


Physical Stability and Molecular Mobility of Resveratrol in a Polyvinylpyrrolidone Matrix

 , , ,
, , ,
Abstract
1. Introduction
2. Results and Discussion
2.1. Effectiveness of the Preparation of Amorphous Solid Dispersion Systems
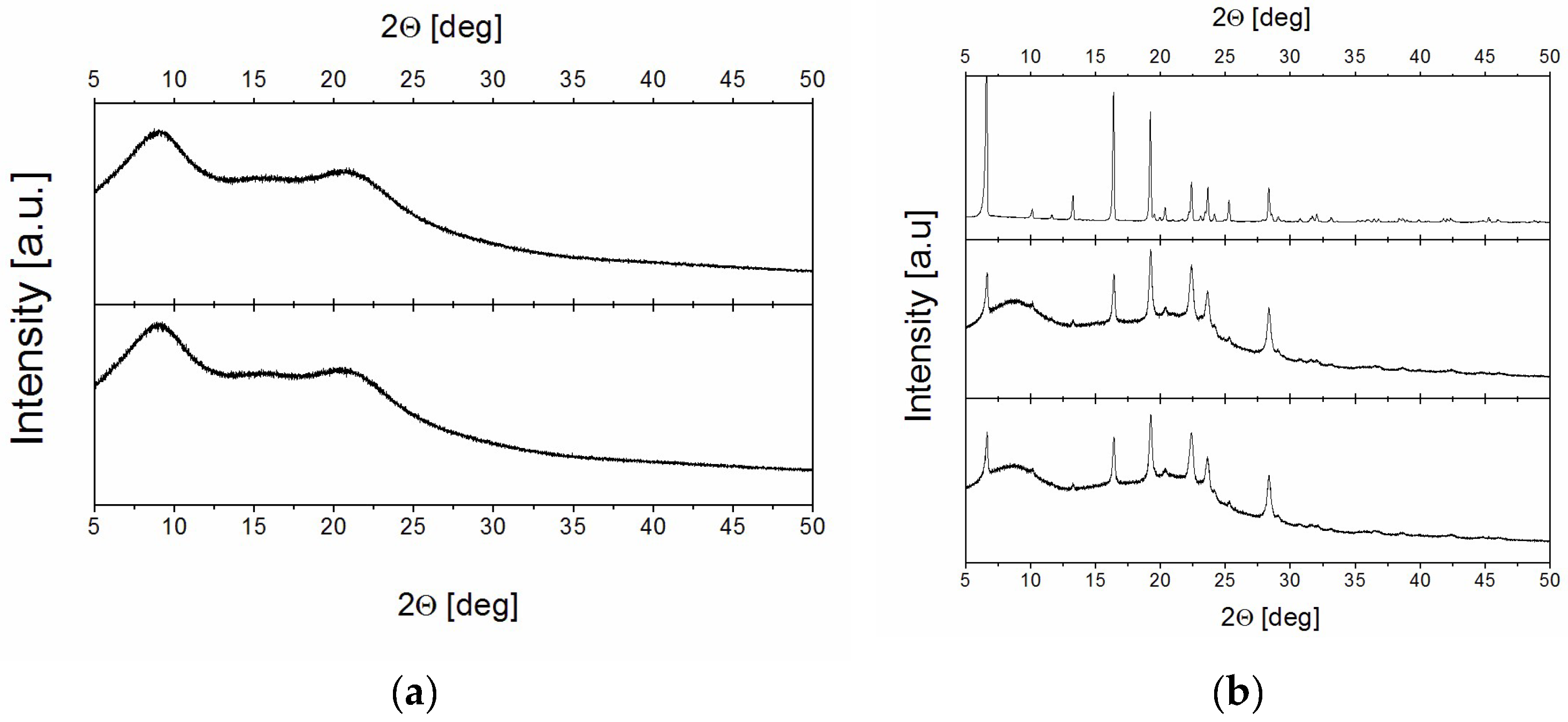
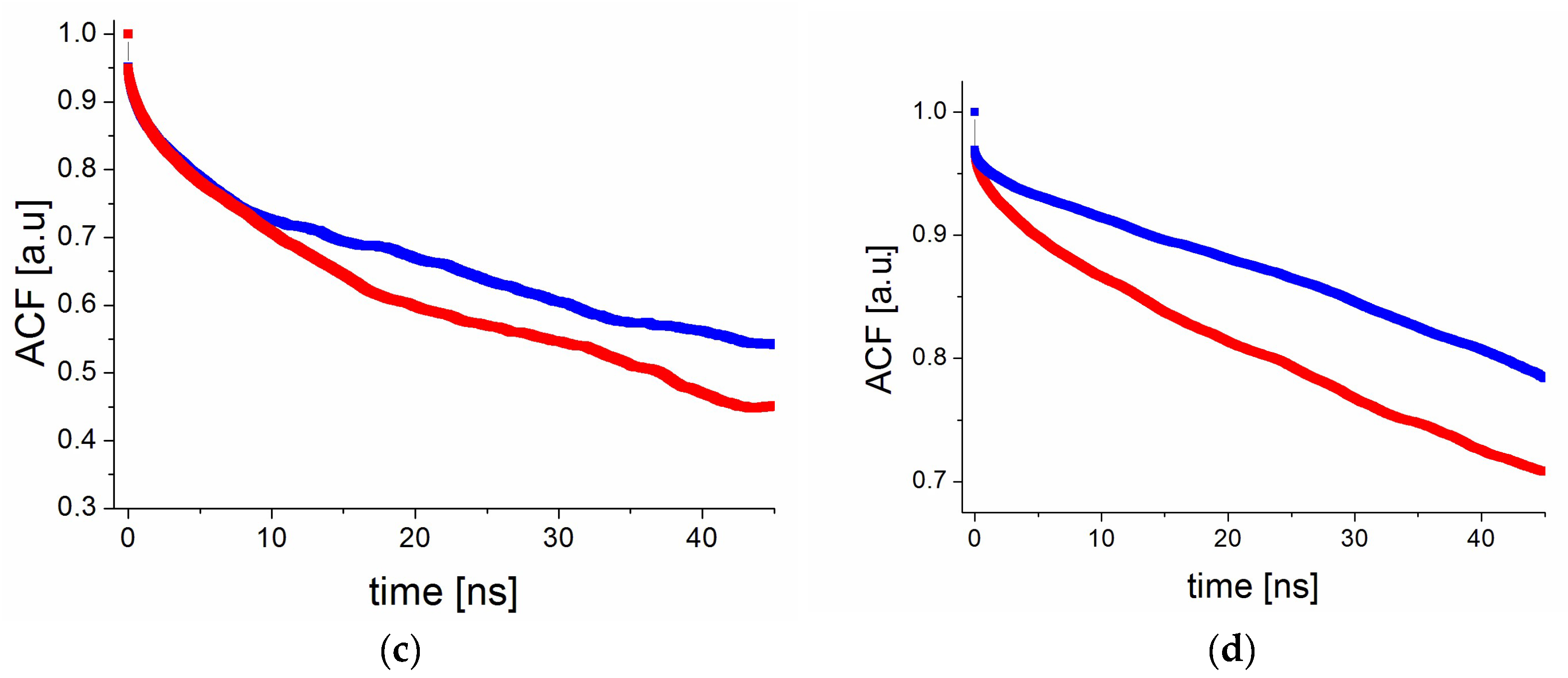
2.2. Systems Stability in 0% and 75% RH
2.2.1. 0% Relative Humidity
2.2.2. 75% Relative Humidity
2.2.3. The Analysis of the Phase Composition in 0% and 75% RH
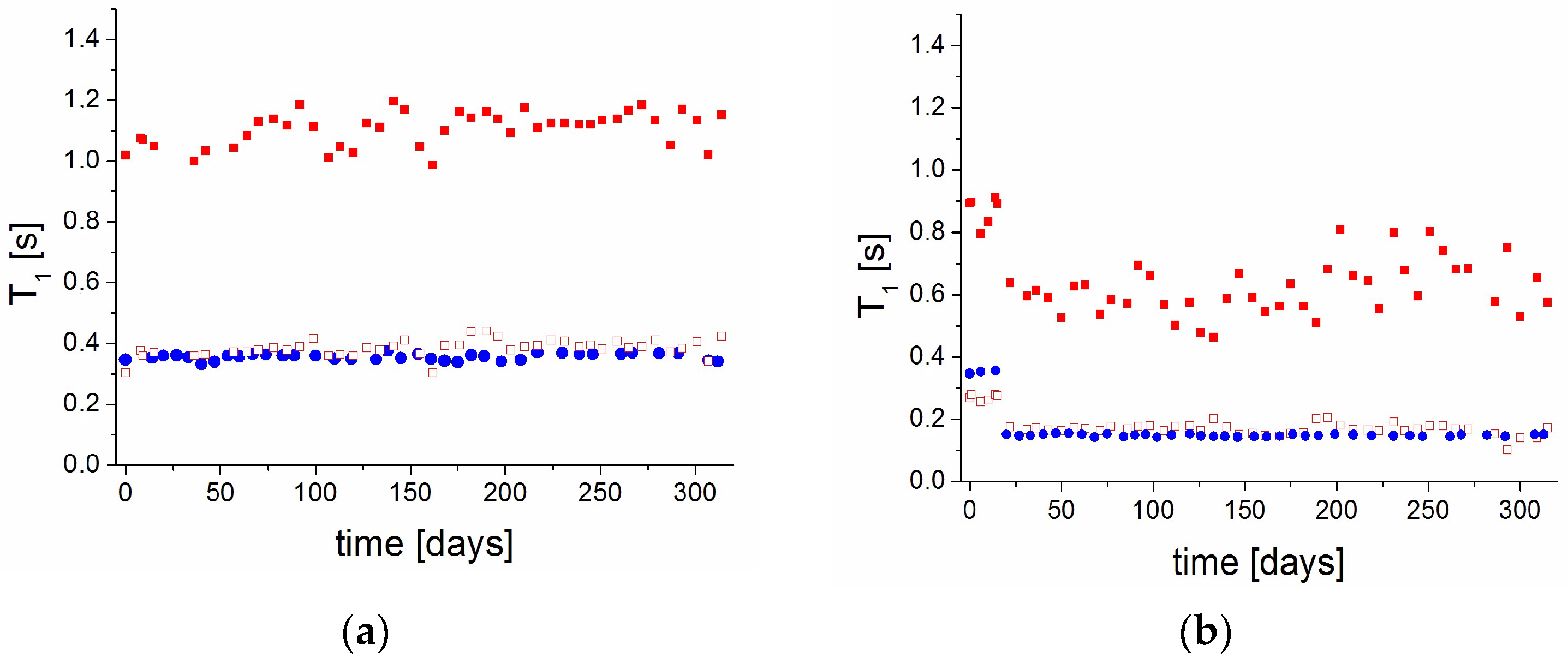
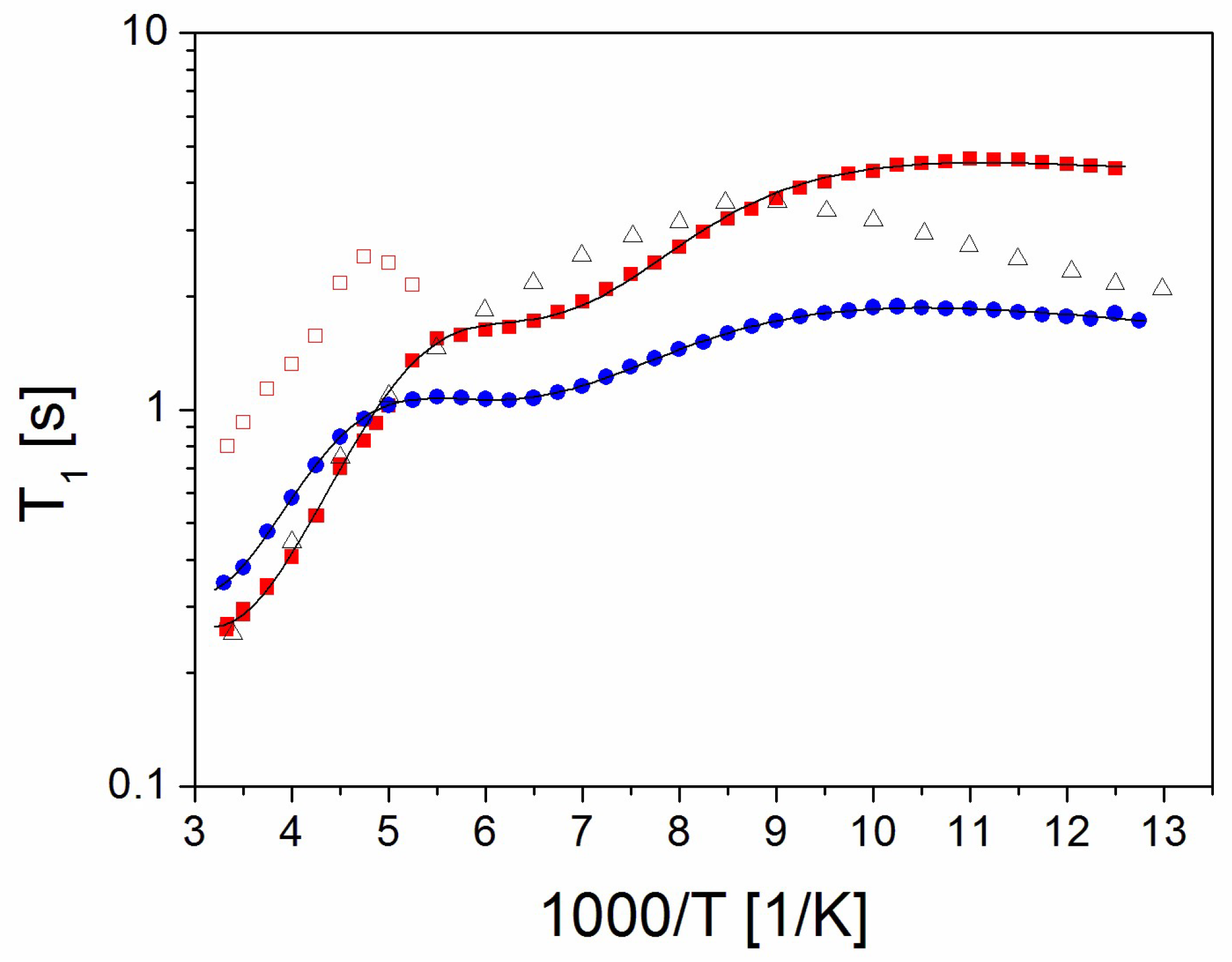
2.3. The Relaxation Processes: The Spin–Lattice Relaxation Time as a Function of Temperature
- (a)
- PVP
- (b)
- PVP70/RSV30 and PVP50/RSV50 Samples
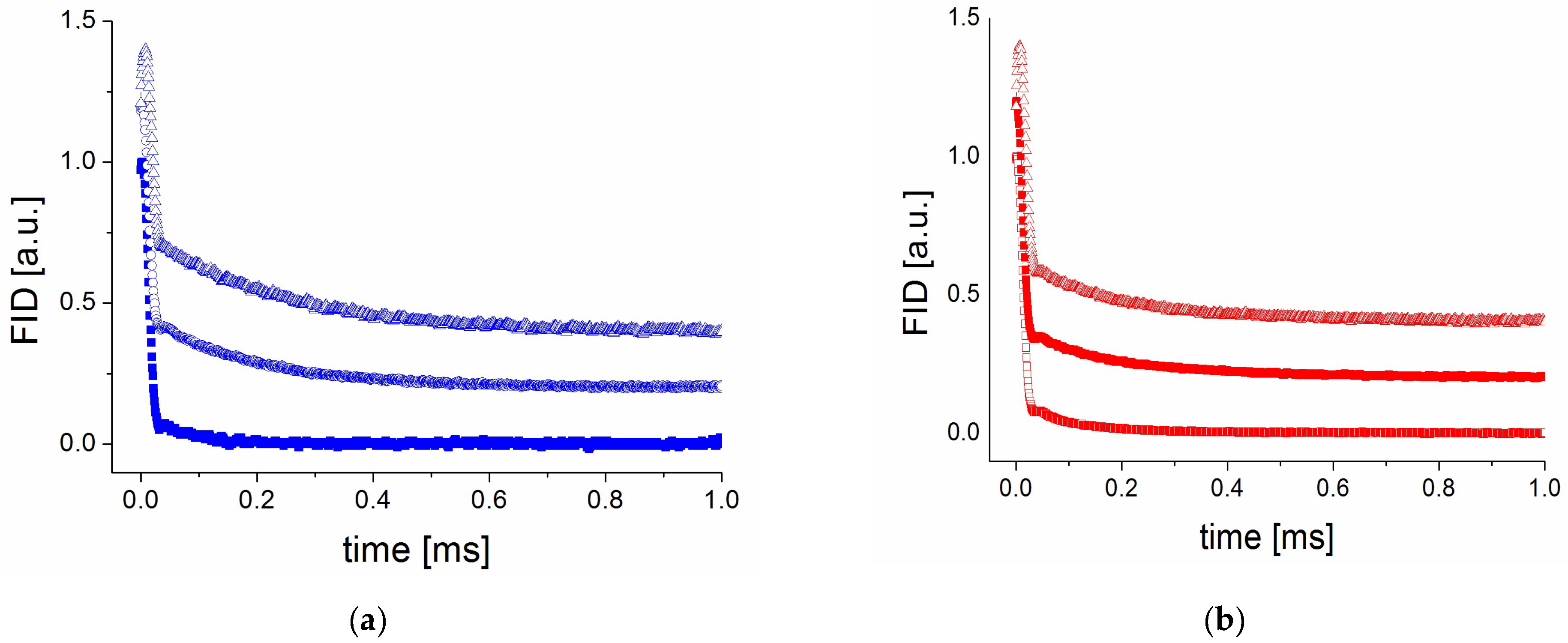
2.4. Characterization of Resveratrol Nanocrystals in PVP50/RSV50 (RH 0%)
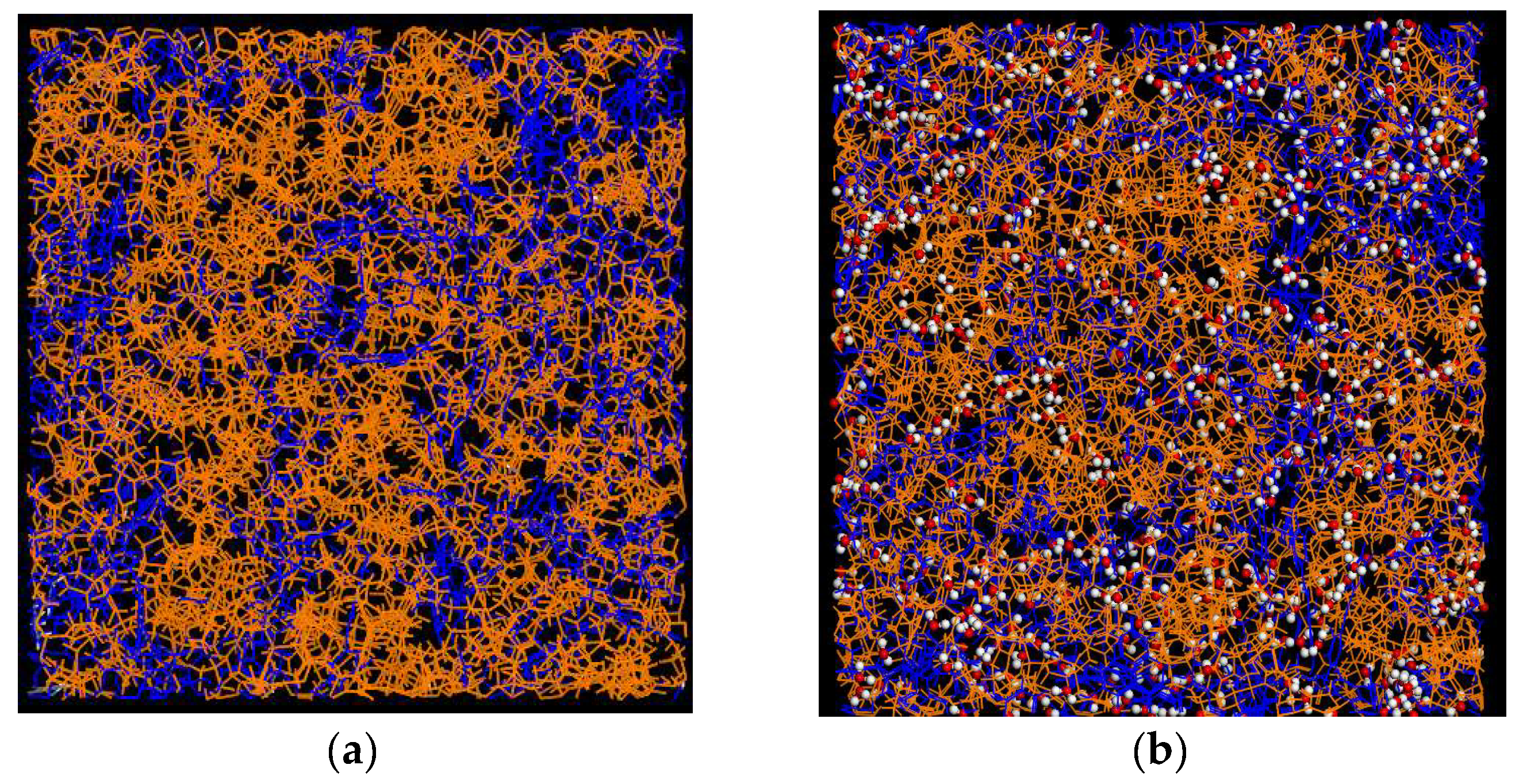
2.5. Intermolecular Interactions and Molecular Mobility–Molecular Dynamics Simulations Study
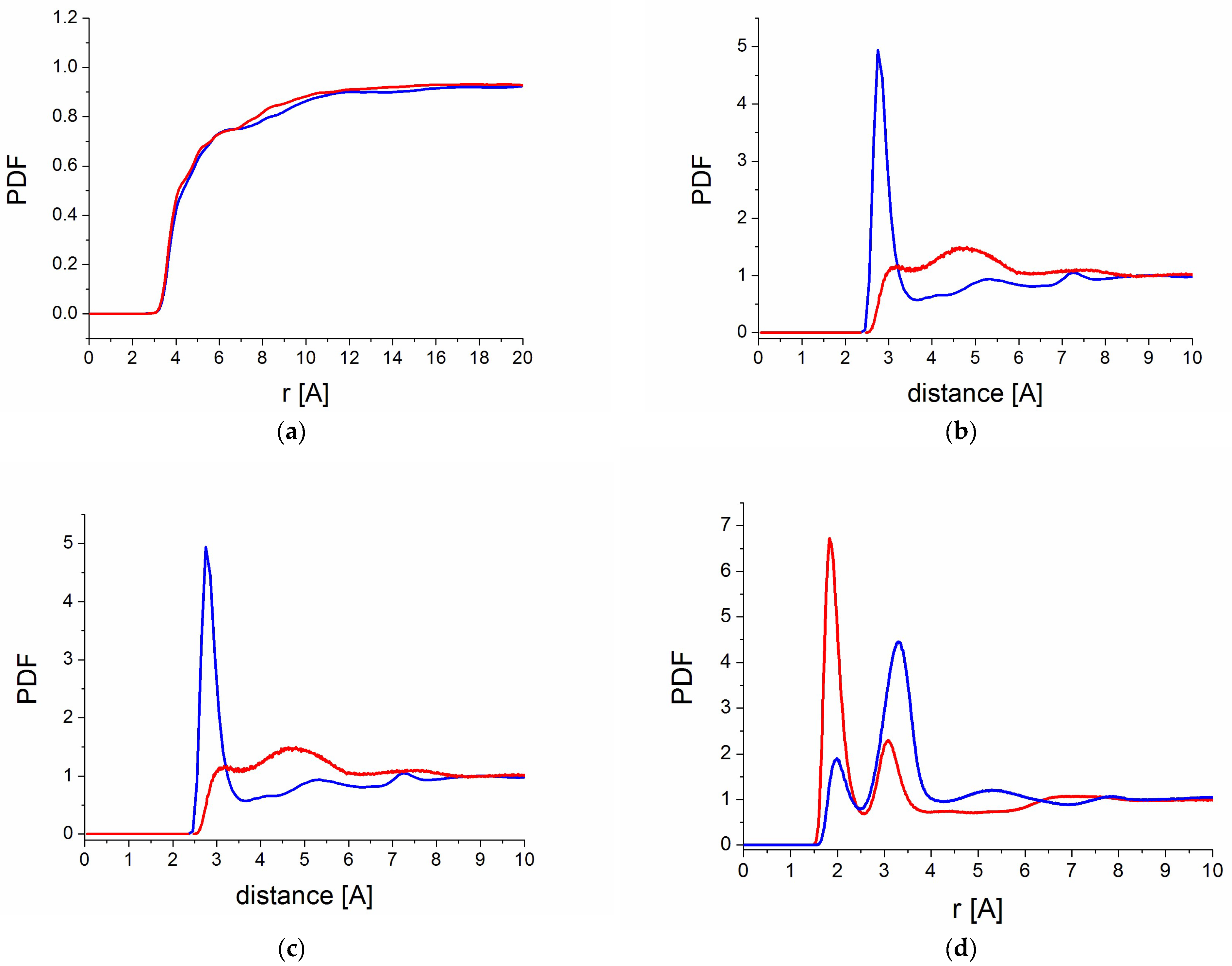
2.5.1. Intermolecular Interactions
2.5.2. Hydrogen Bond Analysis
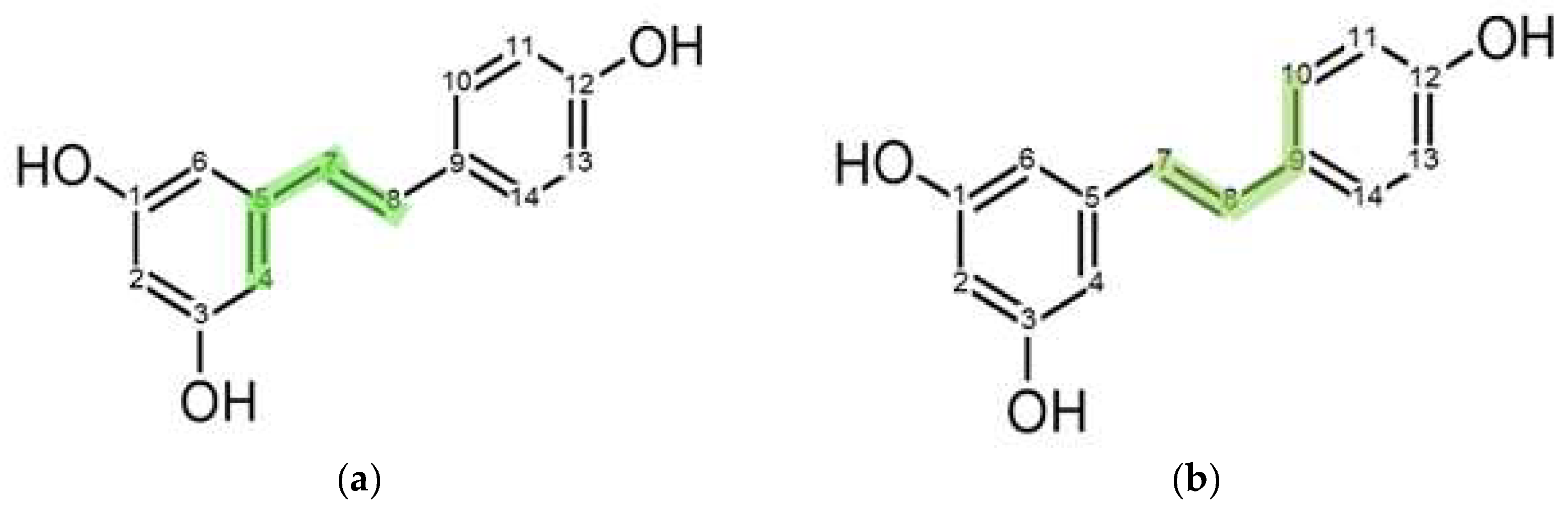
2.5.3. Reorientation Dynamics of RSV in PVP
3. Materials and Methods
4. Conclusions
- The sample PVP70/RSV30 is a homogenous system, meaning it is an amorphous solid dispersion with resveratrol dispersed in the polymer matrix at the molecular level.
- The sample PVP50/RSV50 is a system composed of two subsystems, the first of which is crystalline resveratrol and the second of which is a PVP polymer with a small amount of resveratrol molecules connected to it by hydrogen bonds. Crystalline resveratrol was formed already at the stage of its preparation. Resveratrol crystals occur as nanocrystals with sizes about 40 nm, and their sizes were not affected by humidity (75% RH). To our knowledge, this is the first determination of the size of nanocrystals in a PVP matrix, which was performed via two independent methods: Scherrer analysis and NMR spin diffusion constraints.
- Both samples are stable in 0% RH and 75% RH for at least 300 days from preparation. Humidity influenced mobility and caused plasticization of the studied samples.
- The incorporation of resveratrol into the polymer matrix makes the molecular dynamics reorientations of these systems more complex compared to pure PVP. In addition to the mobility of the main polymer chains (a process effective at high temperatures with high activation energy) and side chains (a process visible at low temperatures with low activation energy) observed in pure PVP, the reorientations of the resveratrol molecules were also noted, and their activation parameters were determined. Additionally, the mobility of resveratrol molecules was confirmed by molecular dynamics simulations.
- In the PVP70/RSV30 and PVP70/RSV30/H2O samples, the hydrogen bond network is highly developed, which ensures that each resveratrol molecule is linked via at least one hydrogen bond. In the PVP70/RSV30 sample, resveratrol molecules are mainly bonded to other resveratrol molecules, while in PVP70/RSV30/H2O they form hydrogen bonds with water molecules. This extensive network of HBs likely contributes to the suppression of recrystallization, even for samples with water.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- López, M.; Martínez, F.; Del Valle, C.; Orte, C.; Miró, M. Analysis of phenolic constituents of biological interest in red wines by high-performance liquid chromatography. J. Chromatogr. A 2001, 922, 359–363. [Google Scholar] [CrossRef] [PubMed]
- Tian, B.; Liu, J. Resveratrol: A review of plant sources, synthesis, stability, modification and food application. J. Sci. Food. Agric. 2020, 100, 1392–1404. [Google Scholar] [CrossRef] [PubMed]
- Frémont, L. Biological effects of resveratrol. Life Sci. 2000, 66, 663–673. [Google Scholar] [CrossRef]
- Akinwumi, B.C.; Bordun, K.A.M.; Anderson, H.D. Biological activities of stilbenoids. Int. J. Mol. Sci. 2018, 19, 792. [Google Scholar] [CrossRef] [PubMed]
- Herrera, D.P.; Chánique, A.M.; Martínez-Márquez, A.; Bru-Martínez, R.; Kourist, R.; Parra, L.P.; Schuller, A. Rational design of resveratrol o-methyltransferase for the production of pinostilbene. Int. J. Mol. Sci. 2021, 22, 4345. [Google Scholar] [CrossRef]
- Varoni, E.M.; Lo Faro, A.F.; Sharifi-Rad, J.; Iriti, M. Anticancer molecular mechanisms of resveratrol. Front. Nutr. 2016, 3, 8. [Google Scholar] [CrossRef]
- Stojanović, S.; Sprinz, H.; Brede, O. Efficiency and mechanism of the antioxidant action of trans-resveratrol and its ana-logues in the radical liposome oxidation. Arch. Biochem. Biophys. 2021, 391, 79–89. [Google Scholar] [CrossRef]
- Bradamante, S.; Barenghi, L.; Piccinini, F.; Bertelli, A.A.; de Jonge, R.; Beemster, P.; de Jong, J.W. Resveratrol provides late-phase cardioprotection by means of a nitric oxide- and adenosine-mediated mechanism. Eur. J. Pharmacol. 2003, 465, 115–123. [Google Scholar] [CrossRef]
- Aguirre, L.; Fernández-Quintela, A.; Arias, N.; Portillo, M.P. Resveratrol: Anti-obesity mechanisms of action. Molecules 2014, 19, 18632–18655. [Google Scholar] [CrossRef]
- Kim, S.; Jin, Y.; Choi, Y.; Park, T. Resveratrol exerts anti-obesity effects via mechanisms involving down-regulation of adipogenic and inflammatory processes in mice. Biochem. Pharmacol. 2011, 81, 1343–1351. [Google Scholar] [CrossRef]
- Wang, K.T.; Chen, L.G.; Tseng, S.H.; Huang, J.S.; Hsieh, M.S.; Wang, C.C. Anti-inflammatory effects of resveratrol and oligostilbenes from Vitis Thunbergii Var. Taiwaniana against lipopolysaccharide-induced arthritis. J. Agric. Food Chem. 2011, 59, 3649–3656. [Google Scholar] [CrossRef] [PubMed]
- Dvorakova, M.; Landa, P. Anti-inflammatory activity of natural stilbenoids: A review. Pharmacol. Res. 2017, 124, 126–145. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Lin, H.S.; Ho, P.C.; Ng, K.Y. The impact of aqueous solubility and dose on the pharmacokinetic profiles of resveratrol. Pharm. Res. 2008, 25, 2593–2600. [Google Scholar] [CrossRef] [PubMed]
- Nyamba, I.; Sombié, C.B.; Yabré, M.; Zimé-Diawara, H.; Yaméogo, J.; Ouédraogo, S.; Lechanteur, A.; Semdé, R.; Evrard, B. Pharmaceutical approaches for enhancing solubility and oral bioavailability of poorly soluble drugs. Eur. J. Pharm. Biopharm. 2024, 204, 114513. [Google Scholar] [CrossRef]
- Pandi, P.; Bulusu, R.; Kommineni, N.; Khan, W.; Singh, M. Amorphous solid dispersions: An update for preparation, characterization, mechanism on bioavailability, stability, regulatory considerations and marketed products. Int. J. Pharm. 2020, 30, 586. [Google Scholar] [CrossRef]
- Janssens, S.; Van den Mooter, G. Review: Physical chemistry of solid dispersions. J. Pharm. Pharmacol. 2009, 61, 1571–1586. [Google Scholar] [CrossRef]
- Kawabata, Y.; Wada, K.; Nakatani, M.; Yamada, S.; Onoue, S. Formulation design for poorly water-soluble drugs based on biopharmaceutics classification system: Basic approaches and practical applications. Int. J. Pharm. 2011, 420, 1–10. [Google Scholar] [CrossRef]
- Litvinov, V.M.; Penning, J.P. Phase composition and molecular mobility in nylon 6 fibers as studied by proton NMR transverse magnetization relaxation. Macromol. Chem. Phys. 2004, 205, 1721–1734. [Google Scholar]
- Vollath, D.; Fischer, F.D.; Holec, D. Surface energy of nanoparticles—Influence of particle size and structure. Beilstein J. Nanotechnol. 2018, 9, 2265–2276. [Google Scholar] [CrossRef]
- Paredes, A.J.; McKenna, P.E.; Ramöller, I.K.; Naser, Y.A.; Volpe-Zanutto, F.; Li, M.; Abbate, M.T.A.; Zhao, L.; Zhang, C.; Abu-Ershaid, J.M.; et al. Microarray patches: Poking a hole in the challenges faced when delivering poorly soluble drugs. Adv. Funct. Mater. 2021, 31, 2005792. [Google Scholar] [CrossRef]
- Yang, X.; Ong, T.C.; Michaelis, V.K.; Heng, S.; Huang, J.; Griffin, R.G.; Myerson, A.S. Formation of organic molecular nanocrystals under rigid confinement with analysis by solid state NMR. CrystEngComm 2014, 16, 9345–9352. [Google Scholar] [CrossRef] [PubMed]
- Dwyer, L.M.; Michaelis, V.K.; O’Mahonya, M.; Griffin, R.G.; Myerson, A.S. Confined crystallization of fenofibrate in nanoporous silica. CrystEngComm 2015, 17, 7922–7929. [Google Scholar] [CrossRef] [PubMed]
- Ju, S.P.; Chang, J.G. A molecular dynamics simulation investigation into the behavior of water molecules inside Au nanotubes of various sizes. Microp. Mesop. Mater. 2004, 75, 81–87. [Google Scholar] [CrossRef]
- Lemanowicz, M.; Mielańczyk, A.; Walica, T.; Kotek, M.; Gierczycki, A. Application of polymers as a tool in crystallization—A review. Polymers 2021, 13, 2695. [Google Scholar] [CrossRef]
- Rengarajan, G.T.; Enke, D.; Steinhart, M.; Beiner, M. Size-dependent growth of polymorphs in nanopores and Ostwald’s step rule of stages. Phys. Chem. Chem. Phys. 2011, 13, 21367–21374. [Google Scholar] [CrossRef]
- Navnit, S.; Harpreet, S.; Choi, D.; Chokshi, H. Amorphous Solid Dispersions Theory and Practice (Advances in Delivery Science and Technology); Springer: Berlin/Heidelberg, Germany, 2014. [Google Scholar]
- Caruso, F.; Tanski, J.; Villegas-Estrada, A.; Rossi, M. Structural basis for antioxidant activity of trans-resveratrol: Ab initio calculations and crystal and molecular structure. J. Agric. Food Chem. 2004, 52, 7279–7285. [Google Scholar] [CrossRef]
- Zarychta, B.; Gianopoulos, C.G.; Pinkerton, A.A. Revised structure of trans-resveratrol: Implications for its proposed antioxidant mechanism. Bioorg. Med. Chem. Lett. 2016, 26, 1416–1418. [Google Scholar] [CrossRef]
- Wegiel, L.A.; Mauer, L.J.; Edgar, K.J.; Taylor, L.S. Crystallization of amorphous solid dispersions of resveratrol during preparation and storage—Impact of different polymers. J. Pharm. Sci. 2013, 102, 171–184. [Google Scholar] [CrossRef]
- Pajzderska, A.; Mielcarek, J.; Wąsicki, J. The physical stability of felodipine and its recrystallization from an amorphous solid dispersion studied by NMR relaxometry. AAPS PharmSciTech 2022, 23, 93. [Google Scholar] [CrossRef]
- Xiang, T.; Anderson, B.D. Molecular dynamics simulation of amorphous hydroxypropylmethylcellulose and its mixtures with felodipine and water. J Pharm. Sci. 2017, 106, 803–816. [Google Scholar] [CrossRef]
- Kothari, K.; Ragoonanan, V.; Suryanarayanan, R. The role of drug—polymer hydrogen bonding interactions on the molecular mobility and physical stability of nifedipine solid dispersions. Mol. Pharm. 2015, 12, 162–170. [Google Scholar] [CrossRef] [PubMed]
- Pajzderska, A.; Fojud, Z.; Jarek, M.; Wąsicki, J. NMR relaxometry in the investigation of the kinetics of the recrystallization of felodipine. Powder Technol. 2019, 347, 35–41. [Google Scholar] [CrossRef]
- Abragam, A. The Principles of Nuclear Magnetism; Clarendon Press: Oxford, UK, 1983. [Google Scholar]
- Rumondor, A.C.F.; Stanford, L.A.; Taylor, L.S. Effects of polymer type and storage relative humidity on the kinetics of felodipine crystallization from amorphous solid dispersions. Pharm. Res. 2009, 26, 2599–2606. [Google Scholar] [CrossRef]
- Wegiel, L.A.; Mosquera-Giraldo, L.I.; Mauer, L.J.; Edgar, K.J.; Taylor, L.S. Phase behavior of resveratrol solid dispersions upon addition to aqueous media. Pharm. Res. 2015, 32, 3324–3337. [Google Scholar]
- Litvinov, V.M. Molecular mobility and phase composition in polyolefins: From fundamental to applied research. In NMR Spectroscopy of Polymers: Innovative Strategies for Complex Macromolecules; Cheng, H., Asakura, T., English, A.D., Eds.; ACS Symposium Series; American Chemical Society: Washington, DC, USA, 2011. [Google Scholar]
- Yoshioka, S.; Aso, Y.; Kojima, S. Molecular mobility of lyophilized poly(vinylpyrrolidone) and methylcellulose as determined by the laboratory and rotating frame spin-lattice relaxation times of 1H and 13C. Chem. Pharm. Bull. 2003, 51, 1289–1292. [Google Scholar] [CrossRef]
- Bloembergen, N.; Purcell, E.M.; Pound, R.V. Relaxation effects in nuclear magnetic resonance absorption. Phys. Rev. 1948, 73, 679–712. [Google Scholar] [CrossRef]
- Weibel, A.; Bouchet, R.; Boulc’H, F.; Knauth, P. The big problem of small particles: A comparison of methods for determination of particle size in nanocrystalline anatase powders. Chem. Mater. 2005, 17, 2378–2385. [Google Scholar] [CrossRef]
- Muniz, F.T.; Miranda, M.A.; Dos Santos, C.S.; Sasaki, J.M. The Scherrer equation and the dynamical theory of X-ray diffraction. Acta Crystallogr. A 2016, 1, 385–390. [Google Scholar] [CrossRef]
- Meyer, H.W.; Schneider, H.; Saalwächter, K. Proton NMR spin-diffusion studies of PS-PB block copolymers at low field: Two- vs. three-phase model and recalibration of spin-diffusion coefficients. Polym. J. 2012, 44, 748–756. [Google Scholar] [CrossRef]
- Ernst, M.; Meier, B.H. Studies in physical and theoretical chemistry. In Solid State NMR of Polymers; Ando, I., Asakura, T., Eds.; Springer: Berlin/Heidelberg, Germany, 1998; Volume 84, pp. 83–121. [Google Scholar]
- Qi, S.; Belton, P.; Nollenberger, K.; Clayden, N.; Reading, M.; Craig, D.Q.M. Characterisation and prediction of phase separation in hot-melt extruded solid dispersions: A thermal, microscopic and NMR relaxometry study. Pharm. Res. 2010, 27, 1869–1883. [Google Scholar] [CrossRef]
- Belton, P.S.; Hills, B.P. The effects of diffusive exchange in heterogeneous systems on N.M.R. line shapes and relaxation processes. Mol. Phys. 1987, 61, 999–1018. [Google Scholar] [CrossRef]
- Clauss, J.; Schmidt-Rohr, K.; Spiess, H.W. Determination of domain sizes in heterogeneous polymers by solid-state NMR. Acta Polym. 1993, 44, 1–17. [Google Scholar] [CrossRef]
- Luzar, A. Resolving the hydrogen bond dynamics conundrum. J. Chem. Phys. 2000, 113, 10663–10675. [Google Scholar] [CrossRef]
- Prada-Gracia, D.; Shevchuk, R.; Rao, F. The quest for self-consistency in hydrogen bond definitions. J. Chem. Phys. 2013, 139, 084501. [Google Scholar] [CrossRef]
- Xiang, T.; Anderson, B.D. Effects of molecular interactions on miscibility and mobility of ibuprofen in amorphous solid dispersions with various polymers. J. Pharm. Sci. 2019, 108, 178–186. [Google Scholar] [CrossRef]
- Zhang, N.; Chen, S.; Yang, B.; Huo, J.; Zhang, X.; Bao, J.; Ruan, Z.; He, G. Effect of hydrogen-bonding interaction on the arrangement and dynamics of water confined in a polyamide membrane: A molecular dynamics simulation. J. Phys. Chem. B 2018, 122, 4719–4728. [Google Scholar] [CrossRef]
- Mohapatra, S.; Samanta, S.; Kothari, K.; Mistry, P.; Suryanarayanan, R. Effect of polymer molecular weight on the crystallization behavior of indomethacin amorphous solid dispersions. Cryst. Growth Des. 2017, 17, 3142–3150. [Google Scholar] [CrossRef]
- Chao, J.; Li, H.; Cheng, K.W.; Yu, M.S.; Chang, R.C.C.; Wang, M. Protective effects of pinostilbene, a resveratrol methylated derivative, against 6-hydroxydopamine-induced neurotoxicity in SH-SY5Y cells. J. Nutr. Biochem. 2010, 21, 482–489. [Google Scholar] [CrossRef]
- Wang, Y.; Ma, R.; Wang, B.; Liu, X.; Zhao, X.; Liu, L.; Zhang, L.; Gao, Y. Influence of number and strength of hydrogen bonds on fracture property and microscopic mechanisms of associative hydrogen-bonded polymers via molecular dynamics simulation. Langmuir 2024, 40, 27501–27510. [Google Scholar] [CrossRef]
- Shi, Q.; Chen, H.; Wang, Y.; Wang, R.; Xu, J.; Zhang, C. Amorphous solid dispersions: Role of the polymer and its importance in physical stability and in vitro performance. Pharmaceutics 2022, 14, 1747. [Google Scholar] [CrossRef]
- Trasi, N.S.; Taylor, L.S. Effect of polymers on nucleation and crystal growth of amorphous acetaminophen. CrystEngComm 2012, 14, 5188–5197. [Google Scholar] [CrossRef]
- Kestur, U.S.; Taylor, L.S. Role of polymer chemistry in influencing crystal growth rates from amorphous felodipine. CrystEngComm 2010, 12, 2390–2397. [Google Scholar] [CrossRef]
- Taylor, L.S.; Zografi, G. Spectroscopic characterization of interactions between pvp and indomethacin in amorphous molecular dispersions. Pharm. Res. 1997, 14, 1691–1698. [Google Scholar] [CrossRef]
- Newman, A.; Zografi, G. What are the important factors that influence api crystallization in miscible amorphous api-excipient mixtures during long-term storage in the glassy state? Mol. Pharm. 2022, 19, 378–391. [Google Scholar] [CrossRef]
- Zhong, H.; Lu, T.; Wang, R.; Ouyang, D. Quantitative analysis of physical stability mechanisms of amorphous solid dispersions by molecular dynamic simulation. AAPS J. 2025, 27, 9. [Google Scholar] [CrossRef]
- Wilke, S.K.; Al-Rubkhi, A.; Benmore, C.J.; Byrn, S.R.; Weber, R. Modeling the structure of ketoprofen-poly(vinylpyrrolidone) amorphous solid dispersions with empirical potential structure refinements of x-ray scattering data. Mol. Pharm. 2024, 21, 3967–3978. [Google Scholar] [CrossRef]
- Aulich, V.; Ludík, J.; Fulem, M.; Červinka, C. Molecular insights into kinetic stabilization of amorphous solid dispersion of pharmaceuticals. Phys. Chem. Chem. Phys. 2024, 1567–1584. [Google Scholar] [CrossRef]
- Luebbert, C.; Wessner, M.; Sadowski, G. Mutual impact of phase separation/crystallization and water sorption in amorphous solid dispersions. Mol. Pharm. 2018, 15, 669–678. [Google Scholar]
- Hancock, B.C.; Zografi, G. The relationship between the glass transition temperature and the water content of amorphous pharmaceutical solids. Pharm. Res. 1994, 11, 471–477. [Google Scholar] [CrossRef]
- Pajzderska, A.; Gonzalez, M.A. Molecular dynamics simulations of selected amorphous stilbenoids and their amorphous solid dispersions with poly(vinylpyrrolidone). J. Pharm. Sci. 2023, 112, 2444–2452. [Google Scholar] [CrossRef]
- Powles, J.G.; Mansfield, P. Double-pulse nuclear-resonance transients in solids. Phys. Lett. 1962, 2, 58–59. [Google Scholar] [CrossRef]
- Martínez, L.; Andrade, R.; Birgin, E.G.; Martinez, J.M. Packmol: A package for building initial configurations for molecular dynamics simulations. J. Comput. Chem. 2009, 30, 2157–2164. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.P.; Aktulga, H.M.; Berger, R.; Bolintineanu, D.S.; Brown, W.M.; Crozier, P.S.; In’t Veld, P.J.; Kohlmeyer, A.; Moore, S.; Nguyen, T.D.; et al. LAMMPS—A flexible simulation tool for particle-based materials modeling at the atomic, meso, and continuum scales. Comp. Phys. Comm. 2022, 271, 10817. [Google Scholar] [CrossRef]
- Banks, J.L.; Beard, H.S.; Cao, Y.; Cho, A.E.; Damm, W.; Farid, R.; Felts, A.K.; Halgren, T.A.; Mainz, D.T.; Maple, J.R.; et al. Integrated modeling program, applied chemical theory (IMPACT). J. Comput. Chem. 2005, 26, 1752–1780. [Google Scholar] [CrossRef]
- Abascal, J.L.F.; Vega, C. A general purpose model for the condensed phases of water: TIP4P/2005. J. Chem. Phys. 2005, 123, 234505. [Google Scholar] [CrossRef]
- Allen, M.P.; Tildesley, D. Computer Simulation of Liquids; Oxford University Press: Oxford, UK, 2002. [Google Scholar]
- Hinsen, K.; Pellegrini, E.; Stachura, S.; Kneller, G.R. NMoldyn 3: Using task farming for a parallel spectroscopy-oriented analysis of molecular dynamics simulations. J. Comput. Chem. 2012, 33, 2043–2048. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Intermediate | High-Temperature | |||
|---|---|---|---|---|
| EA [kJ/mol] | τ0 [s] | C [1/s2] | EA [kJ/mol] | |
| PVP | - | - | - | 8.6 |
| PVP70/RSV30 | 6.8 | 2.4 × 10−11 | 5.7 × 107 | 12.9 |
| PVP50/RSV50 | 8.7 | 0.4 × 10−11 | 3.8 × 107 | 11.9 |
| % Molecules with | HBs RSV-RSV | HBs RSV-PVP | ||
|---|---|---|---|---|
| PVP70/RSV30 | PVP70/RSV30/H2O | PVP70/RSV30 | PVP70/RSV30/H2O | |
| 0 HBs | 18.0 | 77.7 | 18.4 | 79.1 |
| 1 HB | 40.1 | 20.7 | 38.6 | 18.3 |
| 2 HBs | 28.1 | 1.6 | 33.3 | 2.6 |
| 3 HBs | 11.4 | 0.1 | 9.8 | 0.0 |
| % Molecules with | HBs RSV–Water | HBs RSV–Water | HBs Water–PVP |
|---|---|---|---|
| RSV (Acceptor)–Water (Donor) | RSV (Donor)–Water (Acceptor) | Water (Donor)–PVP (Acceptor) | |
| 0 HBs | 51.9 | 3.5 | 40.3 |
| 1 HB | 36.4 | 23.4 | 42.7 |
| 2 HBs | 10.5 | 42.2 | 17.0 |
| 3 HBs | 1.2 | 30.9 | 0.0 |
| PVP70/RSV30 | PVP70/RSV30/H2O | |
|---|---|---|
| Number of PVP chains | 6 | 6 |
| Number of resveratrol molecules | 120 | 120 |
| Number of water molecules | - | 420 |
| Total number of atoms | 13,728 | 15,078 |
| Size of simulation box after equilibration [Å3] | 52.04 × 52.04 × 52.04 | 53.5 × 53.5 × 53.5 |
| Density [g/cm3] | 1.11 | 1.11 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pajzderska, A.; González, M.A.; Jarek, M.; Mielcarek, J.; Wąsicki, J. Physical Stability and Molecular Mobility of Resveratrol in a Polyvinylpyrrolidone Matrix. Molecules 2025, 30, 1909. https://doi.org/10.3390/molecules30091909
Pajzderska A, González MA, Jarek M, Mielcarek J, Wąsicki J. Physical Stability and Molecular Mobility of Resveratrol in a Polyvinylpyrrolidone Matrix. Molecules. 2025; 30(9):1909. https://doi.org/10.3390/molecules30091909
Chicago/Turabian StylePajzderska, Aleksandra, Miguel Angel González, Marcin Jarek, Jadwiga Mielcarek, and Jan Wąsicki. 2025. "Physical Stability and Molecular Mobility of Resveratrol in a Polyvinylpyrrolidone Matrix" Molecules 30, no. 9: 1909. https://doi.org/10.3390/molecules30091909
APA StylePajzderska, A., González, M. A., Jarek, M., Mielcarek, J., & Wąsicki, J. (2025). Physical Stability and Molecular Mobility of Resveratrol in a Polyvinylpyrrolidone Matrix. Molecules, 30(9), 1909. https://doi.org/10.3390/molecules30091909