoxy-5-methyl-3-((1R,2S,3R)-1,2,3-trihydroxy-2-methyloctyl) furan-2(5H)-one, 24
To a solution of 13 (7.6 mg, 0.025 mmol) in 0.7 mL THF, TBAF (11 μL, 0.038 mmol, 1.6 M) was added. After stirring for 1 h at 60 °C, the reaction was quenched with saturated aqueous NaHCO3 and extracted with EA. The combined organic layers were washed with brine, dried over Na2SO4 and concentrated in vacuo. The residue was purified with column chromatography (gradient elution PS–EA 2:1 to 1:2) to afford the isomeric mixture 24 (20%) as orange oil. 24 (major isomer): 1H NMR (500 MHz, CDCl3) δ 4.62 (s, 1H), 4.31 (s, 3H), 4.07 (dd, J = 10.0, 2.3 Hz, 1H), 1.77–1.64 (m, 5H), 1.59 (s, 3H), 1.38 (m, 3H), 1.35 (s, 3H), 0.90 (t, J = 6.9 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 163.1, 161.0, 105.7, 94.5, 84.1, 83.7, 70.5, 62.4, 31.6, 28.7, 25.6, 22.8, 22.5, 21.3, 14.0; ESI-LCMS, positive mode: m/z calcd mass for C15H24NaO7 [M+Na]+ = 341.1576, was found to be 341.20.
(4R,5R,6R,E)-4,5-dihydroxy-3-(1-methoxy-2-oxopropylidene)-5-methyl-6-pentyltetrahydro-2H-pyran-2-one, 25
To a solution of 13 (5.3 mg, 0.018 mmol) in 0.47 mL toluene, DBU (4 μL, 0.026 mmol) was added, and the reaction was stirred overnight at room temperature. The reaction mixture was then concentrated under reduced pressure and the residue subjected to column chromatography (gradient elution PS–EA 2:1 to 1:2) to form 25 (57%) as orange oil. 25: 1H NMR (500 MHz, CDCl3) δ 4.90 (s, 1H), 3.89 (s, 3H), 3.47 (d, J = 9.7 Hz, 1H), 2.51 (s, 3H), 2.07 (br, 1H), 1.64–1.45 (m, 4H), 1.52–1.43 (m, 2H), 1.36 (s, 3H), 1.34–1.27 (m, 2H), 0.89 (t, J = 6.7 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 198.3, 168.6, 164.8, 106.9, 90.4, 76.0, 70.1, 58.1, 31.7, 30.9, 30.7, 25.9, 22.5, 15.3, 14.0; ESI-MS, positive mode: m/z calcd mass for C15H24O6 [M+Na]+ = 323.1471; 323.05 was found. Optical activity: observed rotation αD = +2.27 (c = 0.0044 g/mL, T = 26.4 °C, CHCl3).
(S)-5-((R)-hydroxy((4R,5R)-2,2,4-trimethyl-5-pentyl-1,3-dioxolan-4-yl)methyl)-4-methoxyfuran-2(5H)-one, syn-27
To a solution of diisopropylamine (0.23 mL, 1.624 mmol) in 1 mL THF under argon at −78 °C, a solution of n-BuLi in THF (1.06 mL, 1.6 M) was added. After 45 min, a solution of 18 (110 mg, 0.96 mmol) in 0.8 mL THF was added dropwise over a 5 min period, followed by the addition of 21 (185 mg, 1.624 mmol) in 1.23 mL THF. The reaction mixture was stirred for 3 h at −78 °C. Upon completion, the reaction was quenched with 0.16 mL MeOH followed by 2.5 mL of sat. NH4Cl(aq) and extracted with EA. The combined organic layers were washed with brine, dried over Na2SO4, and concentrated under reduced pressure. The residue was subjected to column chromatography (gradient elution PS–EA 3:1 to 1:2) to yield syn-, anti-27 (49%) as orange oil. Syn-27: 1H NMR (500 MHz, CDCl3) δ 5.13 (s, 1H), 5.08 (s, 1H), 4.00 (d, J = 9.5 Hz, 1H), 3.92 (s, 3H), 3.91 (d, J = 1.8 Hz, 1H), 3.79 (s, 1H), 1.64–1.46 (m, 6H), 1.44 (s, 3H), 1.34 (s, 3H), 1.31 (m, 2H), 1.18 (s, 3H), 0.89 (t, J = 6.2 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 180.6, 172.5, 107.4, 89.4, 82.6, 82.5, 77.5, 74.2, 59.6, 31.8, 30.4, 28.8, 26.9, 26.9, 22.6, 16.8, 14.0; ESI-MS, positive mode: m/z calcd mass for C17H28O6 [M+Na]+ = 351.1784; 350.95 was found. Optical activity: observed rotation αD25 = −16.0 (c = 0.0025 g/mL, CHCl3).
(S)-4-methoxy-5-((1R,2S,3R)-1,2,3-trihydroxy-2-methyloctyl)furan-2(5H)-one, 16
To a round bottom flask containing syn-27 (436.6 mg, 1.33 mmol) a solution of TFA in water (5.85 mL, 77.16 mmol, 1:1 v/v) was added. The reaction was stirred for 2 h at 0 °C, quenched with saturated aqueous NaHCO3 and extracted with DCM. The combined organic layers were washed with brine, dried over Na2SO4, and concentrated in vacuo. The residue was purified with column chromatography (gradient elution PS–EA 1:3 to 1:6) to afford 16 (53%) as white solid. 16: 1H NMR (500 MHz, DMSO-d6) δ 5.32 (s, 1H), 5.11 (s, 1H), 4.81 (d, J = 6.9 Hz, 1H), 4.33 (s, 1H), 4.18 (d, J = 7.6 Hz, 1H), 3.92 (d, J = 6.9 Hz, 1H), 3.86 (s, 3H), 3.81 (br, 1H), 1.50 (m, 2H), 1.25 (m, 6H), 1.03 (s, 3H), 0.85 (t, J = 6.1 Hz, 3H); 13C NMR (126 MHz, DMSO-d6) δ 182.4, 173.2, 89.9, 78.1, 74.8, 74.4, 70.7, 60.0, 32.0, 30.5, 26.3, 22.7, 19.5, 14.5. ESI-HRMS, positive mode: m/z calcd mass for C14H24O6 [M+Na]+ = 311.1471; 311.1465 was found. Optical activity: observed rotation αD20 = +26.1 (c = 0.0023 g/mL, MeOH). Melting range: 175–178 °C.
(3aS,5R,6R,7R,7aS)-6,7-dihydroxy-3a-methoxy-6-methyl-5-pentylhexahydro-2H-furo[3,2-b]pyran-2-one, 10a and (3aR,5R,6R,7R,7aS)-6,7-dihydroxy-3a-methoxy-6-methyl-5-pentylhexahydro-2H-furo[3,2-b]pyran-2-one, 10b
To a solution of 16 (113.7 mg, 0.396 mmol) in 10.5 mL DMF, DBU (0.09 mL, 0.592 mmol) was added, and the rection was stirred overnight at 60 °C. The reaction mixture was then concentrated under reduced pressure and the residue was subjected to column chromatography (gradient elution PS–EA 7:1 to 1:2) to yield 28 (12%) followed by 10 (74%, 10a/10b = 4/1). 10a: 1H NMR (500 MHz, CDCl3) δ 4.72 (d, J = 2.1 Hz, 1H), 4.22 (d, J = 2.1 Hz, 1H), 3.44 (d, J = 8.7 Hz, 2H), 3.38 (s, 3H), 2.87 (d, J = 17.6 Hz, 1H), 2.78 (d, J = 17.6 Hz, 1H), 1.44 (m, 2H), 1.32 (m, 6H), 1.25 (s, 3H), 0.89 (t, J = 6.9 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 173.3, 109.6, 93.5, 90.5, 77.6, 75.9, 51.3, 39.4, 31.7, 30.7, 25.97, 22.6, 16.4, 14.0. 10b: 1H NMR (500 MHz, CDCl3) δ 4.75 (d, J = 2.9 Hz, 1H), 4.37 (d, J = 2.9 Hz, 1H), 3.50 (d, J = 9.6 Hz, 1H), 3.35 (s, 3H), 2.83 (d, J = 17.9 Hz, 1H), 2.78 (d, J = 17.9 Hz, 1H), 1.56 (m, 5H), 1.32 (s, 3H), 1.31 (s, 3H), 0.90 (t, J = 6.4 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 172.7, 109.6, 93.5, 89.4, 78.8, 76.6, 51.2, 39.4, 31.7, 31.0, 26.0, 22.6, 16.3, 14.0; ESI-MS, positive mode: m/z calcd mass for C14H24NaO6 [M+Na]+ = 311.1471, was found 310.90. 28: 1H NMR (500 MHz, acetone-d6) δ 5.79 (d, J = 1.6 Hz, 1H), 5.24 (d, J = 1.6 Hz, 1H), 4.45 (br, 1H), 4.17 (dd, J = 9.9, 2.6 Hz, 1H), 1.95–1.80 (m, 3H), 1.74–1.65 (m, 1H), 1.52–1.44 (m, 1H), 1.41 (s, 3H), 1.39–1.31 (m, 3H), 0.92 (t, J = 6.9 Hz, 3H); 13C NMR (126 MHz, acetone-d6) δ 168.1, 166.7, 144.1, 110.1, 88.1, 87.6, 67.2, 31.5, 28.0, 25.6, 25.2, 22.3, 13.4; ESI-MS, negative mode: m/z calcd mass for C13H18O4 [M-H]− = 237.1127; 236.90 was found. Optical activity: observed rotation αD20 = +64.15° (c = 0.0053 g/mL, MeOH).
(5R,6R)-6-hydroxy-6-methyl-5-pentyl-5,6-dihydro-2H-furo[3,2-b]pyran-2-one, 28
To a solution of mixture 10a,b (42.2 mg, 0.146 mmol) in 3.8 mL DMF, DBU (0.07 mL, 0.430 mmol) was added, and the reaction was stirred overnight at 80 °C. The reaction mixture was then concentrated in vacuo, and its residue subjected to column chromatography (gradient elution PS–EA 7:1 to 4:1) to afford 28 in a 23% yield.
(5R,6R,7aS)-6-hydroxy-6-methyl-5-pentyl-5,6,7,7a-tetrahydro-2H-furo[3,2-b]pyran-2-one, 9 and (3aR,5R,6R,7aS)-6-hydroxy-6-methyl-5-pentylhexahydro-2H-furo[3,2-b]pyran-2-one, 29
To a solution of 28 (12.1 mg, 0.051 mmol) in 1.5 mL MeOH, a catalytic amount of Pd/C (10% w/w) was added under a hydrogen atmosphere. The reaction mixture was stirred for 30 min at room temperature, filtrated over celite and then concentrated under reduced pressure. The residue was subjected to column chromatography (gradient elution PS–EA 2:1 to 1:2) to yield 9 (79%) and 29 (18%) as orange oils. 9: 1H NMR (500 MHz, CDCl3) δ 5.12 (d, J = 0.9 Hz, 1H), 4.93 (t, J= 9.5, 1H), 3.91 (d, J = 9.1 Hz, 1H), 2.62 (dd, J = 14.0, 9.5 Hz, 1H), 2.10 (br, 1H), 1.82 (dd, J = 14.0, 9.5 Hz, 1H), 1.74–1.63 (m, 2H), 1.36 (m, 6H), 1.32 (s, 3H), 0.91 (t, J = 6.8 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 180.5, 173.5, 88.4, 84.9, 71.4, 69.7, 42.4, 31.6, 27.6, 26.8, 25.4, 22.5, 14.0; ESI-MS, positive mode: m/z calcd mass for C13H20O4 [M+Na]+ = 263.1259; 263.15 was found. Optical activity: observed rotation αD = +133.3 (c = 0.0033 g/mL, T = 22.8 °C, MeOH). 29: 1H NMR (500 MHz, CDCl3) δ 1H NMR (500 MHz, CDCl3) δ 4.42 (d, J = 1.9 Hz, 1H), 4.23 (br, 1H), 3.16 (d, J = 8.1 Hz, 1H), 2.77–2.52 (m, 2H), 2.42 (d, J = 15.8 Hz, 1H), 2.34–2.13 (br, 1H), 1.79 (dd, J = 15.8, 4.2 Hz, 1H), 1.55 (m 2H), 1.27 (m, 6H), 1.12 (s, 3H), 0.89 (t, J = 6.8 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 175.2, 82.2, 77.7, 73.9, 67.5, 38.4, 38.3, 31.8, 28.0, 25.6, 24.6, 22.6, 14.0; ESI-MS, positive mode: m/z calcd mass for C13H22O4 [M+H]+ = 243.1596; 243.1572 was found.
(S)-5-((R)-hydroxy((4R,5R)-2,2,4-trimethyl-5-pentyl-1,3-dioxolan-4-yl)methyl)-4-methoxy-1,5-dihydro-2H-pyrrol-2-one, syn-30, (R)-5-((R)-hydroxy((4R,5R)-2,2,4-trimethyl-5-pentyl-1,3-dioxolan-4-yl)methyl)-4-methoxy-1,5-dihydro-2H-pyrrol-2-one, anti-30
To a solution of 19 (17 mg, 0.149 mmol) in 0.7 mL THF, KOH(aq) (0.043 mL, 4M) was added, dropwise, and the reaction was stirred at 60 °C. After one hour, a solution of 21 (20 mg, 0.093 mmol) in 0.6 mL THF was added and the reaction mixture was stirred at room temperature overnight. Upon completion, the reaction was concentrated under reduced pressure and its residue was subjected to column chromatography (gradient elution DCM-MeOH 10:0.4 to 10:0.7) to afford 30 (41%, syn-30/anti-30 = 3/1) as orange oil. Syn-30: 1H NMR (500 MHz, CDCl3) δ 5.69 (s, 1H), 5.07 (s, 1H), 4.39 (s, 1H), 3.84 (1H, overlapping with OMe), 3.83 (s, 3H), 3.76 (d, J = 9.9 Hz, 1H), 2.73 (d, J = 10.4 Hz, 1H), 1.60–1.47 (m, 3H), 1.42 (s, 3H), 1.35 (s, 3H), 1.31 (m, 5H), 1.16 (s, 3H), 0.89 (t, J = 6.9 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 176.7, 175.4, 107.7, 94.7, 84.3, 81.7, 72.5, 58.5, 31.8, 29.7, 28.6, 26.9, 26.8, 22.5, 17.8, 14.0; ESI-MS, negative mode: m/z calcd mass for C17H28NO5 [M-H]- = 326.1967; 326.05 was found. Anti-30: 1H NMR (500 MHz, CDCl3) δ 7.78 (s, 1H), 5.16 (s, 1H), 5.09 (s, 1H), 3.85 (1H, overlapping with OMe), 3.83 (s, 3H), 1.57 (m, 4H), 1.46 (s, 3H), 1.20 (s, 3H), 1.33 (m, 4H), 0.89 (t, J = 6.9 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 170.7, 166.2, 132.4, 108.5, 108.1, 93.0, 83.2, 82.5, 58.2, 32.0, 29.1, 28.6, 26.7, 26.4, 22.7, 22.3, 14.2; ESI-MS, negative mode: m/z calcd mass for C17H29O5 [M-H]− = 326.1973; 326.1971 was found.
tert-butyl (S)-2-((R)-hydroxy((4R,5R)-2,2,4-trimethyl-5-pentyl-1,3-dioxolan-4-yl)methyl)-3-methoxy-5-oxo-2,5-dihydro-1H-pyrrole-1-carboxylate, syn-32, tert-butyl (R)-2-((R)-hydroxy((4R,5R)-2,2,4-trimethyl-5-pentyl-1,3-dioxolan-4-yl)methyl)-3-methoxy-5-oxo-2,5-dihydro-1H-pyrrole-1-carboxylate, anti-32
To a solution of diisopropylamine (0.23 mL, 1.66 mmol) in 1 mL THF under argon at 0 °C, a solution of n-BuLi in THF (1.08 mL, 1.6 M) was added, and the overall mixture was transferred to a dry ice bath (−78 °C) to stir. After 20 min, a solution of 31 (345.9 mg, 1.66 mmol) in 1.47 mL THF was added dropwise over 5 min, followed by the addition of 21 (118.59 mg, 0.55 mmol) in 0.57 mL THF. The reaction mixture was stirred for 2 h at −78 °C. Upon completion, the reaction was quenched with 0.16 mL MeOH followed by sat. NH4Cl(aq) and extracted with EA. The combined organic layers were washed with brine, dried over Na2SO4, and concentrated under reduced pressure. The residue was subjected to column chromatography (gradient elution PS–EA 3:1 to 2:1) to yield the isomeric mixture 32 (55%) as orange oil. Syn-32: 1H NMR (500 MHz, CDCl3) δ 5.26 (d, J = 6.1Hz, 1H), 5.01 (s, 1H), 4.72 (s, 1H), 4.07 (d, J = 6.3Hz, 1H), 3.83 (overlapping with OMe, 1H), 3.80 (s, 3H), 1.68 (m, 1H), 1.54 (s, 9H), 1.53-1.33 (m, 3H), 1.22-1.30 (m, 4H), 1.08 (s, 3H), 0.88 (t, J = 6.8 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 176.0, 168.4, 153.7, 107.1, 94.2, 85.6, 84.4, 83.4, 65.0, 58.5, 32.2, 30.2, 28.9, 28.3, 27.5, 26.5, 22.8, 16.2, 14.2; ESI-MS, positive mode: m/z calcd mass for C22H37O7 [M+Na]+ = 450.2468; 450.2448 was found. Anti-32: 1H NMR (500 MHz, CDCl3) δ 5.06 (s, 1H), 4.80 (s, 1H), 4.20 (m, 2H), 3.84 (overlapping with OMe, 1H), 3.84 (s, 3H), 1.56 (s, 9H), 1.25-1.55 (m, 8H), 1.25 (s, 3H), 0.89 (t, J = 6.5 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 175.1, 168.3, 107.6, 95.2, 83.9, 83.4, 79.5, 70.5, 58.7, 32.2, 29.1, 28.3, 28,1, 26.9, 26.4, 22.7, 18.0, 14.2; ESI-MS, positive mode: m/z calcd mass for C22H37O7 [M+Na]+ = 450.2468; 450.2445 was found.
(S)-4-methoxy-5-((1R,2S,3R)-1,2,3-trihydroxy-2-methyloctyl)-1,5-dihydro-2H-pyrrol-2-one, 17
To a round bottom flask containing syn-30 (8 mg, 0.024 mmol), a solution of TFA in water (0.22 mL, 2.81 mmol, 1:1 v/v) was added. The reaction was stirred for 30 min at 0 °C quenched with saturated aqueous NaHCO3 and extracted with DCM. The combined organic layers were washed with brine, dried over Na2SO4, and concentrated in vacuo. The residue was purified with column chromatography (gradient elution DCM–MeOH 10:0.2 to 10:0.7) to afford 17 (50%). 17: 1H NMR (500 MHz, CDCl3) δ 6.90 (s, 1H), 5.03 (s, 1H), 4.48 (br, 1H), 4.34 (br, 1H), 4.23 (s, 1H), 3.82 (overlapping with OMe, 1H), 3.80 (s, 3H), 3.60 (d, J = 9.40 Hz, 1H), 1.25-1.41 (m, 8H), 1.26 (s, 3H), 0.89 (t, J = 6.75Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 171.2, 166.5, 133.8, 110.7, 93.0, 78.0, 58.5, 32.2, 32.0, 30.1, 26.7, 23.5, 23.0, 14.4; ESI-MS, positive mode: m/z calcd mass for C14H25O5 [M+Na]+ = 310.1630; 310.1618 was found.
(3aR,5R,6R,7R,7aS)-6,7-dihydroxy-3a-methoxy-6-methyl-5-pentylhexahydropyrano[3,2-b]pyrrol-2(1H)-one, 33a
To a solution of 17 (6.8 mg, 0.024 mmol) in 0.63 mL toluene was added DBU (5 μL, 0.036 mmol) and the reaction was stirred overnight at 60 °C. The reaction mixture was then concentrated under reduced pressure and the residue subjected to column chromatography (gradient elution DCM–MeOH 10:0.2 to 10:0.5) to afford 33a (62%) as yellow solid. 33a: 1H NMR (500 MHz, CDCl3) δ 6.65 (s, 1H), 4.39 (d, J = 5.6 Hz, 1H), 4.10 (d, J = 5.6 Hz, 1H), 3.39 (t, J = 6.7 Hz, 1H), 3.36 (s, 3H), 2.70 (br, 2H), 2.44 (br, 1H), 1.74–1.49 (m, 2H), 1.33 (m, 6H), 1.19 (s, 3H), 0.90 (t, J = 6.7 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 174.1, 110.3, 91.6, 76.8, 73.7, 66.6, 51.3, 41.2, 31.9, 31.4, 26.2, 22.6, 17.3, 14.0; ESI-MS, positive mode: m/z calcd mass for C14H26O5 [M+H]+ = 288.1811; 287.90 was found.
Cell Toxicity
Cell Culture: HeLa and MOLT-4 cancer cell lines were kindly provided by Professor Eleni Nikolakaki (Laboratory of Biochemistry, Department of Chemistry of Aristotle University of Thessaloniki). HeLa cells were maintained at 37 °C with 5% CO2 in DMEM, while MOLT-4 cells were maintained in RPMI medium, both supplemented with 10% (v/v) fetal bovine serum (FBS) and antibiotics/antimycotics.
MTT Assays: Cells (HeLa and MOLT-4) were seeded in 96-well plates (3 × 103 cells for HeLa and 15 × 103 per well) and one day after were exposed to increasing concentrations of the inhibitors for 48 h. The viability of the cells was estimated by a (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) metabolic colorimetric assay as described previously [22]. The absorbance values (the means ± SE of three independent experiments) of treated cells were normalized to the untreated cells, which was set as 100% viability. Half maximal effective concentration where then calculated (EC50 values).
5. Conclusions
In summary, a synthetic route towards the synthesis of several synthetic analogues of phaeosphaerides employing vinylogous aldol and intramolecular Michael reactions was explored. Further optimization of the vinylogous reaction steps is necessary; however, it was possible to create sp3-rich scaffolds with multiple stereogenic centres related to the natural phaeosphaerides, worthy of more chemical and pharmacological exploration. These findings highlight the challenges in optimizing phaeosphaerides for anticancer applications and provide insights into future structural modifications to improve their therapeutic potential.
Supplementary Materials
The following supporting information can be downloaded at https://www.mdpi.com/article/10.3390/molecules30092016/s1: Figures S1–S51: 1H-NMR and 13C-NMR spectra for 13; ESI-LCMS analysis for 13; 1H-NMR and 13C-NMR spectra for 24; ESI-LCMS analysis for 24; 1H-NMR and 13C-NMR spectra for 25; 1H-1H COSY and 1H-1H NOESY spectra for 25; 1H-13C HSQC and 1H-13C HMBC spectra for 25; ESI-LCMS analysis of 25; 1H-NMR spectrum for syn-,anti-27; ESI-LCMS analysis for syn-,anti-27; 1H-NMR and 13C-NMR spectra for syn-27; 1H-NMR and 13C-NMR spectra for 16; 1H-1H COSY and 1H-13C HMBC spectra for 16; 1H-13C HSQC spectrum 16; ESI-LCMS analysis of 16; 1H-NMR spectrum for 10a; 13C-NMR and 1H-1H NOESY spectra for 10a; 1H-NMR and 13C-NMR spectra for 10b; 1H-1H NOESY spectrum for 10b; 1H-13C HSQC and 1H-13C HMBC spectra for 10b; ESI-LCMS analysis of 10b; 1H-NMR and 13C-NMR spectra for 28; 1H-1H COSY and 1H-13C HMBC spectra for 28; 1H-13C HSQC spectrum for 28; ESI-LCMS analysis of 28; 1H-NMR and 13C-NMR spectra for 9; 1H-1H COSY and 1H-13C HSQC spectra for 9; 1H-13C HMBC and 1H-1H NOESY spectra for 9; ESI-LCMS analysis of 9; 1H-NMR and 13C-NMR spectra for 29; 1H-1H COSY and 1H-1H NOESY spectra for 29; 1H-13C HSQC and 1H-13C HMBC spectra for 29; ESI-LCMS analysis of 29; 1H-NMR and 13C-NMR spectra for syn-30; 1H-1H COSY and 1H-13C HMBC spectra for syn-30; 1H-13C HSQC spectrum for syn-30; MS analysis for syn-30; 1H-NMR and 13C-NMR spectra for anti-30; 1H-1H COSY and 1H-1H NOESY spectra for anti-30; 1H-13C HMBC and 1H-13C HSQC spectra for anti-30; HRMS analysis for anti-30; 1H-NMR and 13C-NMR spectra for syn-32; 1H-NMR and 13C-NMR spectra for anti-32; HRMS analysis for anti-32; 1H-NMR and 13C-NMR spectra for 17; ESI-LCMS analysis of 17; 1H-NMR and 13C-NMR spectra for 33a; 1H-1H COSY spectrum and HMBC spectrum for 33a; 1H-13C HSQC and 1H-1H NOESY spectra for 33a; 1H-1H NOESY spectrum for 33a; ESI-LCMS analysis of 33a; HPLC-MS parameters and method development. Table S1: Conditions for the elution of 33a.
Author Contributions
Conceptualization, V.S.; methodology, V.S. and I.S.; investigation, K.R., O.-E.C., J.G. and I.S.; resources, V.S.; data curation, K.R. and O.-E.C.; writing—original draft preparation, V.S.; writing—review and editing, V.S. and G.L.; supervision, V.S. and G.L. All authors have read and agreed to the published version of the manuscript.
Funding
The work was also supported by the project ‘An Open-Access Research Infrastructure of Chemical Biology and Target-Based Screening Technologies for Human and Animal Health, Agriculture, and the Environment (OPENSCREEN-GR)’ (MIS5002691 to V.S.) under the Action “Reinforcement of the Research and Innovation Infrastructure” (NSRF 2014–2020).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are contained within the article and Supplementary Materials.
Acknowledgments
We thank Eleni Nikolakaki (Laboratory of Biochemistry, Department of Chemistry of Aristotle University of Thessaloniki) for kindly providing HeLa and MOLT-4 cancer cell lines.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| STAT3 | Signal Transducer and Activator of Transcription 3 |
| SAR | Structure Activity Relationships |
| DMEM | Dulbecco’s Modified Eagle Medium |
| TBAF | Tetrabutylammonium fluoride |
| DBU | 1,8-Diazabicyclo(5.4.0)undec-7-ene |
| TFA | Trifluoroacetic acid |
| pTSA | p-Toluenesulfonic acid |
References
- Butler, M.S.; Robertson, A.A.; Cooper, M.A. Natural product and natural product derived drugs in clinical trials. Nat. Prod. Rep. 2014, 31, 1612–1661. [Google Scholar] [CrossRef] [PubMed]
- Atanasov, A.G.; Zotchev, S.B.; Dirsch, V.M.; Supuran, C.T.; International Natural Product Sciences Taskforce. Natural products in drug discovery: Advances and opportunities. Nat. Rev. Drug Discov. 2021, 20, 200–216. [Google Scholar] [CrossRef] [PubMed]
- Mathur, S.; Hoskins, C. Drug development: Lessons from nature. Biomed. Rep. 2017, 6, 612–614. [Google Scholar] [CrossRef] [PubMed]
- Wilson, B.A.P.; Thornburg, C.C.; Henrich, C.J.; Grkovic, T.; O’Keefe, B.R. Creating and screening natural product libraries. Nat. Prod. Rep. 2020, 37, 893–918. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, S.; Zhao, S.; Wang, X.; Liu, B.; Xu, H. Click Chemistry in Natural Product Modification. Front. Chem. 2021, 9, 774977. [Google Scholar] [CrossRef]
- Singh, K.; Gupta, J.K.; Chanchal, D.K.; Shinde, M.G.; Kumar, S.; Jain, D.; Almarhoon, Z.M.; Alshahrani, A.M.; Calina, D.; Sharifi-Rad, J.; et al. Natural products as drug leads: Exploring their potential in drug discovery and development. Naunyn Schmiedebergs Arch. Pharmacol. 2024, 398, 4673–4687. [Google Scholar] [CrossRef]
- Maloney, K.N.; Hao, W.; Xu, J.; Gibbons, J.; Hucul, J.; Roll, D.; Brady, S.F.; Schroeder, F.C.; Clardy, J. Phaeosphaeride A, an inhibitor of STAT3-dependent signaling isolated from an endophytic fungus. Org. Lett. 2006, 8, 4067–4070. [Google Scholar] [CrossRef]
- Chatzimpaloglou, A.; Yavropoulou, M.P.; Rooij, K.E.; Biedermann, R.; Mueller, U.; Kaskel, S.; Sarli, V. Total synthesis and biological activity of the proposed structure of phaeosphaeride A. J. Org. Chem. 2012, 77, 9659–9667. [Google Scholar] [CrossRef]
- Chatzimpaloglou, A.; Kolosov, M.; Eckols, T.K.; Tweardy, D.J.; Sarli, V. Synthetic and biological studies of phaeosphaerides. J. Org. Chem. 2014, 79, 4043–4054. [Google Scholar] [CrossRef]
- Kobayashi, K.; Okamoto, I.; Morita, N.; Kiyotani, T.; Tamura, O. Synthesis of the proposed structure of phaeosphaeride A. Org. Biomol. Chem. 2011, 9, 5825–5832. [Google Scholar] [CrossRef]
- Poluektova, E.; Tokarev, Y.; Sokornova, S.; Chisty, L.; Evidente, A.; Berestetskiy, A. Curvulin and Phaeosphaeride A from Paraphoma sp. VIZR 1.46 Isolated from Cirsium arvense as Potential Herbicides. Molecules 2018, 23, 2795. [Google Scholar] [CrossRef] [PubMed]
- Abzianidze, V.; Beltyukov, P.; Zakharenkova, S.; Moiseeva, N.; Mejia, J.; Holder, A.; Trishin, Y.; Berestetskiy, A.; Kuznetsov, V. Synthesis and Biological Evaluation of Phaeosphaeride A Derivatives as Antitumor Agents. Molecules 2018, 23, 3043. [Google Scholar] [CrossRef] [PubMed]
- Abzianidze, V.V.; Efimova, K.P.; Poluektova, E.V.; Trishin, Y.G.; Kuznetsov, V.A. Synthesis of natural phaeosphaeride A and semi-natural phaeosphaeride B derivatives. Mendeleev Commun. 2017, 27, 490–492. [Google Scholar] [CrossRef]
- Kobayashi, K.; Tanaka, K.; Kogen, H. Total Synthesis and Biological Evaluation of Phaeosphaerides. Catalysts 2018, 8, 206. [Google Scholar] [CrossRef]
- Li, C.S.; Ding, Y.; Yang, B.J.; Miklossy, G.; Yin, H.Q.; Walker, L.A.; Turkson, J.; Cao, S. A New Metabolite with a Unique 4-Pyranone-γ-Lactam-1,4-Thiazine Moiety from a Hawaiian-Plant Associated Fungus. Org. Lett. 2015, 17, 3556–3559. [Google Scholar] [CrossRef]
- Silva, G.H.; Zeraik, M.L.; de Oliveira, C.M.; Teles, H.L.; Trevisan, H.C.; Pfenning, L.H.; Nicolli, C.P.; Young, M.C.M.; Mascarenhas, Y.P.; Abreu, L.M.; et al. Lactone Derivatives Produced by a Phaeoacremonium sp., an Endophytic Fungus from Senna spectabilis. J. Nat. Prod. 2017, 80, 1674–1678. [Google Scholar] [CrossRef]
- Trenti, F.; Cox, R.J. Structural Revision and Biosynthesis of the Fungal Phytotoxins Phyllostictines A and B. J. Nat. Prod. 2017, 80, 1235–1240. [Google Scholar] [CrossRef]
- Hirayama, Y.; Matsunaga, M.; Fukao, A.; Kobayashi, K. Biological evaluation of signal transducer and activator of transcrip-tion 3 (STAT3) targeting by phaeosphaeride A and its analogs. Bioorg. Med. Chem. Lett. 2024, 114, 130004. [Google Scholar] [CrossRef]
- Karak, M.; Barbosa, L.C.; Acosta, J.A.; Sarotti, A.M.; Boukouvalas, J. Thermodynamically driven, syn-selective vinylogous aldol reaction of tetronamides. Org. Biomol. Chem. 2016, 14, 4897–4907. [Google Scholar] [CrossRef]
- Abzianidze, V.; Moiseeva, N.; Suponina, D.; Zakharenkova, S.; Rogovskaya, N.; Laletina, L.; Holder, A.A.; Krivorotov, D.; Bogachenkov, A.; Garabadzhiu, A.; et al. Natural Phaeosphaeride A Derivatives Overcome Drug Resistance of Tumor Cells and Modulate Signaling Pathways. Pharmaceuticals 2022, 15, 395. [Google Scholar] [CrossRef]
- Abzianidze, V.; Kadochnikov, V.; Suponina, S.D.; Skvortsov, V.N.; Beltyukov, P.P.; Babakov, N.V.; Krivorotov, V.D.; Barysheva, M.E.; Garabadzhiu, V.A. X-ray structure and in silico molecular docking of a natural phaeosphaeride A derivative for targets associated with kinase cascade. Mendeleev Commun. 2023, 33, 534. [Google Scholar] [CrossRef]
- Leonidis, G.; Koukiali, A.; Sigala, I.; Tsimaratou, K.; Beis, D.; Giannakouros, T.; Nikolakaki, E.; Sarli, V. Synthesis and Anti-Angiogenic Activity of Novel c(RGDyK) Peptide-Based JH-VII-139-1 Conjugates. Pharmaceutics 2023, 15, 381. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).