

Microwave-Assisted Enantioselective Synthesis of (2R,5S)-Theaspirane: A Green Chemistry Approach

 , , ,
, , , 
Abstract

1. Introduction
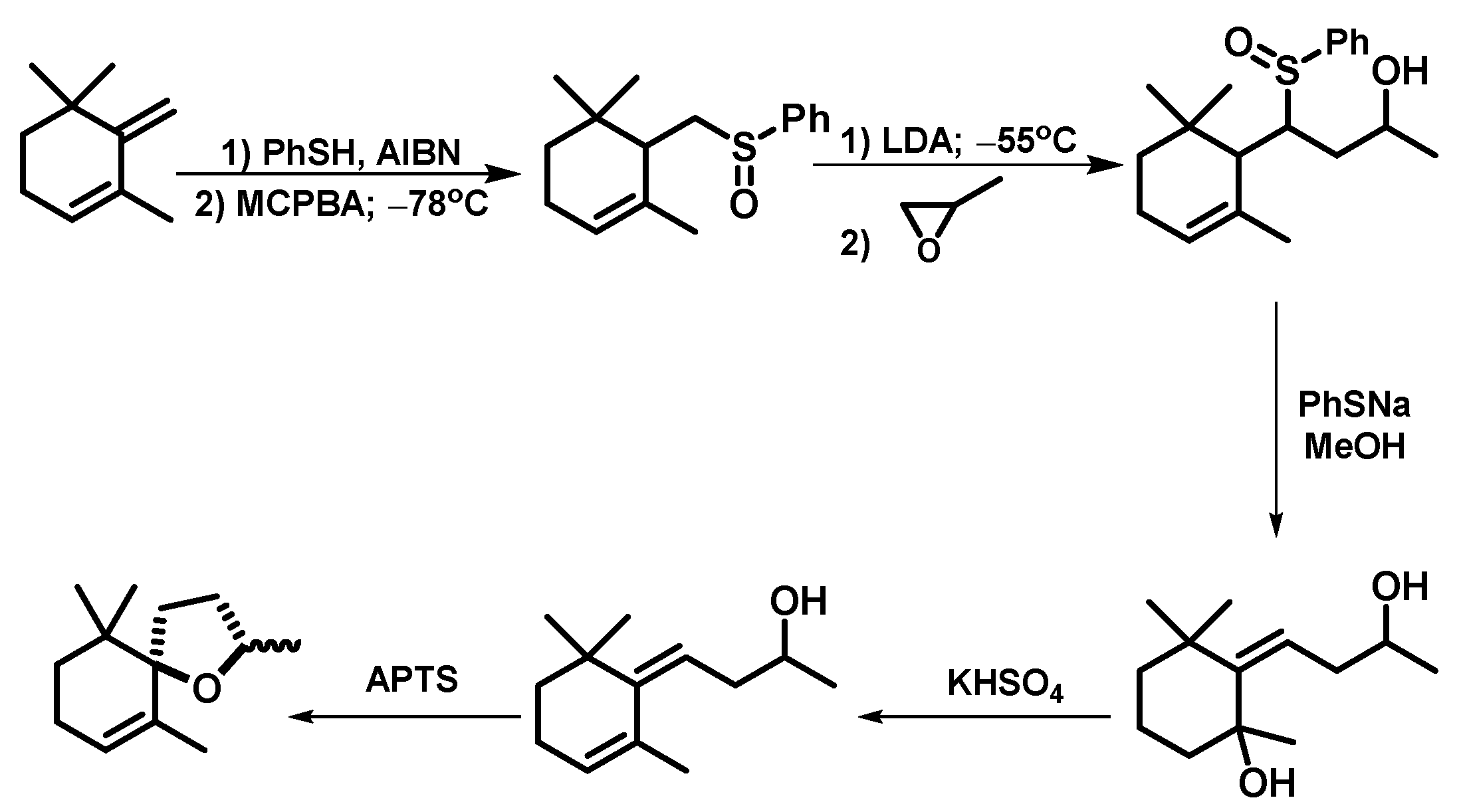
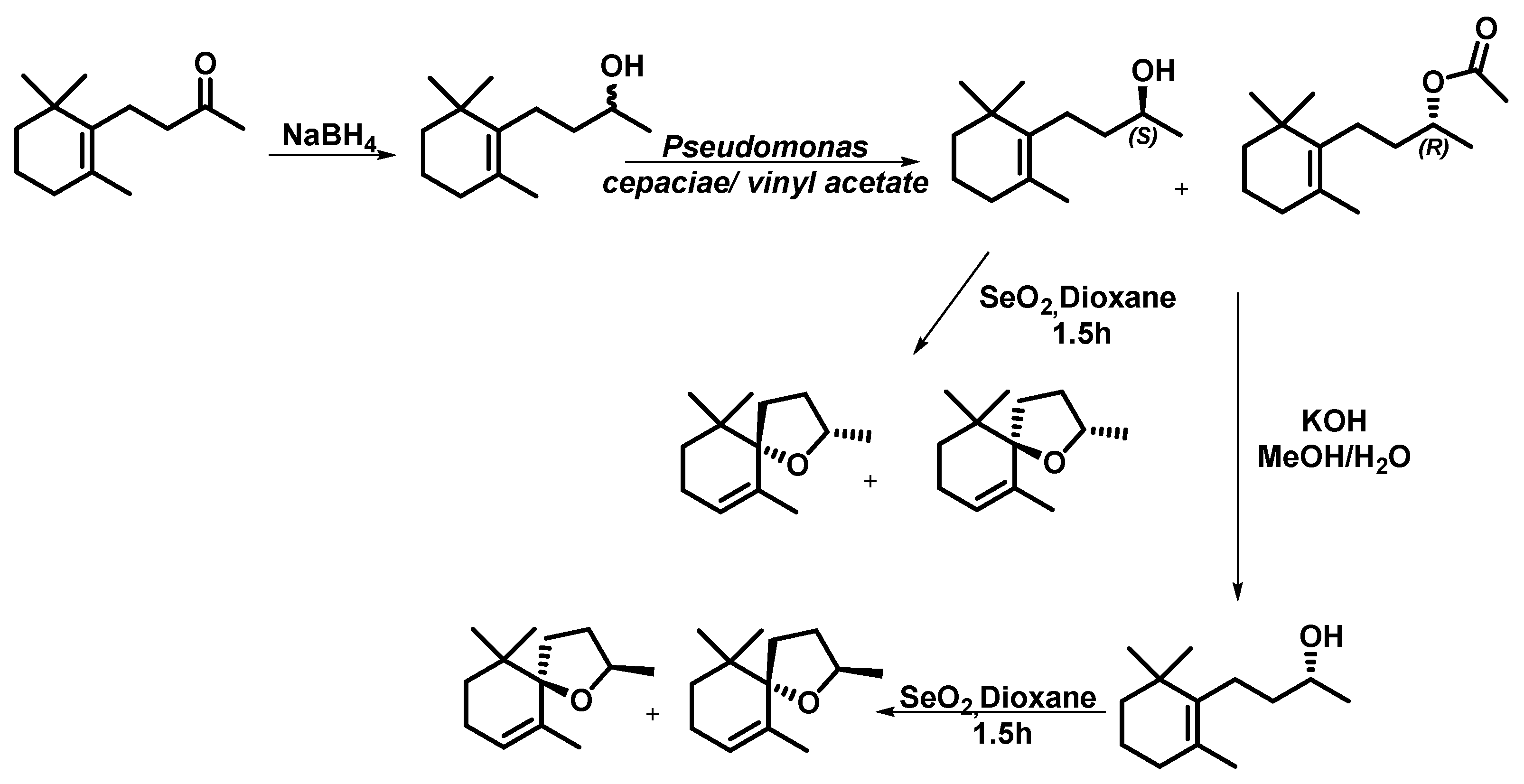
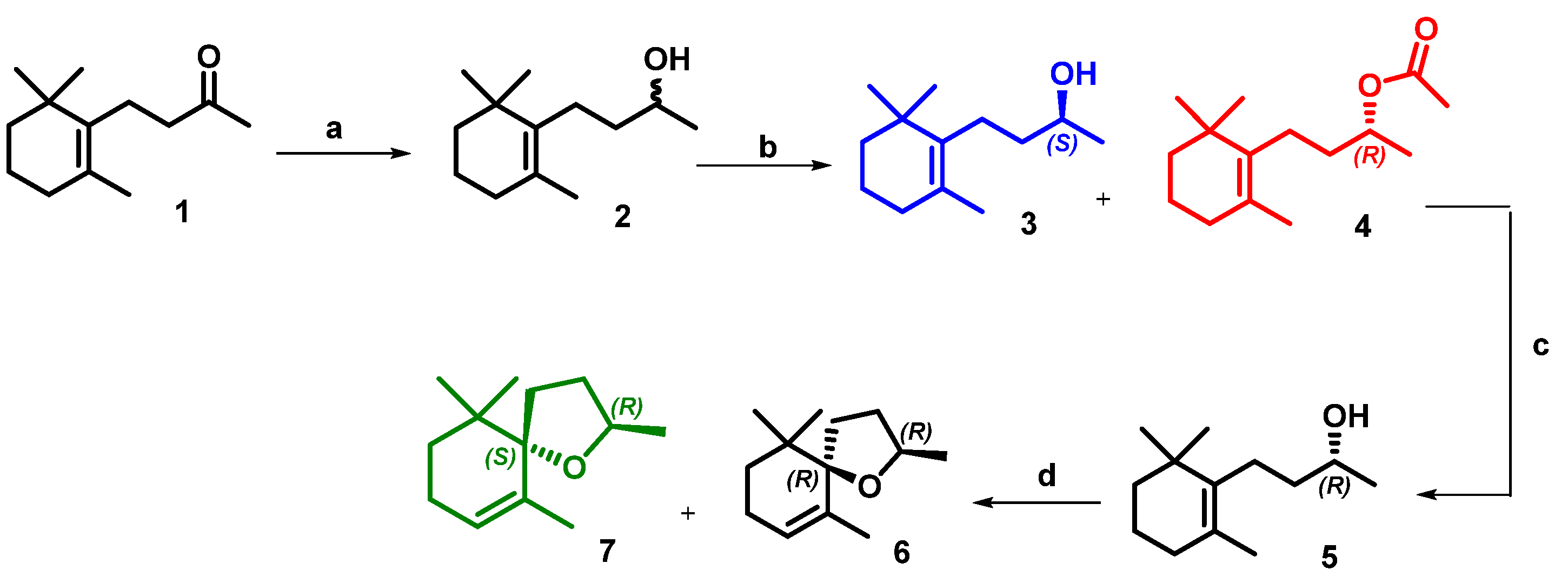
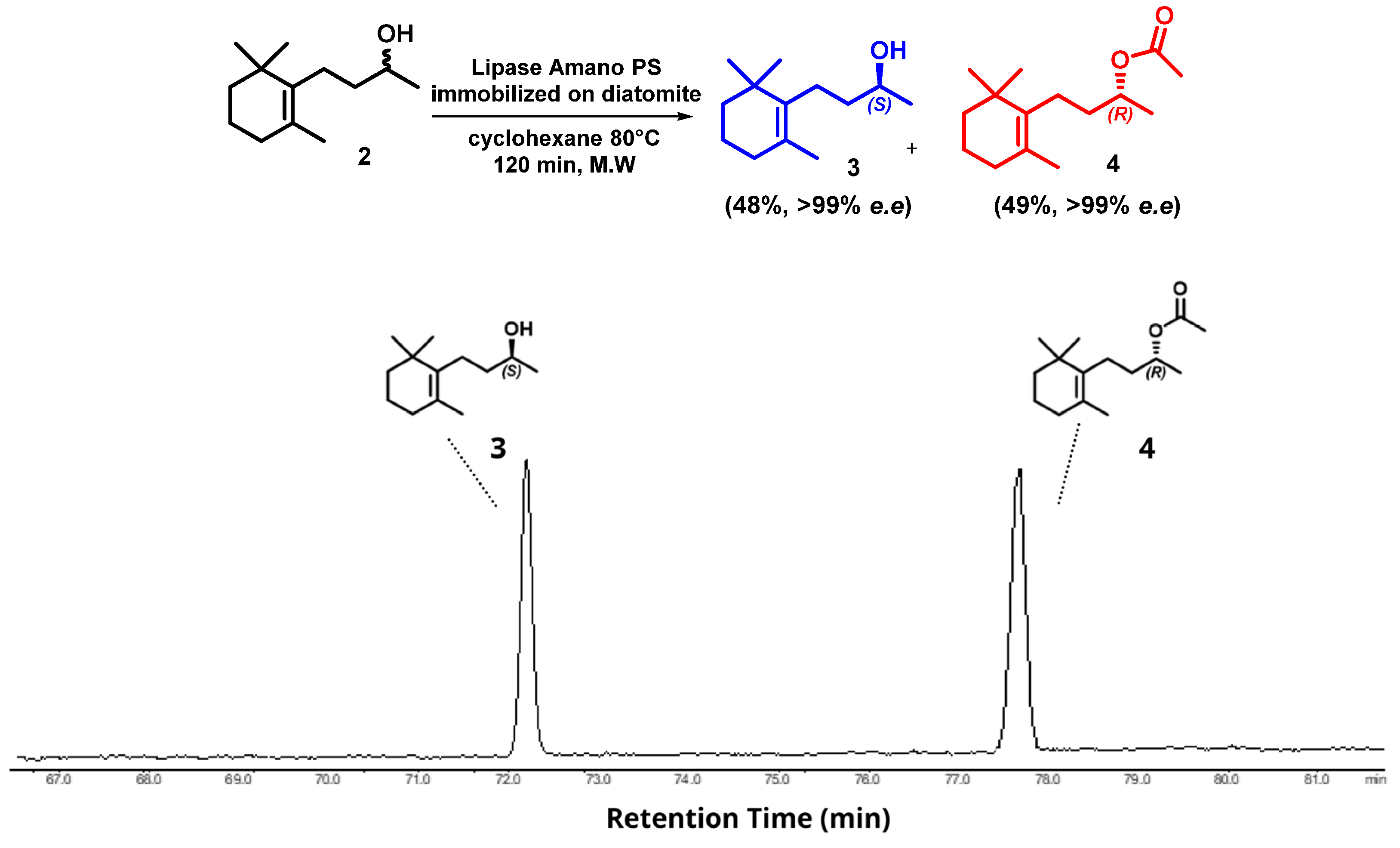
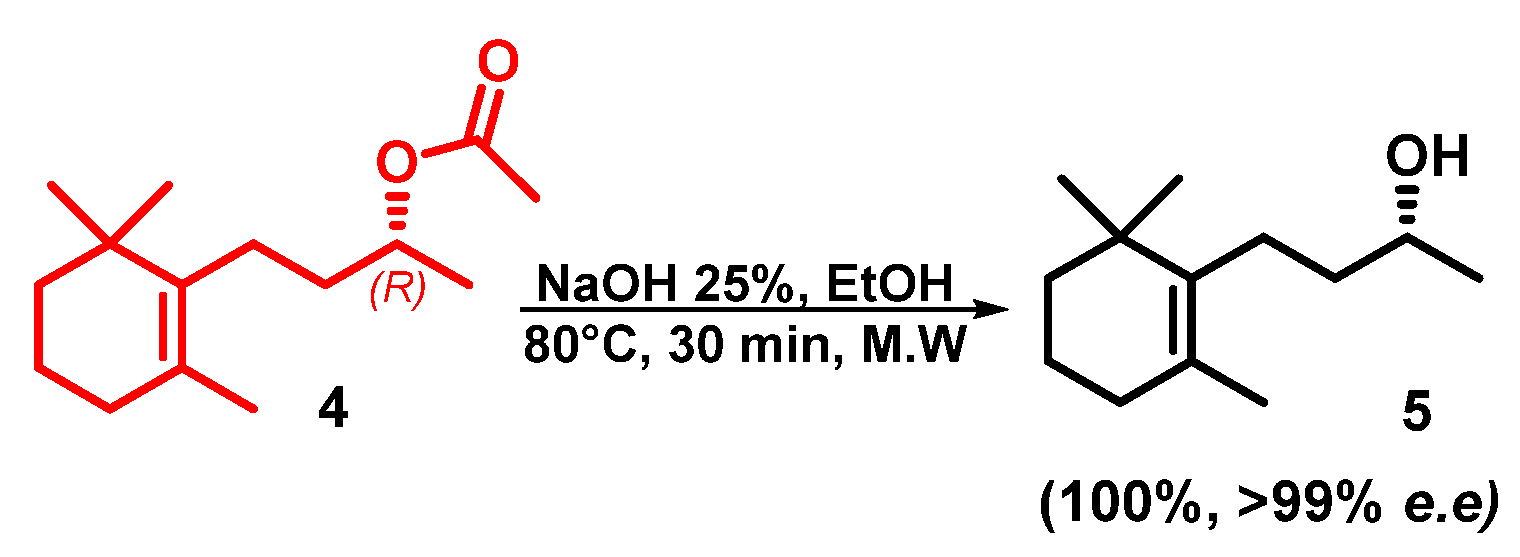
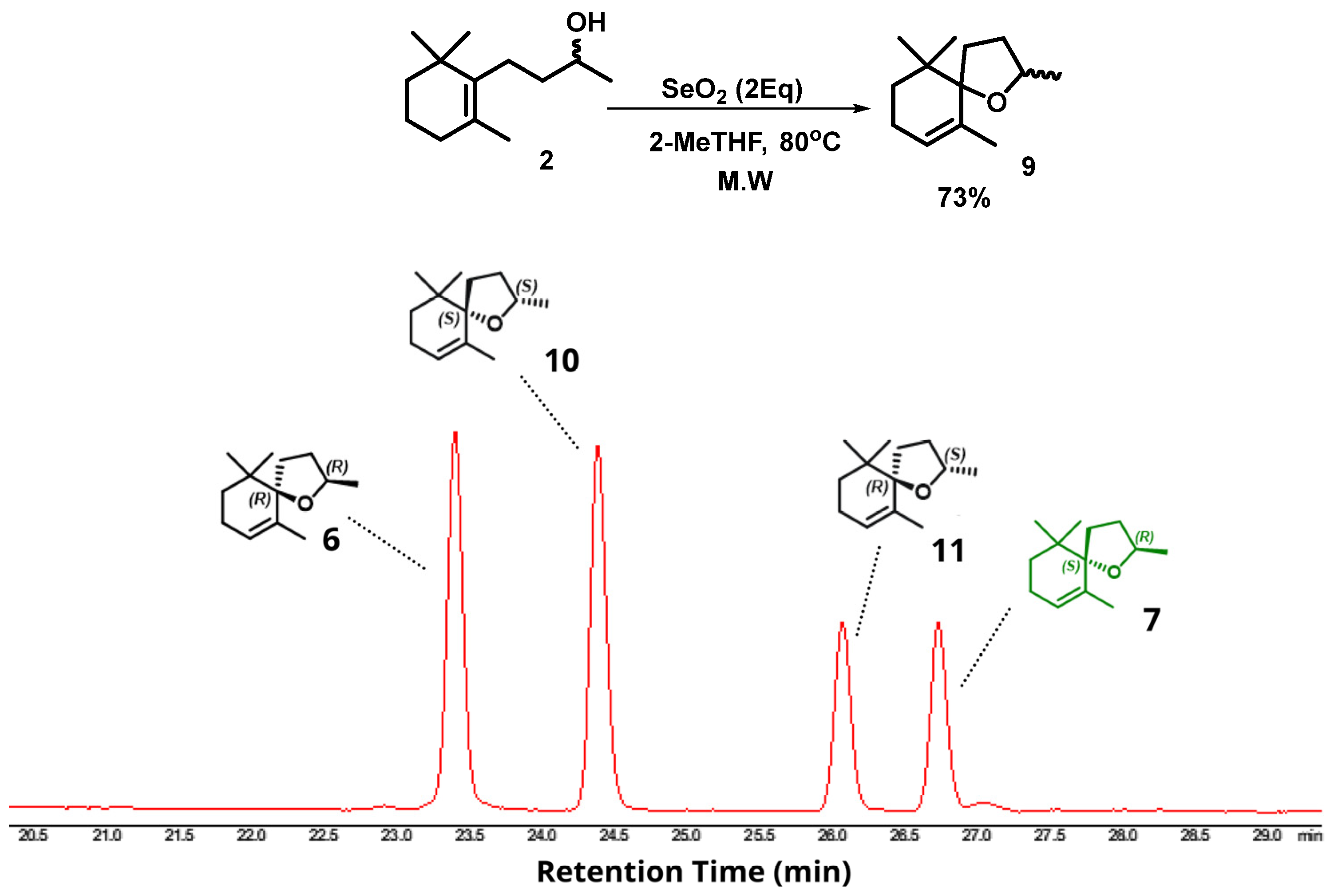
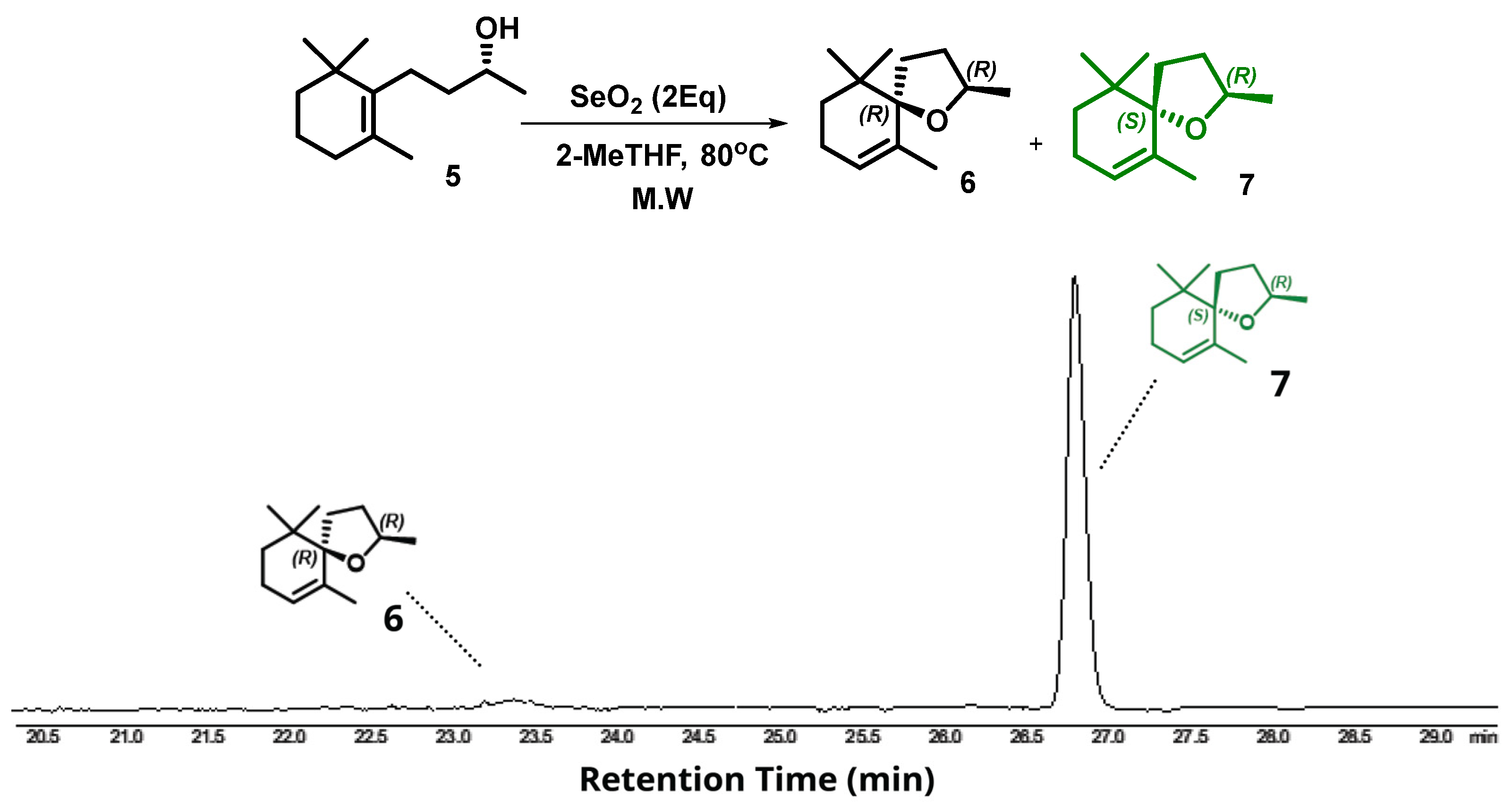
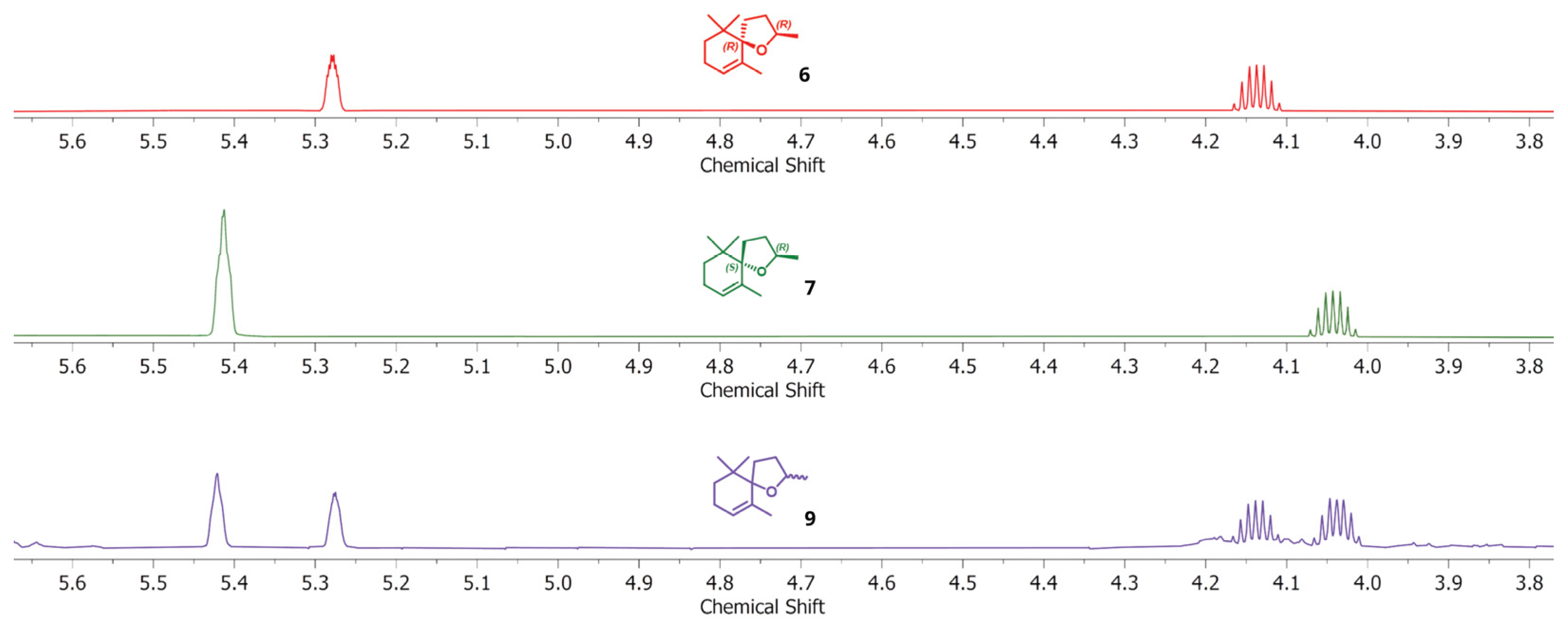
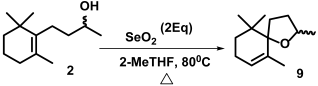
2. Results and Discussion
Microwave-Assisted Stereoselective Synthesis of (2R,5S)-Theaspirane
3. Materials and Methods
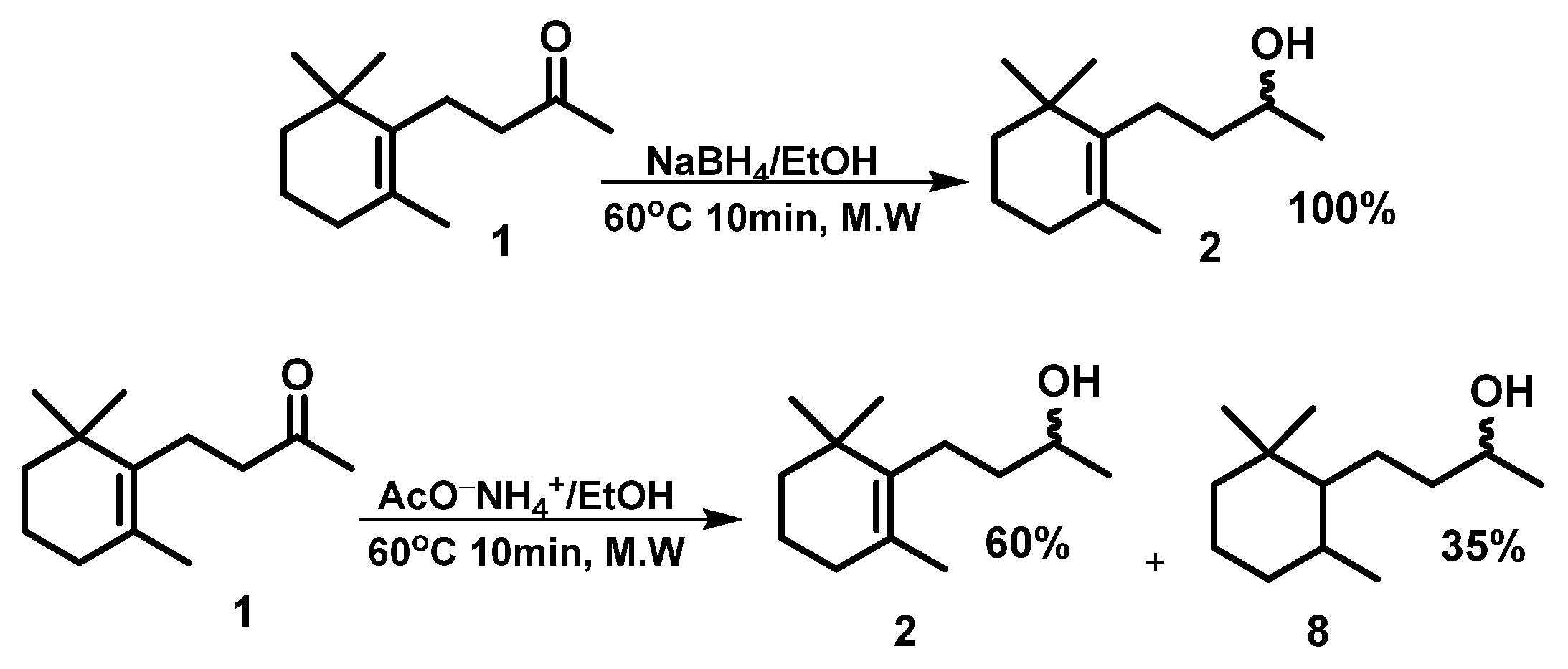
3.1. Microwave-Assisted Synthesis of Dihydro-β-ionol
3.2. Kinetic Resolution of Dihydro-β-ionol by Lipase from Amano Lipase PS Under Microwave-Assisted Conditions
3.3. Microwave-Assisted Deacetylation of (R)-Dihydro-β-ionol Acetate
3.4. Intramolecular Microwave-Assisted Cyclization for the Synthesis of (2R,5S)-Theaspirane
3.5. Intramolecular Microwave-Assisted Cyclization for the Synthesis of Racemic Theaspirane—Scale up
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 1HNMR | proton nuclear magnetic resonance |
| 13CNMR | Carbon-13 nuclear magnetic resonance |
| GC-FID | gas chromatography with flame ionization detection |
References
- Sharma, A.; Sandhi, R.K.; Reddy, G.V.P. A Review of Interactions between Insect Biological Control Agents and Semiochemicals. Insects 2019, 10, 439–455. [Google Scholar] [CrossRef] [PubMed]
- Moraes, M.C.B.; Laumann, R.A.; Sujii, E.R.; Borges, M. Induced Plant Volatiles in Biological Control of Insect Pests: A Review. Neotrop. Entomol. 2009, 38, 303–310. [Google Scholar]
- Serra, S. Recent Advances in the Synthesis of Carotenoid-Derived Flavours and Fragrances. Molecules 2015, 20, 12817–12840. [Google Scholar] [CrossRef] [PubMed]
- Abagale, S.A.; Woodcock, C.M.; Chamberlain, K.; Acquaah, S.O.; Van Emden, H.; Birkett, M.A.; Pickett, J.A.; Braimah, H. Attractiveness of Host Banana Leaf Materials to the Banana Weevil, Cosmopolites sordidus, in Ghana for Development of Field Management Strategies. Pest Manag Sci. 2018, 75, 549–555. [Google Scholar] [CrossRef]
- Anastas, P.T.; Eghbali, N. Green chemistry: Principles and practice. Chem. Soc. Rev. 2010, 39, 301–312. [Google Scholar] [CrossRef]
- Gawande, M.B.; Bonifácio, V.D.B.; Luque, R.; Branco, P.S.; Varma, R.S. Solvent-free and catalysts-free chemistry: A benign pathway to sustainability. Chem. Soc. Rev. 2013, 42, 5522–5551. [Google Scholar] [CrossRef]
- Das, S.; Banik, R.; Kumar, B.; Roy, S.; Noorussabah; Amhad, K.; Sukul, P.K. A Green Approach for Organic Transformations Using Microwave Reactor. Curr. Org. Synth. 2019, 16, 730–764. [Google Scholar] [CrossRef]
- Kappe, C.O. Controlled microwave heating in modern organic synthesis. Angew. Chem. Int. Ed. 2004, 43, 6250–6284. [Google Scholar] [CrossRef]
- Perreux, L.; Loupy, A. A tentative rationalization of microwave effects in organic synthesis according to the reaction medium, and mechanistic considerations. Tetrahedron 2001, 57, 9199–9223. [Google Scholar] [CrossRef]
- Kappe, C.O.; Dallinger, D. The impact of microwave synthesis on drug discovery. Nat. Rev. Drug Discov. 2009, 8, 697–707. [Google Scholar]
- Masuda, H.; Mihara, S. A Short-Step Synthesis of Theaspirane. Agric. Biol. Chem. 1985, 49, 861–862. [Google Scholar]
- Gutiérrez, A.; Palos, R. Green Chemistry: From Wastes to Value-Added Products. Processes 2023, 11, 2131. [Google Scholar] [CrossRef]
- Uneyama, K.; Fujibayashi, S.; Torii, S. A Novel Selenium-Mediated Spiroannelation: One-Step Preparation of dl-Theaspirane from α-Dihydroionol. Tetrahedron Lett. 1985, 26, 4637–4638. [Google Scholar] [CrossRef]
- Lemir, I.D.; Castro-Godoy, W.D.; Heredia, A.A.; Schmidt, L.C.; Argüello, J.E. Metal- and photocatalyst-free synthesis of 3-selenylindoles and asymmetric diarylselenides promoted by visible light. RSC Adv. 2019, 9, 22685–22694. [Google Scholar] [CrossRef] [PubMed]
- University of California, Berkeley. Guidelines for Explosive and Potentially Explosive Chemicals: Safe Storage and Handling. University of California: Berkeley, CA, USA, 2015; pp. 1–17. [Google Scholar]
- Schmidt, G.; Full, G.; Winterhalter, P.; Schreier, P. Synthesis and Enantiodifferentiation of Isomeric Theaspiranes. J. Agric. Food Chem. 1992, 40, 1188–1191. [Google Scholar] [CrossRef]
- Young, J.-J.; Jung, L.-J.; Cheng, K.-M. Amberlyst-15-Catalyzed Intramolecular SN2 Oxaspirocyclization of Secondary Allylic Alcohols: Application to the Total Synthesis of Spirocyclic Ethers Theaspirane and Theaspirone. Tetrahedron Lett. 2000, 41, 3415–3418. [Google Scholar] [CrossRef]
- Kumasaki, M. An explosion of a tank car carrying waste hydrogen peroxide. J. Loss Prev. Process Ind. 2006, 19, 307–311. [Google Scholar] [CrossRef]
- Boulin, B.; Arreguy-San Miguel, B.; Delmond, B. Nouvelles Voies d’Accès aux Theaspiranes, Megastigma-5,7,9-trién-4-one et Megastigma-5,8-dièn-4-one à partir du γ-Pyronène. Tetrahedron 2000, 56, 3927–3932. [Google Scholar] [CrossRef]
- Zhang, X.; Hu, A.; Pan, C.; Zhao, Q.; Wang, X.; Lu, J. Safer preparation of m-CPBA/DMF solution in pilot plant. Org. Process Res. Dev. 2013, 17, 1591–1596. [Google Scholar] [CrossRef]
- Abagale, S.A.; Woodcock, C.M.; Hooper, A.M.; Caulfield, J.C.; Withall, D.; Chamberlain, K.; Acquaah, S.O.; Van Emden, H.; Braimah, H.; Pickett, J.A.; et al. (2R,5S)-Theaspirane Identified as the Kairomone for the Banana Weevil, Cosmopolites sordidus, from Attractive Senesced Leaves of the Host Banana, Musa spp. Chem. Eur. J. 2018, 24, 9217–9219. [Google Scholar] [CrossRef]
- Andrade, C.K.Z.; Silva, W.A. One-Step Reduction of Chalcones to Saturated Alcohols by Ammonium Formate/Palladium on Carbon: A Versatile Method. Lett. Org. Chem. 2006, 3, 39–41. [Google Scholar] [CrossRef]
- Gawley, R.E. Do the Terms “% ee” and “% de” Make Sense as Expressions of Stereoisomer Composition or Stereoselectivity? J. Org. Chem. 2006, 71, 2411–2416. [Google Scholar] [CrossRef] [PubMed]
- Komaki, R.; Ikawa, T.; Saito, K.; Hattori, K.; Ishikawa, N.; Fukawa, H.; Egi, M.; Akai, S. Discovery of aromatic components with excellent fragrance properties and biological activities: β-Ionols with antimelanogenetic effects and their asymmetric syntheses. Chem. Pharm. Bull. 2013, 61, 310–314. [Google Scholar] [CrossRef]
- Zhou, Z.; Walleser, P.M.; Tius, M.A. Isotetronic Acids from an Oxidative Cyclization. Chem. Commun. 2015, 51, 10858–10860. [Google Scholar] [CrossRef]
- Zheng, H.; Wang, K.; Zhang, W.; Liu, R. Selenium Dioxide–Mediated Synthesis of Fused 1,2,4-Triazoles as Cytotoxic Agents. Synth. Commun. 2015, 45, 2849–2856. [Google Scholar] [CrossRef]
- Saravanan, S.; Purushothaman, S.; Amali, I.B.; Muthusubramanian, S. Chemoselective Selenium Dioxide Oxidation of 1,4-Adducts Derived from Substituted Arylidene Acetophenones. Synth. Commun. 2009, 39, 2882–2888. [Google Scholar] [CrossRef]
- Liu, Y.; Sun, F.; An, Z.; Miao, M.; Guo, X.; Li, C. Selenium Dioxide-Promoted Regioselective Synthesis of Multi-Substituted Furans from Terminal Alkynes and Aldehydes. Adv. Synth. Catal. 2023, 366, 43–48. [Google Scholar] [CrossRef]
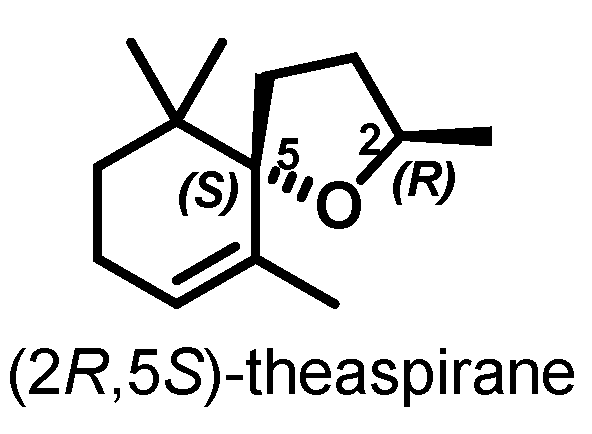

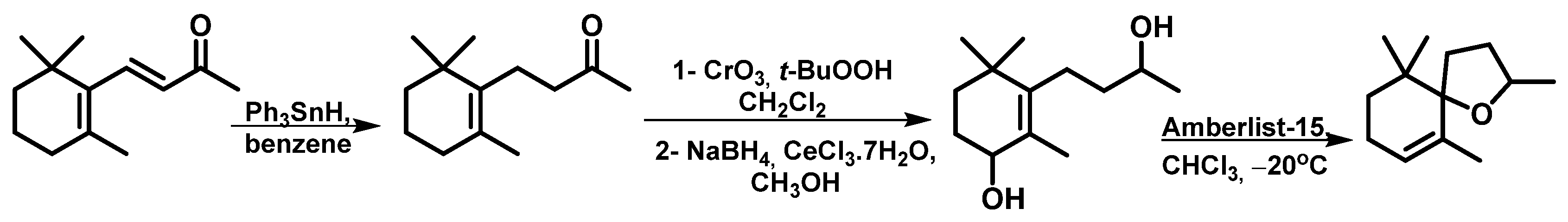

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||
|---|---|---|---|---|---|
| Entry | Temp. (°C) | Time (min) | Solvent | SeO2(Equivalents) | Yield (%) b |
| 1 | 60 a | 90 | dioxane | 1 | 12 |
| 2 | 60 a | 90 | dioxane | 2 | 28 |
| 3 | 60 | 15 | 2-MeTHF | 1 | 15 |
| 4 | 60 | 30 | 2-MeTHF | 1 | 23 |
| 5 | 60 | 15 | 2-MeTHF | 2 | 31 |
| 6 | 60 | 30 | 2-MeTHF | 2 | 35 |
| 7 | 80 | 15 | 2-MeTHF | 1,1 | 30 |
| 8 | 80 | 30 | 2-MeTHF | 1,1 | 33 |
| 9 | 80 | 15 | 2-MeTHF | 2 | 54 |
| 10 | 80 | 30 | 2-MeTHF | 2 | 73 |
| 11 | 80 | 60 | 2-MeTHF | 2 | 40 |
| 12 | 80 | 30 | 2-MeTHF | 3 | 56 |
| 13 | 100 | 15 | 2-MeTHF | 1,1 | 12 |
| 14 | 100 | 15 | 2-MeTHF | 2 | 29 |
| 15 | 100 | 15 | 2-MeTHF | 4 | 25 |
| 16 | 100 | 30 | 2-MeTHF | 2 | 0 |
| Entry | Reagents and Conditions | Overall Yield (%) | e.e (%) | Enantiomeric Ratio (E) c | Reference |
|---|---|---|---|---|---|
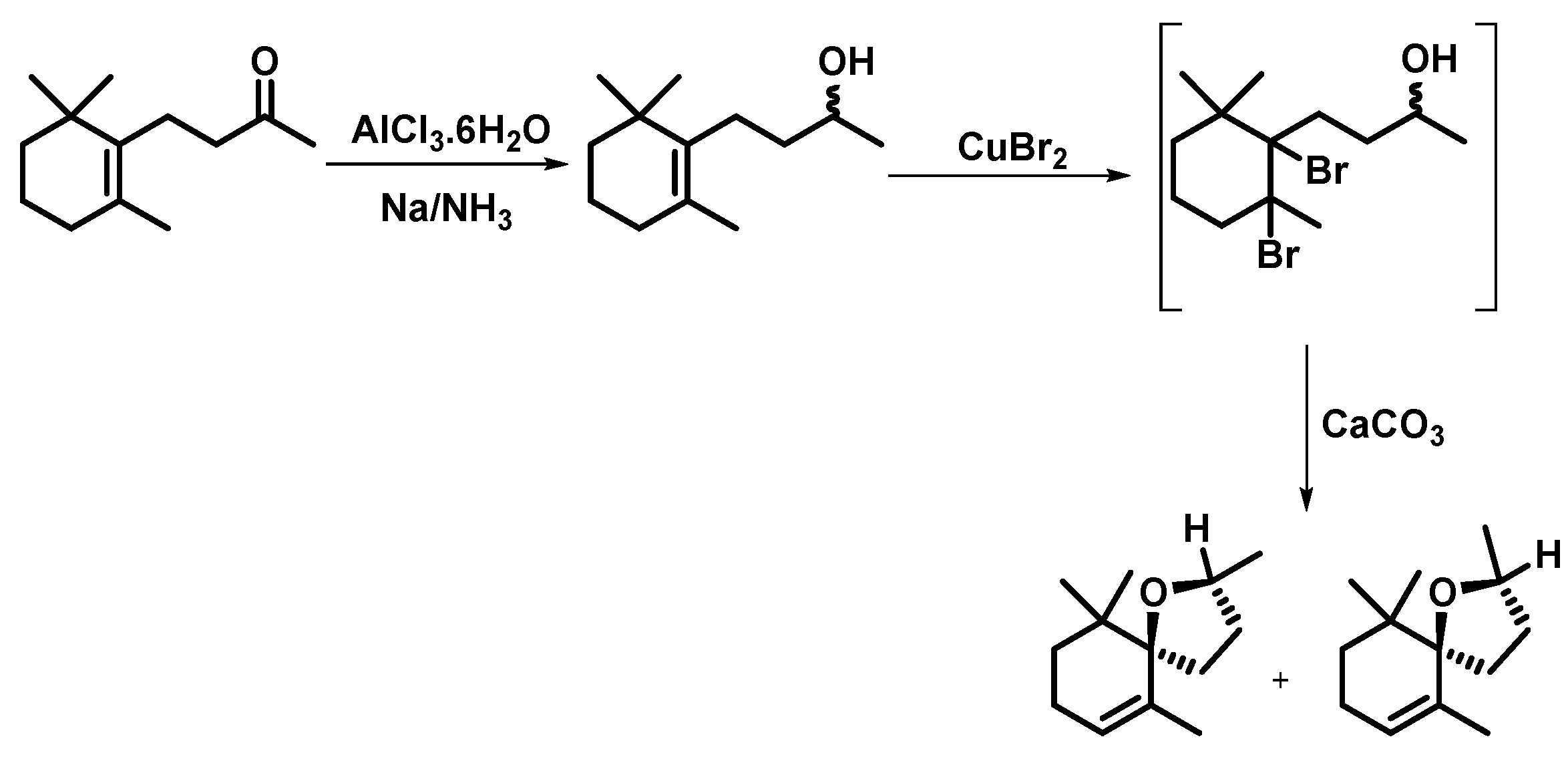
| 1 | AlCl3.6H2O; Na/NH3; CuBr2, CaCO3. a | 20 | Mixture isomers | - | [11] |
| 2 | bis(p-chloro phenyl diselenide), Et4NCl4/electrode Pt. a | 70 | Mixture isomers | - | [13] |
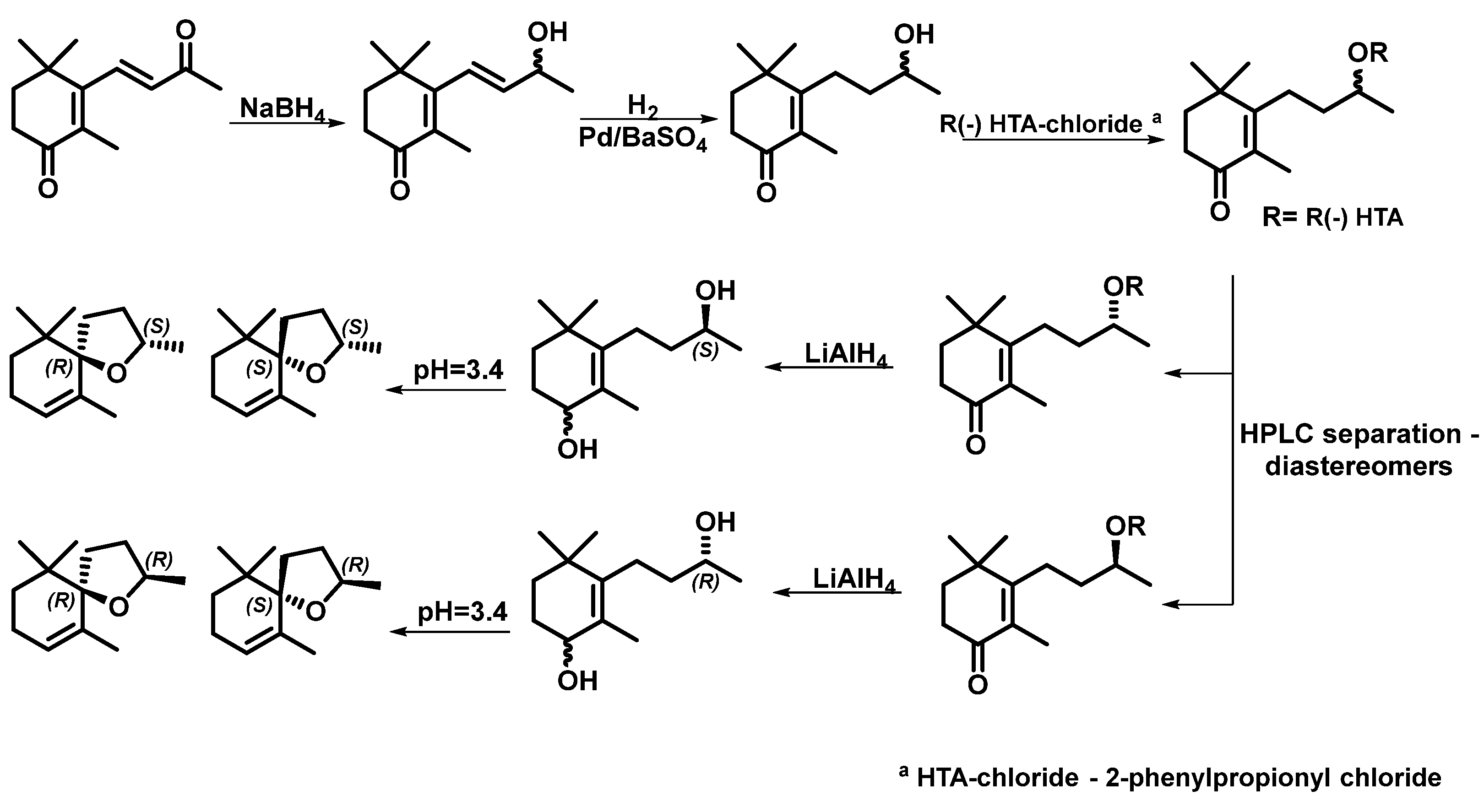
| 3 | LiAlH4, CCl4, H2, Pd/BaSO4, acidic conditions, energy-intensive, techniques, prolonged reaction times. | did not report the yields for all products. | 98% | - | [16] |
| 4 | Ph3SnH, CrO3, t-BuOOH, benzene, NaBH4, CeCl3.7H2O, amberlyst 15, −20 °C. a | 46 | Mixture isomers | - | [17] |
| 5 | AIBN, PhSH, MCPBA, propylene oxide, LDA, cryogenic temperatures. a | 21 | Mixture isomers | - | [19] |
| 6 | Dioxane, SeO2, (No quantities or methodologies described), prolonged reaction times. a | 41 * | 98.7 * | 1377 | [21] |
| 7 | 2-MeTHF, optimized methodologies for kinetic resolution and cyclization (SeO2), decreased total reaction time. b | 71 | 99.9 | 15,198 | This work |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takada, S.C.S.; Blassioli-Moraes, M.C.; Borges, M.; Laumann, R.A.; Maravalho, I.V.; Silva, W.A. Microwave-Assisted Enantioselective Synthesis of (2R,5S)-Theaspirane: A Green Chemistry Approach. Molecules 2025, 30, 1519. https://doi.org/10.3390/molecules30071519
Takada SCS, Blassioli-Moraes MC, Borges M, Laumann RA, Maravalho IV, Silva WA. Microwave-Assisted Enantioselective Synthesis of (2R,5S)-Theaspirane: A Green Chemistry Approach. Molecules. 2025; 30(7):1519. https://doi.org/10.3390/molecules30071519
Chicago/Turabian StyleTakada, Sayuri Cristina Santos, Maria Carolina Blassioli-Moraes, Miguel Borges, Raul Alberto Laumann, Izabella Vitória Maravalho, and Wender Alves Silva. 2025. "Microwave-Assisted Enantioselective Synthesis of (2R,5S)-Theaspirane: A Green Chemistry Approach" Molecules 30, no. 7: 1519. https://doi.org/10.3390/molecules30071519
APA StyleTakada, S. C. S., Blassioli-Moraes, M. C., Borges, M., Laumann, R. A., Maravalho, I. V., & Silva, W. A. (2025). Microwave-Assisted Enantioselective Synthesis of (2R,5S)-Theaspirane: A Green Chemistry Approach. Molecules, 30(7), 1519. https://doi.org/10.3390/molecules30071519