

Synthesis of 1,4-Benzodiazepines via Intramolecular C–N Bond Coupling and Ring Opening of Azetidines


Abstract
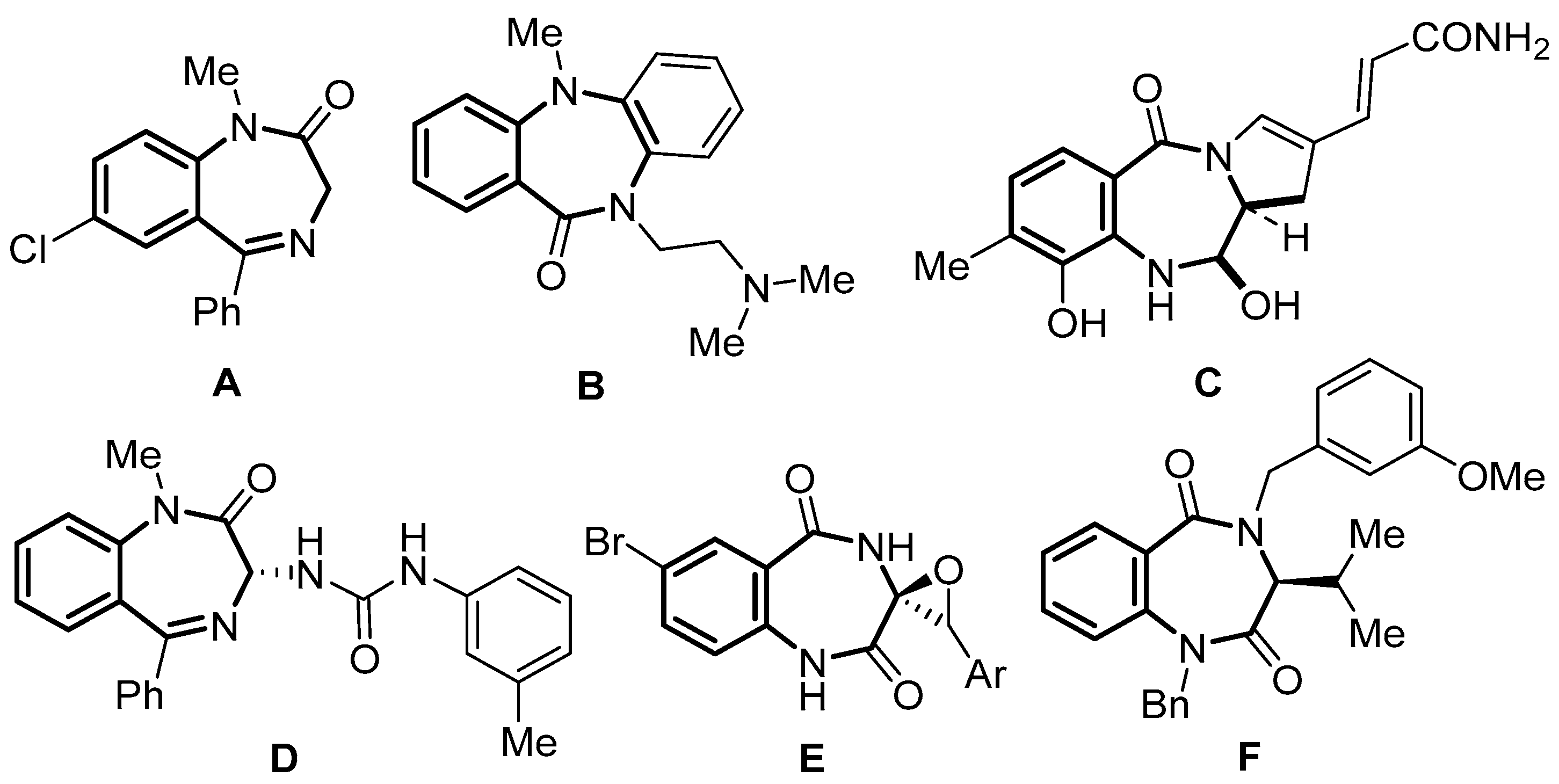
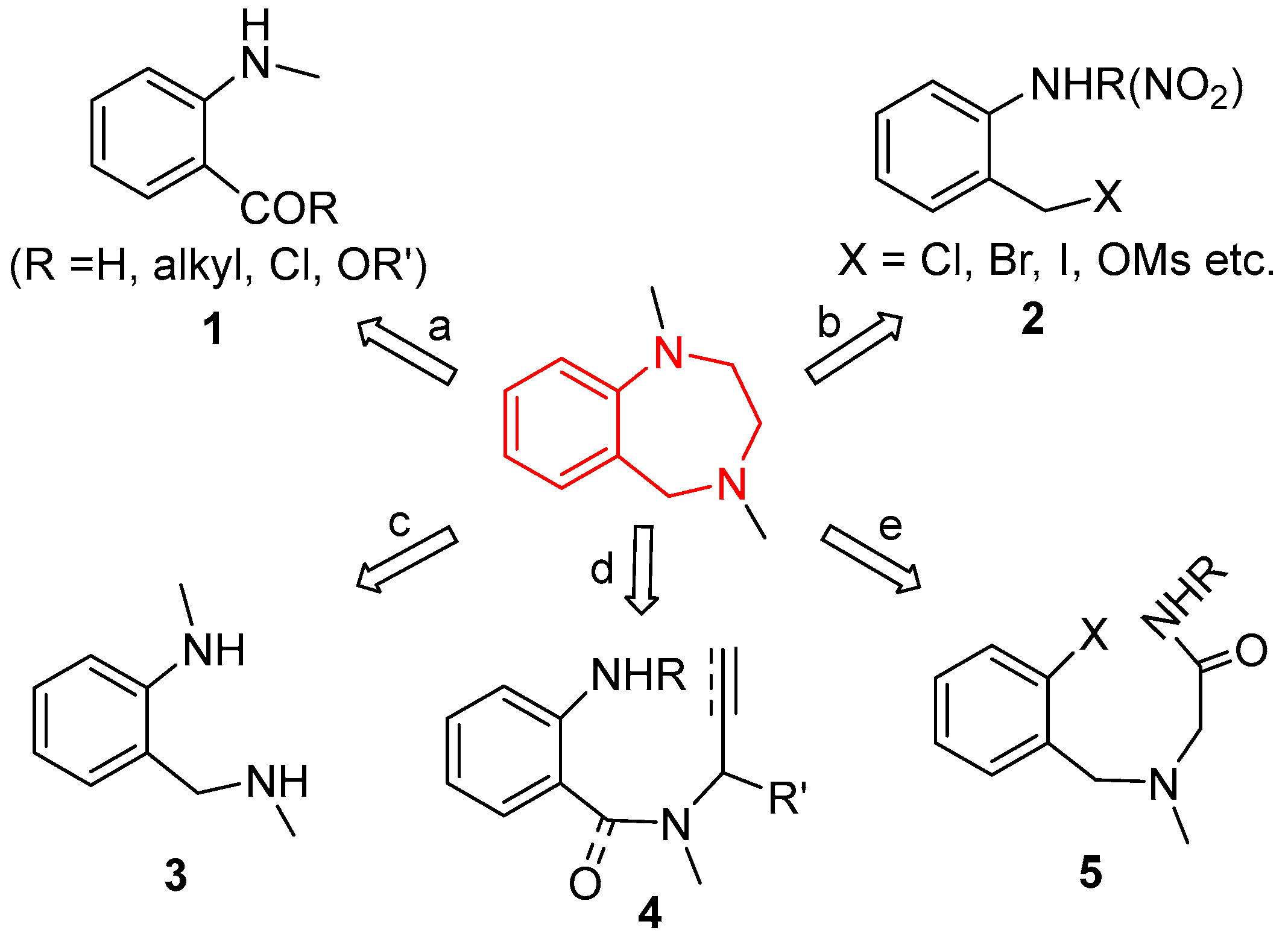
1. Introduction
2. Results and Discussion
3. Conclusions
4. Materials and Methods
4.1. General Information
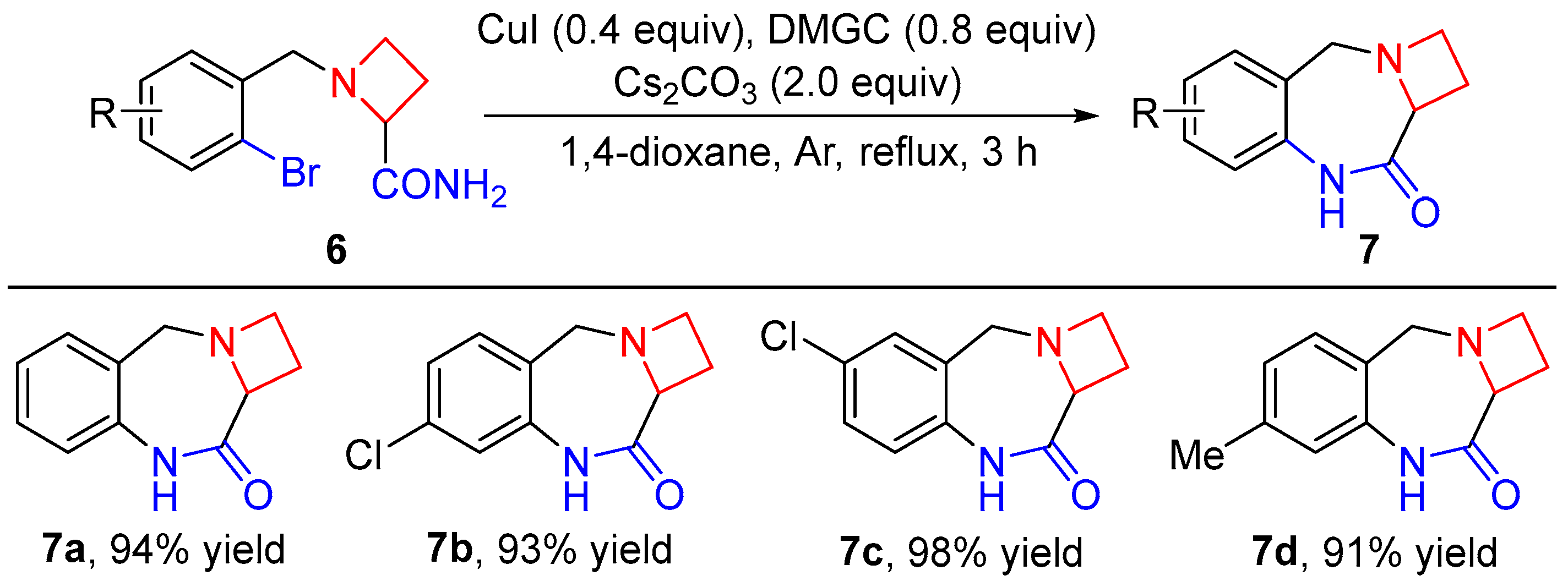
4.2. General Procedure for the Synthesis of Compounds 6a–d
4.3. General Procedure for the Synthesis of Compounds 7a–d
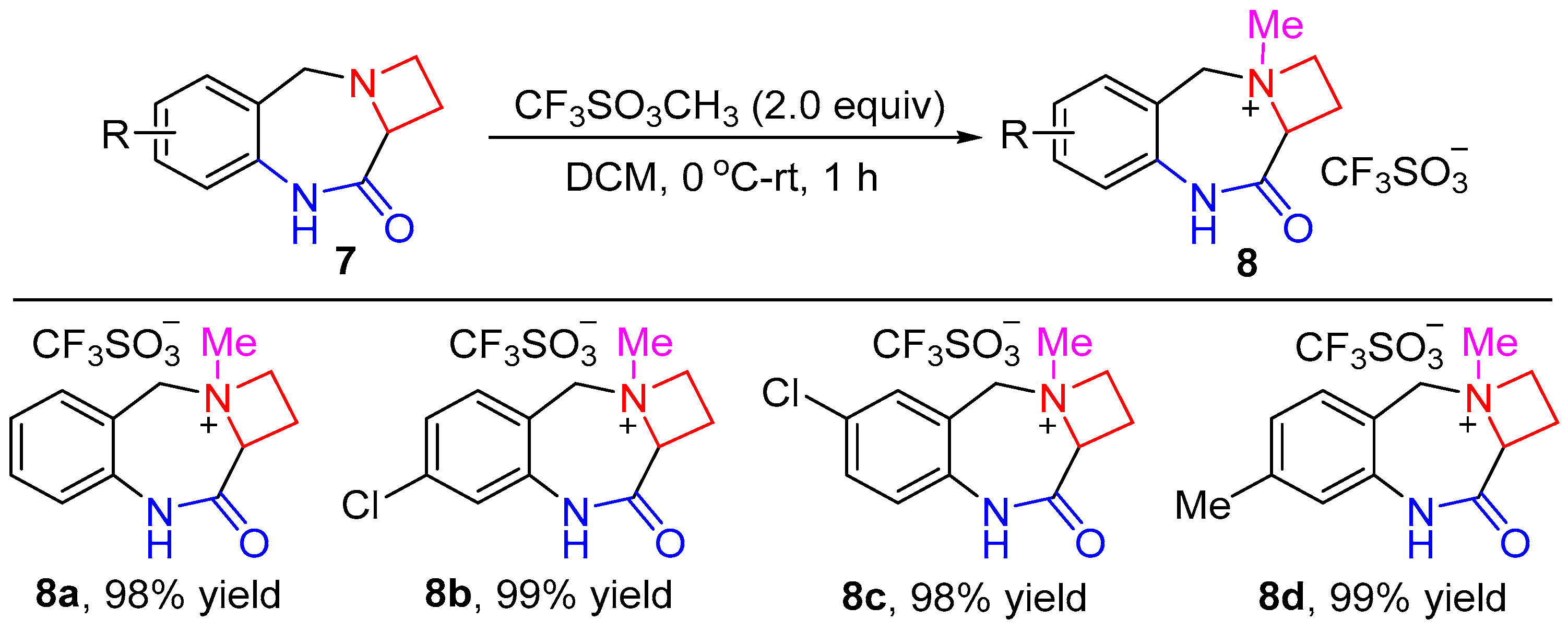
4.4. General Procedure for the Synthesis of Compounds 8a–d
4.5. General Procedure for the Ring-Opening Reaction of Compounds 8
4.6. General Procedure for the Ring-Opening Reaction of Compounds 7
4.7. General Procedure for the Scale-Up Synthesis of Compound 9ad
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Costantino, L.; Barlocco, D. Privileged Structures as Leads in Medicinal Chemistry. Curr. Med. Chem. 2006, 13, 65–85. [Google Scholar] [CrossRef] [PubMed]
- Landquist, J.K. Comprehensive Heterocyclic Chemistry; Katritzky, A.R., Rees, C.W., Eds.; Pergamon: Oxford, UK, 1984; Volume 1. [Google Scholar]
- Khan, I.; Anupama; Singh, B. 1,4-benzodiazepine: An overview of biological properties. Sci. Rev. Chem. Commun. 2015, 5, 13–20. [Google Scholar]
- Yet, L. (Ed.) Benzodiazepines. In Privileged Structures in Drug Discovery; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2018; pp. 15–58. [Google Scholar]
- Sternbach, L.H. The Benzodiazepine Story. J. Med. Chem. 1979, 22, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Herrero, S.; García-López, M.T.; Herranz, R. Expedient One-pot Synthesis of Novel Chiral 2-Substituted 5-phenyl-1,4-Benzodiazepine Scaffolds from Amino Acid-derived Amino Nitriles. J. Org. Chem. 2003, 68, 4582–4585. [Google Scholar] [CrossRef]
- Hanley, D.F.; Pozo, M. Treatment of Satus Epilepticus with Midazolam in the Critical Care Setting. Int. J. Clin. Pract. 2000, 54, 30–35. [Google Scholar] [CrossRef]
- Selnick, H.G.; Liverton, N.J.; Baldwin, J.J.; Butcher, J.W.; Claremon, D.A.; Elliott, J.M.; Freidinger, R.M.; King, S.A.; Libby, B.E.; McIntyre, C.J.; et al. Class III Antiarrhythmic Activity in vivo by Selective Blockade of the Slowly Activating Cardiac Delayed Rectifier Potassium Current IKs by (R)-2-(2,4-trifluoromethyl)-N-[2-oxo-5phenyl-1-(2,2,2-trifluoroethyl)-2,3-Dihydro-1Hbenzo[e][1,4]diazepin-3-yl]acetamide. J. Med. Chem. 1997, 40, 3865–3868. [Google Scholar]
- Hsu, M.C.; Schutt, A.D.; Holly, M.; Slice, L.W.; Sherman, M.I.; Richman, D.D.; Potash, M.J.; Volsky, D.J. Inhibition of HIV Replication in Acute and Chronic Infections in vitro by a TAT Antagonist. Science 1991, 254, 1799–1802. [Google Scholar] [CrossRef]
- Zahradník, I.; Minarovič, I.; Zahradníková, A. Inhibition of the Cardiac L-Type Calcium Channel Current by Antidepressant Drugs. J. Pharmacol. Exp. Ther. 2008, 324, 977–984. [Google Scholar] [CrossRef]
- Semple, G.; Ryder, H.; Rooker, D.P.; Batt, A.R.; Kendrick, D.A.; Szelke, M.; Ohta, M.; Satoh, M.; Nishiha, A.; Akuzawa, S.; et al. (3R)-N-(1-(tert-Butylcarbonylmethyl)-2,3-dihydro-2-oxo-5-(2-pyridyl)-1H-1,4-benzodiazepin-3-yl)-N’-(3-(methylamino)phenyl)urea (YF476): A Potent and Orally Active Gastrin/CCK-B Antagonist. J. Med. Chem. 1997, 40, 331–341. [Google Scholar] [CrossRef]
- Leimgruber, W.; Stefanovic, V.; Schenker, F.; Karr, A.; Berger, J. Isolation and Characterization of Anthramycin, a New Antitumor Antibiotic. J. Am. Chem. Soc. 1965, 87, 5791–5793. [Google Scholar] [CrossRef]
- Hu, Y.; Phelan, V.; Ntai, I.; Farnet, C.M.; Zazopoulos, E.; Bachmann, B.O. Benzodiazepine Biosynthesis in Streptomyces refuineus. Chem. Biol. 2007, 14, 691–701. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, L.W.; El-yamany, M.F. Design and Synthesis of Novel 1,4-Benzodiazepine Derivatives and Their Biological Evaluation as Cholinesterase Inhibitors. Arch. Pharmacol. Res. 2012, 35, 1369–1377. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.-F.; Yu, H.-M.; Ko, B.-W.; Chang, Y.; Chen, M.-Y.; Ho, T.-I.; Tsai, Y.-M.; Fang, J.-M. Practical Synthesis of Potential Endothelin Receptor Antagonists of 1,4-Benzodiazepine-2,5-dione Derivatives Bearing Substituents at the C3-, N1-and N4-Positions. Org. Biomol. Chem. 2006, 4, 510–518. [Google Scholar] [CrossRef] [PubMed]
- Kaur, N.; Kishore, D. Synthetic Strategies Applicable in the Synthesis of Privileged Scaffold: 1,4-Benzodiazepine. Synth. Commun. 2014, 44, 1375–1413. [Google Scholar] [CrossRef]
- Keating, T.A.; Armstrong, R.W. A Remarkable Two-Step Synthesis of Diverse 1,4-Benzodiazepine-2,5-diones Using the Ugi Four-Component Condensation. J. Org. Chem. 1996, 61, 8935–8939. [Google Scholar] [CrossRef]
- Huang, Y.; Khoury, K.; Chanas, T.; Dömling, A. Multicomponent Synthesis of Diverse 1,4-Benzodiazepine Scaffolds. Org. Lett. 2012, 14, 5916–5919. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, M.; Ding, M.-W. A Simple and One-pot Synthesis of 2,3,4,5-Tetrasubstituted 4,5-dihydro-3H-1,4-Benzodiazepines. Tetrahedron 2013, 69, 9056–9062. [Google Scholar] [CrossRef]
- Nadin, A.; Sanchez Lopez, J.M.; Owens, A.P.; Howells, D.M.; Talbot, A.C.; Harrison, T. New Synthesis of 1,3-Dihydro-1,4-benzodiazepin-2(2H)-ones and 3-Amino-1,3-dihydro-1,4-benzodiazepin-2(2H)-ones: Pd-Catalyzed Cross-Coupling of Imidoyl Chlorides with Organoboronic Acids. J. Org. Chem. 2003, 68, 2844–2852. [Google Scholar] [CrossRef]
- Xu, X.-M.; Chen, D.-M.; Wang, Z.-L. Recent Advances in Sulfenylation of C(sp3)-H Bond Under Transition Metal-Free Conditions. Chin. Chem. Lett. 2020, 31, 49–57. [Google Scholar]
- Liu, S.; Zhao, T.; Qu, J.; Wang, B. Expedient Synthesis of 1,4-Benzodiazepines via a Tandem Condensation/[1,5]-Hydride Transfer/Cyclization Process. Adv. Synth. Catal. 2018, 360, 4094–4098. [Google Scholar] [CrossRef]
- Nunewar, S.N.; Sangu, K.G.; Kotla, N.K.; Tangellamudi, N.D. Oxidative Dearomatization as a Strategy for a Facile, Metal-Free Synthesis of Vanillyl Benzodiazepines. ChemistrySelect 2020, 5, 10015–10021. [Google Scholar] [CrossRef]
- Wang, J.-Y.; Guo, X.-F.; Wang, D.-X.; Huang, Z.-T.; Wang, M.-X. A New Strategy for the Synthesis of 1,4-Benzodiazepine Derivatives Based on the Tandem N-Alkylation-Ring Opening-Cyclization Reactions of Methyl 1-Arylaziridine-2-carboxylates with N-[2-Bromomethyl(phenyl)]trifluoroacetamides. J. Org. Chem. 2008, 73, 1979–1982. [Google Scholar] [CrossRef] [PubMed]
- Jumde, V.R.; Cini, E.; Porcheddu, A.; Taddei, M. A Metal-Catalyzed Tandem 1,4-Benzodiazepine Synthesis Based on Two Hydrogen-Transfer Reactions. Eur. J. Org. Chem. 2015, 2015, 1068–1074. [Google Scholar] [CrossRef]
- Popp, T.A.; Uhl, E.; Ong, D.N.; Dittrich, S.; Bracher, F. A New Approach to Monoprotected 1,4-Benzodiazepines via a One-pot N-Deprotection/Reductive Cyclization Procedure. Tetrahedron 2016, 72, 1668–1674. [Google Scholar] [CrossRef]
- Mishra, J.K.; Garg, P.; Dohare, P.; Kumar, A.; Siddiqi, M.I.; Ray, M.; Panda, G. Amino Acid-based Enantiomerically Pure 3-substituted 1,4-Benzodiazepin-2-ones: A New Class of Anti-ischemic Agents. Bioorg. Med. Chem. Lett. 2007, 17, 1326–1331. [Google Scholar] [CrossRef]
- Massa, S.; Mai, A.; Artico, M. Syntheses of 3b,4,6,7-Tetrahydro-5h,9h-pyrazino [2,1-c] phyrolo [1,2-a] [1,4] benzodiazepine, A Valuable Precursor of Potential Central Nervous System Agents. Tetrahedron 1989, 45, 2763–2772. [Google Scholar] [CrossRef]
- Hone, N.D.; Wilson, W.; Reader, J.C. Solid Phase Synthesis of Tetrahydro-1,4-benzodiazepin-2-ones. Tetrahedron Lett. 2003, 44, 8493–8495. [Google Scholar] [CrossRef]
- Hamann, L.G.; Ding, C.Z.; Miller, A.V.; Madsen, C.S.; Wang, P.; Stein, P.D.; Pudzianowski, A.T.; Green, D.W.; Monshizadegan, H.; Atwal, K.S. Benzodiazepine-based Selective Inhibitors of Mitochondrial F1F0 ATP Hydrolase. Bioorg. Med. Chem. Lett. 2004, 14, 1031–1034. [Google Scholar] [CrossRef]
- Molteni, G.; Buttero, P.D. A One-Step Synthesis of Enantiopure 2-Substituted 4,5-Dihydro-1,4-benzodiazepine-3-ones via Intramolecular Azide Cycloaddition. Tetrahedron Asymmetry 2007, 18, 1197–1201. [Google Scholar] [CrossRef]
- Manick, A.D.; Duret, G.; Tran, D.N.; Berhal, F.; Prestat, G. Synthesis of 1,4-Benzodiazepinones and 1,4-Benzoxazepinones via Palladium-Catalyzed Amino and Oxyacetoxylation. Org. Chem. Front. 2014, 1, 1058–1061. [Google Scholar] [CrossRef]
- Zhang, Z.; Qin, Z.; Chang, W.; Li, J.; Fan, R.; Wu, X.; Guo, R.; Xie, X.; Zhou, L. Nicke-Catalyzed Decarboxylative Cyclization of Isatoic Anhydrides with Carbodiimides: Synthesis of 2,3-Dihydroquinazolin-4(1H)-ones. Adv. Synth. Catal. 2020, 362, 2864–2869. [Google Scholar] [CrossRef]
- Neukom, J.D.; Aquino, A.S.; Wolfe, J.P. Synthesis of Saturated 1,4-Benzodiazepines via Pd-Catalyzed Carboamination Reactions. Org. Lett. 2011, 13, 2196–2199. [Google Scholar] [CrossRef] [PubMed]
- Beccalli, E.M.; Broggini, G.; Paladino, G.; Penoni, A.; Zoni, C. Regioselective Formation of Six- and Seven-Membered Ring by Intramolecular Pd-Catalyzed Amination of N-Allyl-anthranilamides. J. Org. Chem. 2004, 69, 5627–5630. [Google Scholar] [CrossRef] [PubMed]
- Herrero, M.T.; Tellitu, I.; Dominguez, E.; Moreno, I.; SanMartin, R. A Novel and Efficient Iodine(III)-Mediated Access to 1,4-Benzodiazepin-2-ones. Tetrahedron Lett. 2002, 43, 8273–8275. [Google Scholar] [CrossRef]
- Li, X.; Yang, L.; Zhang, X.; Zhang, D.; Du, Y.; Zhao, K. Construction of 1,4-Benzodiazepine Skeleton From 2-(Arylamino)benzamides Through PhI(OAc)2-Mediated Oxidative C-N Bond Formation. J. Org. Chem. 2014, 79, 955–962. [Google Scholar] [CrossRef]
- Ghorai, M.K.; Shahi, C.K.; Bhattacharyya, A.; Sayyad, M.; Mal, A.; Wani, I.A.; Chauhan, N. Syntheses of Tetrahydrobenzodiazepines via SN2-Type Ring-Opening of Activated Aziridines with 2-Bromobenzylamine Followed by Copper-Powder-Mediated C-N Bond Formation. Asian J. Org. Chem. 2015, 4, 1103–1111. [Google Scholar] [CrossRef]
- Leng, D.-H.; Wang, D.-X.; Pan, J.; Huang, Z.-T.; Wang, M.-X. Highly Efficient and Enantioselective Biotransformations of Racemic Azetidine-2-carbonitriles and Their Synthetic Applications. J. Org. Chem. 2009, 74, 6077–6082. [Google Scholar] [CrossRef]
- Agami, C.; Couty, F.; Evano, G. A Straightforward Synthesis of Enantiopure 2-Cyano Azetidines from β-Amino Alcohols. Tetrahedron Asymmetry 2002, 13, 297–302. [Google Scholar] [CrossRef]
- Sulmon, P.; De Kimpe, N.; Schamp, N.; Tinant, B.; Declercq, J.-P. Synthesis of Azetidines From β-Chloro Imines. Tetrahedron 1988, 44, 3653–3670. [Google Scholar] [CrossRef]
- Wang, L.; Sullivan, G.M.; Hexamer, L.A.; Hasvold, L.A.; Thalji, R.; Przytulinska, M.; Tao, Z.-F.; Li, G.; Chen, Z.; Xiao, Z.; et al. Design, Synthesis, and Biological Activity of 5,10-Dihydro-dibenzo[b,e][1,4]diazepin-11-one-Based Potent and Selective Chk-1 Inhibitors. J. Med. Chem. 2007, 50, 4162–4176. [Google Scholar] [CrossRef]
- Hasvold, L.A.; Wang, L.; Przytulinska, M.; Xiao, Z.; Chen, Z.; Gu, W.Z.; Merta, P.J.; Xue, J.; Kovar, P.; Zhang, H.; et al. Investigation of Novel 7,8-Disubstituted-5,10-dihydro-dibenzo[b,e][1,4]diazepin-11-ones as Potent Chk1 Inhibitors. Bioorg. Med. Chem. Lett. 2008, 18, 2311–2315. [Google Scholar] [CrossRef] [PubMed]
- Couty, F.; David, O.; Larmanjat, B.; Marrot, J. Strained Azetidinium Ylides: New Reagents for Cyclopropanation. J. Org. Chem. 2007, 72, 1058–1061. [Google Scholar] [CrossRef] [PubMed]
- Gaertner, V.R. Ring-opening Alkylations of 1,1-Dialkyl-3-substituted Azetidinium Cations. Substituent Entropy-controlled Strained Ring-chain Equilibria. J. Org. Chem. 1968, 33, 523–530. [Google Scholar] [CrossRef]
- Vargas-Sanchez, M.; Couty, F.; Evano, G.; Prim, D.; Marrot, J. 3-Aminopyrrolidines via Ring Rearrangement of 2-Aminomethylazetidines. Synthesis of (−)-Absouline. Org. Lett. 2005, 7, 5861–5864. [Google Scholar] [CrossRef]
- Ghorai, M.K.; Kumar, A.; Das, K. Lewis Acid-Mediated Unprecedented Ring-Opening Rearrangement of 2-Aryl-N-Tosylazetidines to Enantiopure (E)-Allylamines. Org. Lett. 2007, 9, 5441–5444. [Google Scholar] [CrossRef]
- Feng, B.; Tang, M.; Xiao, R.; Wang, Q.; Zhu, G.; Zhang, Z.; Yuan, Z.; Wang, Y. Photocatalytic Three-Component Reductive Coupling Synthesis of gem-Difluorohomoallyl Secondary Amines. J. Org. Chem. 2025, 90, 2118–2125. [Google Scholar] [CrossRef]
- Vargas-Sanchez, M.; Lakhdar, S.; Couty, F.; Evano, G. Reaction of Azetidines with Chloroformates. Org. Lett. 2006, 8, 5501–5504. [Google Scholar] [CrossRef]
- Ma, S.; Yoon, D.H.; Ha, H.-J.; Leeb, W.K. Preparation of Enantiopure 2-Acylazetidines and Their Reactions with Chloroformates. Tetrahedron Lett. 2007, 48, 269–271. [Google Scholar] [CrossRef]

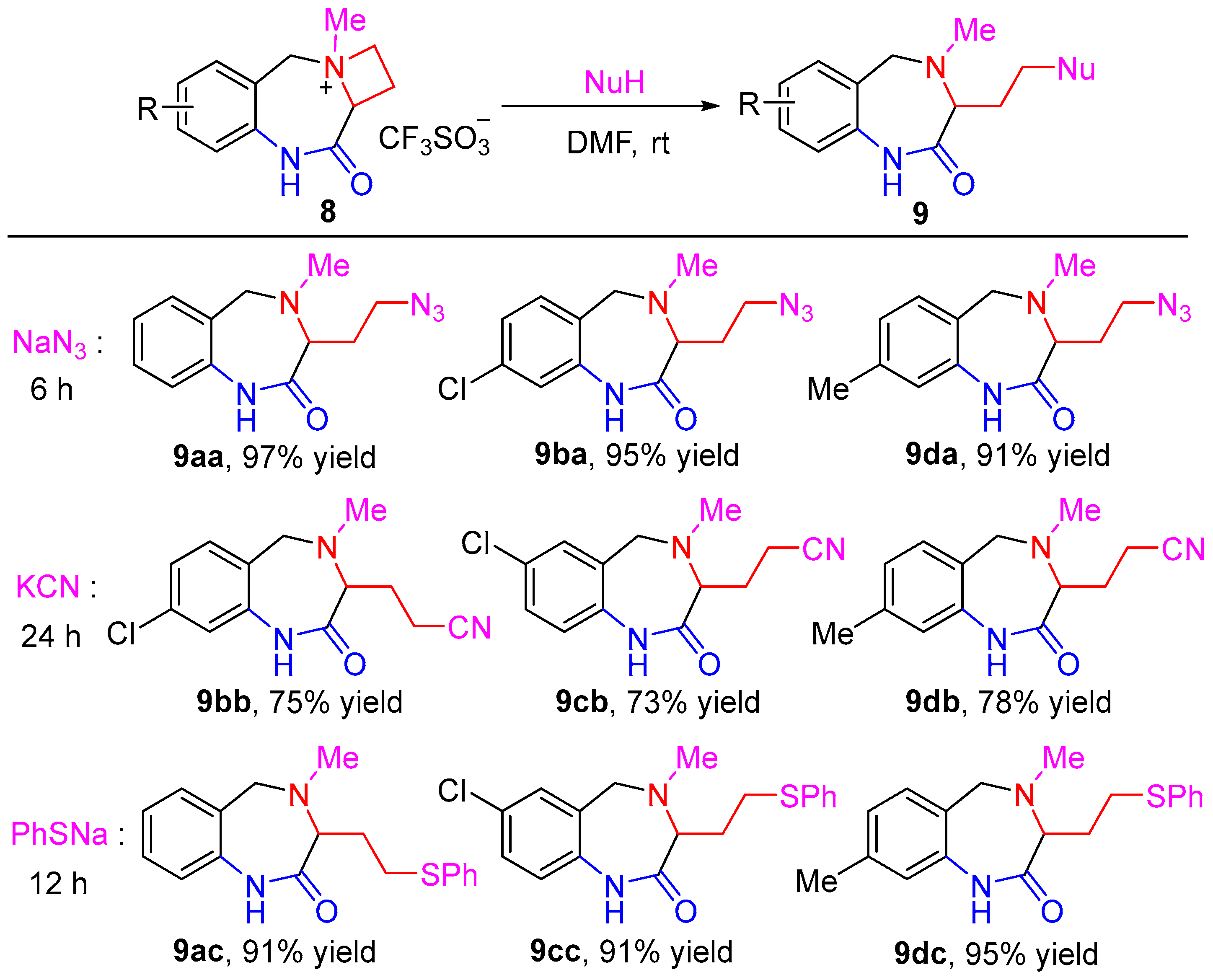
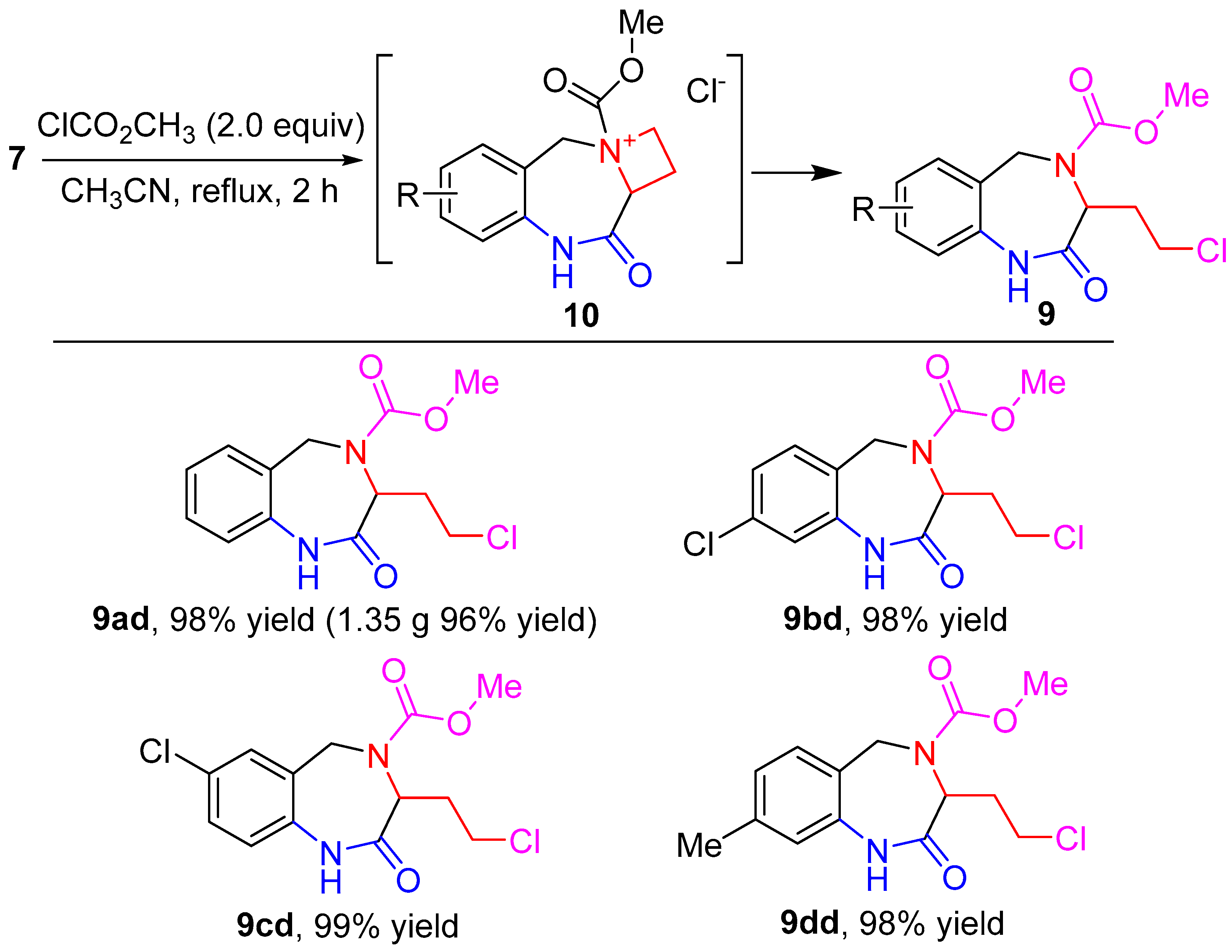

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
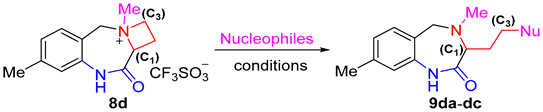 | |||||
|---|---|---|---|---|---|
| Entry |
Nucleophile
(2.0 equiv) | Solvent |
T (°C) |
t (h) |
Yield 2 (%) |
| 1 | NaN3 | DMF | rt | 6 | 91 |
| 2 | NaN3 | THF | rt | 12 | 84 |
| 3 | NaN3 | DCM | rt | 12 | 62 |
| 4 | KCN | DMF | rt | 12 | 64 |
| 5 | KCN | DMF | rt | 24 | 78 |
| 6 | PhSNa | DMF | rt | 12 | 95 |
| 7 | PhOH | DMF | rt | 24 | N.R. 3 |
| 8 | PhOH | DMF | reflux | 24 | trace |
| 9 | PhONa | DMF | rt | 24 | N.R. 3 |
| 10 | PhONa | DMF | reflux | 24 | trace |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, X.-M.; Chen, S.; Duan, S.-L.; Wang, X.-M.; Liu, Q.; Sun, K. Synthesis of 1,4-Benzodiazepines via Intramolecular C–N Bond Coupling and Ring Opening of Azetidines. Molecules 2025, 30, 2014. https://doi.org/10.3390/molecules30092014
Xu X-M, Chen S, Duan S-L, Wang X-M, Liu Q, Sun K. Synthesis of 1,4-Benzodiazepines via Intramolecular C–N Bond Coupling and Ring Opening of Azetidines. Molecules. 2025; 30(9):2014. https://doi.org/10.3390/molecules30092014
Chicago/Turabian StyleXu, Xin-Ming, Sen Chen, Shao-Lei Duan, Xiang-Min Wang, Qian Liu, and Kai Sun. 2025. "Synthesis of 1,4-Benzodiazepines via Intramolecular C–N Bond Coupling and Ring Opening of Azetidines" Molecules 30, no. 9: 2014. https://doi.org/10.3390/molecules30092014
APA StyleXu, X.-M., Chen, S., Duan, S.-L., Wang, X.-M., Liu, Q., & Sun, K. (2025). Synthesis of 1,4-Benzodiazepines via Intramolecular C–N Bond Coupling and Ring Opening of Azetidines. Molecules, 30(9), 2014. https://doi.org/10.3390/molecules30092014