
Research Progress on the Structure–activity Relationship and Mechanism of Flavonoid Derivatives in the Treatment of Lung Cancer

 and
and
Abstract

1. Introduction
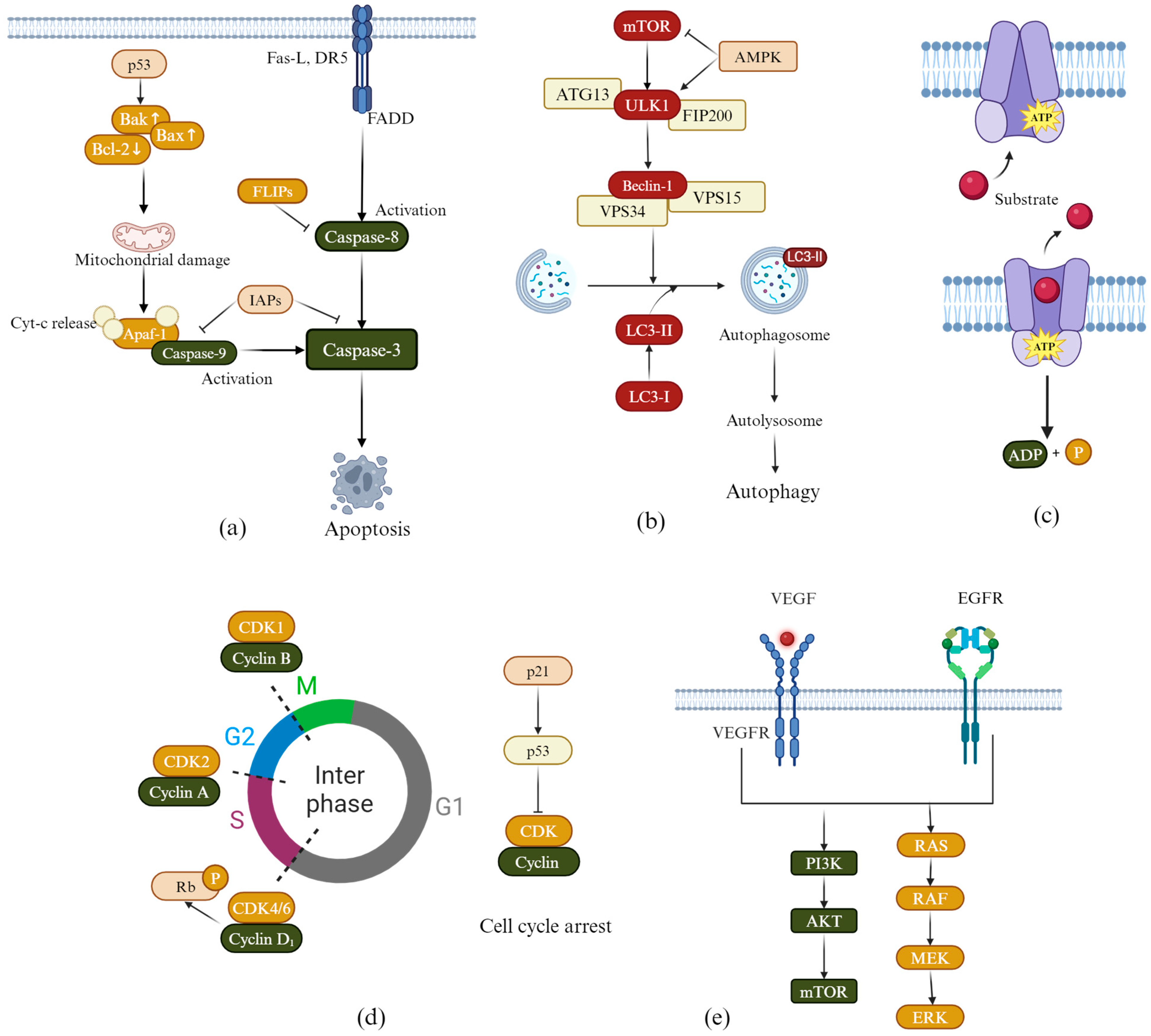
2. Anti-Lung Cancer Mechanism of Flavonoid Derivatives
2.1. Induction of Apoptosis
2.2. Induction of Autophagy
2.3. Overcoming Multidrug Resistance
2.4. Induction of Cell Cycle Arrest
2.5. Inhibiting Tumor Cell Angiogenesis
2.6. Inhibiting EGFR Kinase Activity
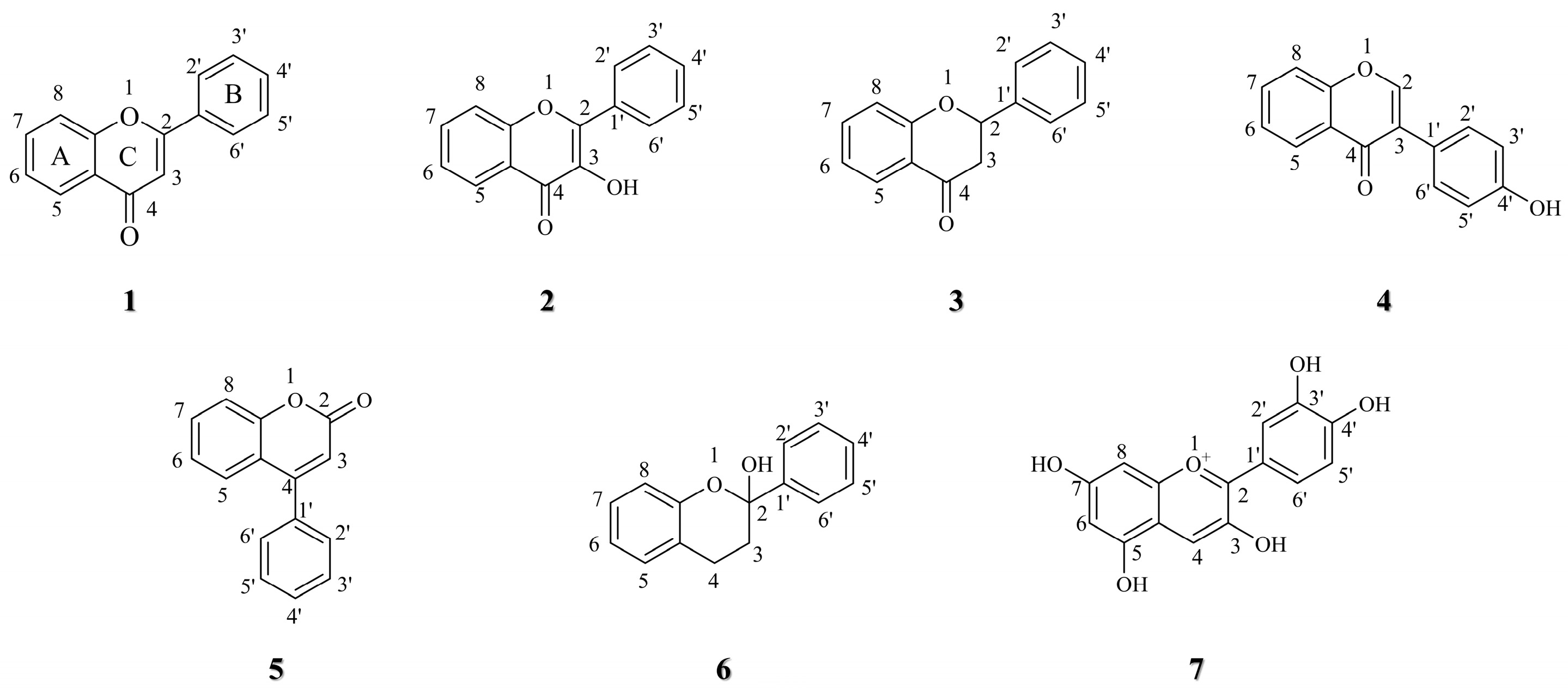
3. Structure–activity Relationship of Flavonoid Derivatives
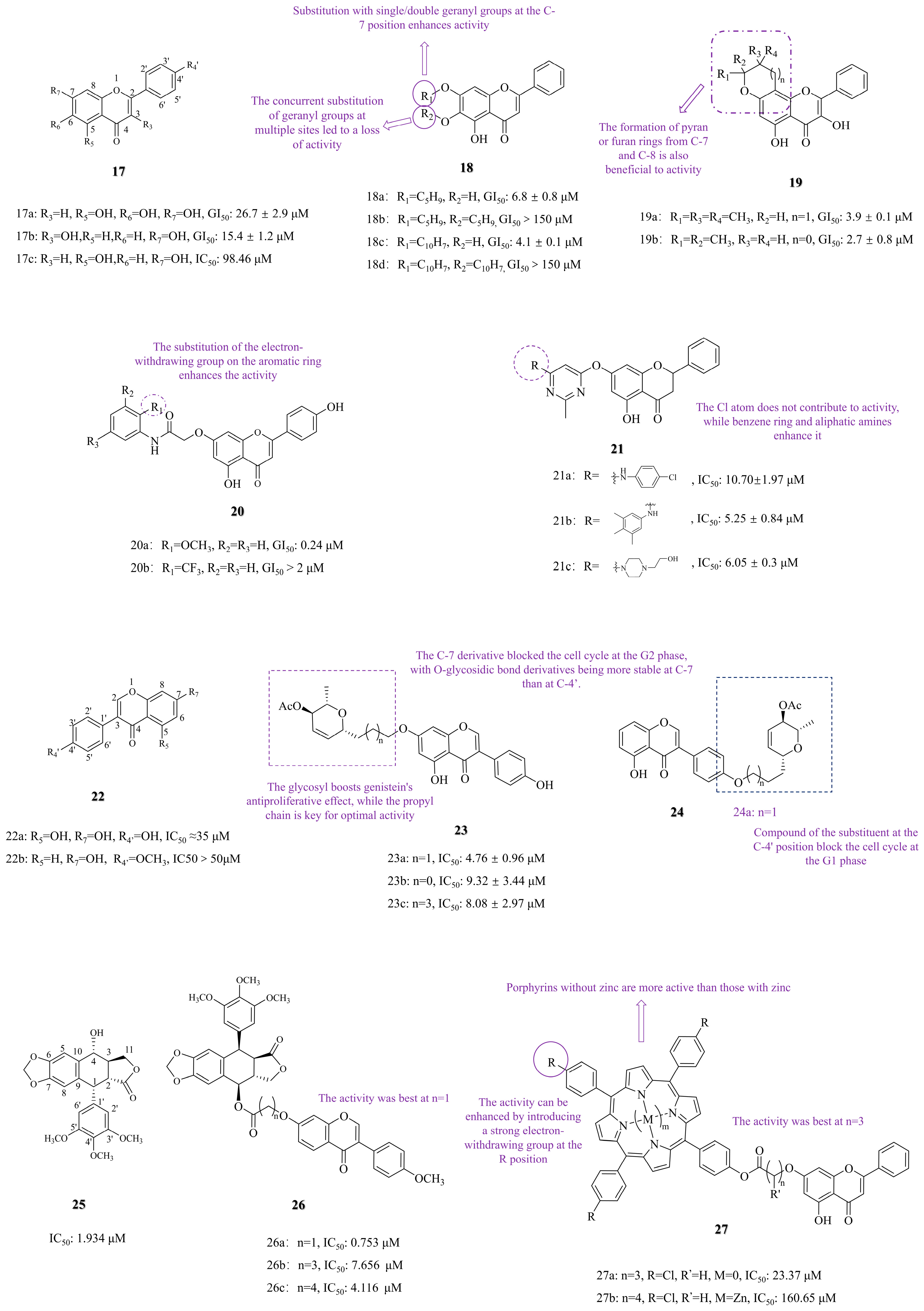
3.1. C-7 Substituted
3.1.1. Functional Group Substitution
3.1.2. Photosensitizer Conjugation and Photodynamic Therapy
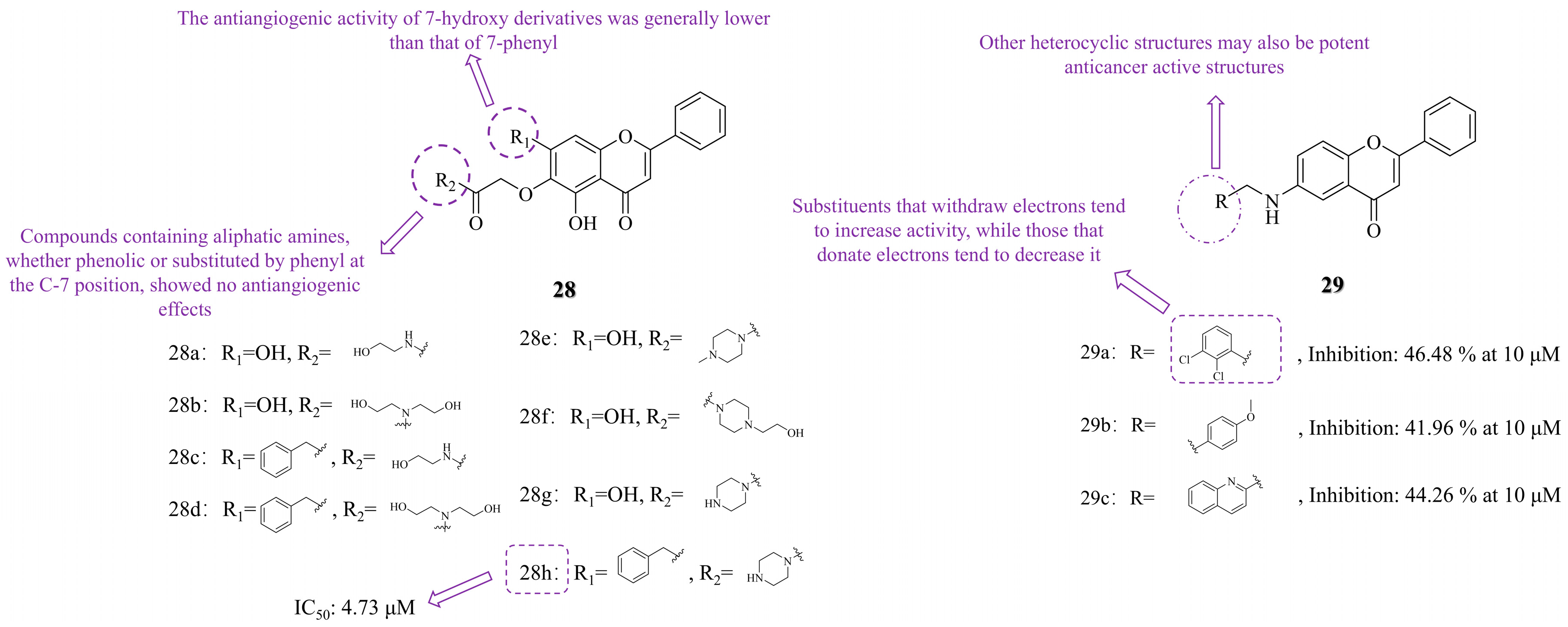
3.2. C-6 Substituted
3.3. Other Modifications
3.3.1. Functional Group Substitution
3.3.2. Heterocyclization
3.4. The SARs Summary of Flavonoid Derivatives in Anti-Lung Cancer Activity
4. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Thai, A.A.; Solomon, B.J.; Sequist, L.V.; Gainor, J.F.; Heist, R.S. Lung cancer. Lancet 2021, 398, 535–554. [Google Scholar] [CrossRef]
- Su, C. Emerging insights to lung cancer drug resistance. Cancer Drug Resist. 2022, 5, 534–540. [Google Scholar] [CrossRef]
- Rabbani, M.; Kanevsky, J.; Kafi, K.; Chandelier, F.; Giles, F.J. Role of artificial intelligence in the care of patients with nonsmall cell lung cancer. Eur. J. Clin. Investig. 2018, 48, e12901. [Google Scholar] [CrossRef]
- Li, M.; Hu, M.; Jiang, L.; Pei, J.; Zhu, C. Trends in cancer incidence and potential associated factors in China. JAMA Netw. Open 2024, 7, e2440381. [Google Scholar] [CrossRef]
- Romani, A.M.P. Cisplatin in cancer treatment. Biochem. Pharmacol. 2022, 206, 115323. [Google Scholar] [CrossRef]
- Sunder, S.S.; Sharma, U.C.; Pokharel, S. Adverse effects of tyrosine kinase inhibitors in cancer therapy: Pathophysiology, mechanisms and clinical management. Signal Transduct. Target. Ther. 2023, 8, 262. [Google Scholar] [CrossRef]
- Huang, M.; Lu, J.J.; Ding, J. Natural Products in Cancer Therapy: Past, Present and Future. Nat. Prod. Bioprospect. 2021, 11, 5–13. [Google Scholar] [CrossRef]
- Jeon, Y.D.; Lee, J.H.; Lee, Y.M.; Kim, D.K. Puerarin inhibits inflammation and oxidative stress in dextran sulfate sodium-induced colitis mice model. Biomed. Pharmacother. 2020, 124, 109847. [Google Scholar] [CrossRef]
- Hu, Q.; Liu, Z.; Guo, Y.; Lu, S.; Du, H.; Cao, Y. Antioxidant capacity of flavonoids from Folium Artemisiae Argyi and the molecular mechanism in Caenorhabditis elegans. J. Ethnopharmacol. 2021, 279, 114398. [Google Scholar] [CrossRef]
- Hou, B.-Y.; Zhao, Y.-R.; Ma, P.; Xu, C.-Y.; He, P.; Yang, X.-Y.; Zhang, L.; Qiang, G.-F.; Du, G.-H. Hypoglycemic activity of puerarin through modulation of oxidative stress and mitochondrial function via AMPK. Chin. J. Nat. Med. 2020, 18, 818–826. [Google Scholar] [CrossRef]
- Zhao, H.; Kong, L.; Shao, M.; Liu, J.; Sun, C.; Li, C.; Wang, Y.; Chai, X.; Wang, Y.; Zhang, Y.; et al. Protective effect of flavonoids extract of Hippophae rhamnoides L. on alcoholic fatty liver disease through regulating intestinal flora and inhibiting TAK1/p38MAPK/p65NF-κB pathway. J. Ethnopharmacol. 2022, 292, 115225. [Google Scholar] [CrossRef] [PubMed]
- Ren, Q.; Guo, F.; Tao, S.; Huang, R.; Ma, L.; Fu, P. Flavonoid fisetin alleviates kidney inflammation and apoptosis via inhibiting Src-mediated NF-κB p65 and MAPK signaling pathways in septic AKI mice. Biomed. Pharmacother. 2020, 122, 109772. [Google Scholar] [CrossRef] [PubMed]
- Unno, K.; Nakamura, Y. Green tea suppresses brain aging. Molecules 2021, 26, 4897. [Google Scholar] [CrossRef]
- Calis, Z.; Mogulkoc, R.; Baltaci, A.K. The Roles of Flavonols/Flavonoids in Neurodegeneration and Neuroinflammation. Mini Rev. Med. Chem. 2020, 20, 1475–1488. [Google Scholar] [CrossRef]
- Badshah, S.L.; Faisal, S.; Muhammad, A.; Poulson, B.G.; Emwas, A.H.; Jaremko, M. Antiviral activities of flavonoids. Biomed. Pharmacother. 2021, 140, 111596. [Google Scholar] [CrossRef]
- Nabavi, S.M.; Šamec, D.; Tomczyk, M.; Milella, L.; Russo, D.; Habtemariam, S.; Suntar, I.; Rastrelli, L.; Daglia, M.; Xiao, J.; et al. Flavonoid biosynthetic pathways in plants: Versatile targets for metabolic engineering. Biotechnol. Adv. 2020, 38, 107316. [Google Scholar] [CrossRef]
- Zhuang, W.-B.; Li, Y.-H.; Shu, X.-C.; Pu, Y.-T.; Wang, X.-J.; Wang, T.; Wang, Z. The classification, molecular structure and biological biosynthesis of flavonoids, and their roles in biotic and abiotic stresses. Molecules 2023, 28, 3599. [Google Scholar] [CrossRef]
- Kopustinskiene, D.M.; Jakstas, V.; Savickas, A.; Bernatoniene, J. Flavonoids as Anticancer Agents. Nutrients 2020, 12, 457. [Google Scholar] [CrossRef]
- Hazafa, A.; Rehman, K.U.; Jahan, N.; Jabeen, Z. The Role of Polyphenol (Flavonoids) Compounds in the Treatment of Cancer Cells. Nutr. Cancer 2020, 72, 386–397. [Google Scholar] [CrossRef]
- Wang, R.; Wang, C.; Lu, L.; Yuan, F.; He, F. Baicalin and baicalein in modulating tumor microenvironment for cancer treatment: A comprehensive review with future perspectives. Pharmacol. Res. 2024, 199, 107032. [Google Scholar] [CrossRef]
- Singh, D.; Dhanawat, M.; Verma, I.; Gupta, S. The Fascinating Effects of Flavonoids on Lung Cancer: A Review. Curr. Nutr. Food Sci. 2024, 20, 1231–1251. [Google Scholar] [CrossRef]
- Zanoaga, O.; Braicu, C.; Jurj, A.; Rusu, A.; Buiga, R.; Berindan-Neagoe, I. Progress in Research on the Role of Flavonoids in Lung Cancer. Int. J. Mol. Sci. 2019, 20, 4291. [Google Scholar] [CrossRef] [PubMed]
- Dev, S.S.; Farghadani, R.; Abidin, S.A.Z.; Othman, I.; Naidu, R. Flavonoids as receptor tyrosine kinase inhibitors in lung cancer. J. Funct. Foods 2023, 110, 105845. [Google Scholar] [CrossRef]
- Li, C.; Dai, T.; Chen, J.; Chen, M.; Liang, R.; Liu, C.; Du, L.; McClements, D.J. Modification of flavonoids: Methods and influences on biological activities. Crit. Rev. Food Sci. Nutr. 2023, 63, 10637–10658. [Google Scholar] [CrossRef]
- Vazhappilly, C.G.; Amararathna, M.; Cyril, A.C.; Linger, R.; Matar, R.; Merheb, M.; Ramadan, W.S.; Radhakrishnan, R.; Rupasinghe, H.P.V. Current methodologies to refine bioavailability, delivery, and therapeutic efficacy of plant flavonoids in cancer treatment. J. Nutr. Biochem. 2021, 94, 108623. [Google Scholar] [CrossRef]
- Alizadeh, S.R.; Savadkouhi, N.; Ebrahimzadeh, M.A. Drug design strategies that aim to improve the low solubility and poor bioavailability conundrum in quercetin derivatives. Expert. Opin. Drug Discov. 2023, 18, 1117–1132. [Google Scholar] [CrossRef]
- Szabó, R.; Rácz, C.P.; Dulf, F.V. Bioavailability Improvement Strategies for Icariin and Its Derivates: A Review. Int. J. Mol. Sci. 2022, 23, 7519. [Google Scholar] [CrossRef]
- Zhang, Q.; Yu, W.; Liu, Z.; Li, H.; Liu, Y.; Liu, X.; Han, Z.; He, J.; Zeng, Y.; Guo, Y.; et al. Design, synthesis, antitumor activity and ct-DNA binding study of photosensitive drugs based on porphyrin framework. Int. J. Biol. Macromol. 2023, 230, 123147. [Google Scholar] [CrossRef]
- Papaj, K.; Kasprzycka, A.; Góra, A.; Grajoszek, A.; Rzepecka, G.; Stojko, J.; Barski, J.J.; Szeja, W.; Rusin, A. Structure-bioavailability relationship study of genistein derivatives with antiproliferative activity on human cancer cell. J. Pharm. Biomed. Anal. 2020, 185, 113216. [Google Scholar] [CrossRef]
- Nkoana, J.K.; Mphahlele, M.J.; More, G.K.; Choong, Y.S. Exploring the 3,5-Dibromo-4,6-dimethoxychalcones and Their Flavone Derivatives as Dual α-Glucosidase and α-Amylase Inhibitors with Antioxidant and Anticancer Potential. Antioxidants 2024, 13, 1255. [Google Scholar] [CrossRef]
- Wong, R.S. Apoptosis in cancer: From pathogenesis to treatment. J. Exp. Clin. Cancer Res. CR 2011, 30, 87. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, G.; Samal, D.; Khandayataray, P.; Murthy, M.K. A Review on Caspases: Key Regulators of Biological Activities and Apoptosis. Mol. Neurobiol. 2023, 60, 5805–5837. [Google Scholar] [CrossRef] [PubMed]
- Jia, W.-D.; Bai, X.; Ma, Q.-Q.; Bian, M.; Bai, C.-M.; Li, D.; Li, L.-F.; Wei, C.-X.; Yu, L.-J. Synthesis, molecular docking studies of formononetin derivatives as potent Bax agonists for anticancer activity. Nat. Prod. Res. 2025, 39, 423–437. [Google Scholar] [CrossRef]
- Li, M.; Zha, G.; Chen, R.; Chen, X.; Sun, Q.; Jiang, H. Anticancer effects of myricetin derivatives in non-small cell lung cancer in vitro and in vivo. Pharmacol. Res. Perspect. 2022, 10, e00905. [Google Scholar] [CrossRef]
- Zhou, H.; Xu, L.; Shi, Y.; Gu, S.; Wu, N.; Liu, F.; Huang, Y.; Qian, Z.; Xue, W.; Wang, X.; et al. A novel myricetin derivative with anti-cancer properties induces cell cycle arrest and apoptosis in A549 cells. Biol. Pharm. Bull. 2023, 46, 42–51. [Google Scholar] [CrossRef]
- Bak, Y.; Kim, H.; Kang, J.-W.; Lee, D.H.; Kim, M.S.; Park, Y.S.; Kim, J.-H.; Jung, K.-Y.; Lim, Y.; Hong, J.; et al. A synthetic naringenin derivative, 5-hydroxy-7,4′-diacetyloxyflavanone-N-phenyl hydrazone (N101-43), induces apoptosis through up-regulation of Fas/FasL expression and inhibition of PI3K/Akt signaling pathways in non-small-cell lung cancer cells. J. Agric. Food Chem. 2011, 59, 10286–10297. [Google Scholar] [CrossRef]
- Debnath, J.; Gammoh, N.; Ryan, K.M. Autophagy and autophagy-related pathways in cancer. Nat. Rev. Mol. Cell Biol. 2023, 24, 560–575. [Google Scholar] [CrossRef]
- Li, Y.R.; Li, S.; Ho, C.T.; Chang, Y.H.; Tan, K.T.; Chung, T.W.; Wang, B.Y.; Chen, Y.K.; Lin, C.C. Tangeretin derivative, 5-acetyloxy-6,7,8,4′-tetramethoxyflavone induces G2/M arrest, apoptosis and autophagy in human non-small cell lung cancer cells in vitro and in vivo. Cancer Biol. Ther. 2016, 17, 48–64. [Google Scholar] [CrossRef]
- Tian, Y.; Lei, Y.; Wang, Y.; Lai, J.; Wang, J.; Xia, F. Mechanism of multidrug resistance to chemotherapy mediated by P-glycoprotein (Review). Int. J. Oncol. 2023, 63, 119. [Google Scholar] [CrossRef]
- Feng, S.; Zhou, H.; Wu, D.; Zheng, D.; Qu, B.; Liu, R.; Zhang, C.; Li, Z.; Xie, Y.; Luo, H.B. Nobiletin and its derivatives overcome multidrug resistance (MDR) in cancer: Total synthesis and discovery of potent MDR reversal agents. Acta Pharm. Sin. B 2020, 10, 327–343. [Google Scholar] [CrossRef]
- Suski, J.M.; Braun, M.; Strmiska, V.; Sicinski, P. Targeting cell-cycle machinery in cancer. Cancer Cell 2021, 39, 759–778. [Google Scholar] [CrossRef] [PubMed]
- Morris, E.J.; Dyson, N.J. Retinoblastoma protein partners. Adv. Cancer Res. 2001, 82, 1–54. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Leone, G.W.; Wang, H. Cyclin D-CDK4/6 functions in cancer. Adv. Cancer Res. 2020, 148, 147–169. [Google Scholar] [CrossRef] [PubMed]
- Peng, B.; Cao, J.; Yi, S.; Wang, C.; Zheng, G.; He, Z. Inhibition of proliferation and induction of G1-phase cell-cycle arrest by dFMGEN, a novel genistein derivative, in lung carcinoma A549 cells. Drug Chem. Toxicol. 2013, 36, 196–204. [Google Scholar] [CrossRef]
- Le, X.; Nilsson, M.; Goldman, J.; Reck, M.; Nakagawa, K.; Kato, T.; Ares, L.P.; Frimodt-Moller, B.; Wolff, K.; Visseren-Grul, C.; et al. Dual EGFR-VEGF Pathway Inhibition: A Promising Strategy for Patients With EGFR-Mutant NSCLC. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2021, 16, 205–215. [Google Scholar] [CrossRef]
- Manzo, A.; Montanino, A.; Carillio, G.; Costanzo, R.; Sandomenico, C.; Normanno, N.; Piccirillo, M.C.; Daniele, G.; Perrone, F.; Rocco, G.; et al. Angiogenesis Inhibitors in NSCLC. Int. J. Mol. Sci. 2017, 18, 2021. [Google Scholar] [CrossRef]
- Jiang, X.; Zhou, J.; Lin, Q.; Gong, G.; Sun, H.; Liu, W.; Guo, Q.; Feng, F.; Qu, W. Anti-angiogenic and anticancer effects of baicalein derivatives based on transgenic zebrafish model. Bioorganic Med. Chem. 2018, 26, 4481–4492. [Google Scholar] [CrossRef]
- Harrison, P.T.; Vyse, S.; Huang, P.H. Rare epidermal growth factor receptor (EGFR) mutations in non-small cell lung cancer. Semin. Cancer Biol. 2020, 61, 167–179. [Google Scholar] [CrossRef]
- Wang, J.; Sun, P.; Wang, Q.; Zhang, P.; Wang, Y.; Zi, C.; Wang, X.; Sheng, J. (-)-Epigallocatechin-3-gallate derivatives combined with cisplatin exhibit synergistic inhibitory effects on non-small-cell lung cancer cells. Cancer Cell Int. 2019, 19, 266. [Google Scholar] [CrossRef]
- Chandrashekar, N.; Pandi, A. Baicalein: A review on its anti-cancer effects and mechanisms in lung carcinoma. J. Food Biochem. 2022, 46, e14230. [Google Scholar] [CrossRef]
- Monasterio, A.; Urdaci, M.C.; Pinchuk, I.V.; López-Moratalla, N.; Martínez-Irujo, J.J. Flavonoids induce apoptosis in human leukemia U937 cells through caspase- and caspase-calpain-dependent pathways. Nutr. Cancer 2004, 50, 90–100. [Google Scholar] [CrossRef]
- Panek-Krzyśko, A.; Stompor-Gorący, M. The Pro-Health Benefits of Morusin Administration-An Update Review. Nutrients 2021, 13, 3043. [Google Scholar] [CrossRef]
- Wen, L.; Zhou, T.; Jiang, Y.; Chang, S.K.; Yang, B. Prenylated flavonoids in foods and their applications on cancer prevention. Crit. Rev. Food Sci. Nutr. 2022, 62, 5067–5080. [Google Scholar] [CrossRef]
- Neves, M.P.; Cidade, H.; Pinto, M.; Silva, A.M.; Gales, L.; Damas, A.M.; Lima, R.T.; Vasconcelos, M.H.; de São José Nascimento, M. Prenylated derivatives of baicalein and 3,7-dihydroxyflavone: Synthesis and study of their effects on tumor cell lines growth, cell cycle and apoptosis. Eur. J. Med. Chem. 2011, 46, 2562–2574. [Google Scholar] [CrossRef]
- Mayer, S.; Keglevich, P.; Ábrányi-Balogh, P.; Szigetvári, Á.; Dékány, M.; Szántay, C., Jr.; Hazai, L. Synthesis and In Vitro Anticancer Evaluation of Novel Chrysin and 7-Aminochrysin Derivatives. Molecules 2020, 25, 888. [Google Scholar] [CrossRef]
- Yu, Q.; Huang, B.; Ling, Y. Design, Synthesis, and In vitro Anticancer Activity of Novel Chrysin Derivatives. Lett. Drug Des. Discov. 2023, 20, 854–862. [Google Scholar] [CrossRef]
- Xu, H.; Ma, H.; Zha, L.; Li, Q.; Pan, H.; Zhang, L. Genistein promotes apoptosis of lung cancer cells through the IMPDH2/AKT1 pathway. Am. J. Transl. Res. 2022, 14, 7040–7051. [Google Scholar]
- Fu, Z.; Cao, X.; Liu, L.; Cao, X.; Cui, Y.; Li, X.; Quan, M.; Ren, K.; Chen, A.; Xu, C.; et al. Genistein inhibits lung cancer cell stem-like characteristics by modulating MnSOD and FoxM1 expression. Oncol. Lett. 2020, 20, 2506–2515. [Google Scholar] [CrossRef]
- Fu, J.; Yang, J.; Seeberger, P.H.; Yin, J. Glycoconjugates for glucose transporter-mediated cancer-specific targeting and treatment. Carbohydr. Res. 2020, 498, 108195. [Google Scholar] [CrossRef]
- Vaidya, S.P.; Patra, M. Platinum glycoconjugates: “Sweet bullets” for targeted cancer therapy? Curr. Opin. Chem. Biol. 2023, 72, 102236. [Google Scholar] [CrossRef]
- Rusin, A.; Zawisza-Puchałka, J.; Kujawa, K.; Gogler-Pigłowska, A.; Wietrzyk, J.; Świtalska, M.; Głowala-Kosińska, M.; Gruca, A.; Szeja, W.; Krawczyk, Z.; et al. Synthetic conjugates of genistein affecting proliferation and mitosis of cancer cells. Bioorganic Med. Chem. 2011, 19, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Viegas-Junior, C.; Danuello, A.; da Silva Bolzani, V.; Barreiro, E.J.; Fraga, C.A. Molecular hybridization: A useful tool in the design of new drug prototypes. Curr. Med. Chem. 2007, 14, 1829–1852. [Google Scholar] [CrossRef] [PubMed]
- Oh, H.N.; Kwak, A.W.; Lee, M.H.; Kim, E.; Yoon, G.; Cho, S.S.; Liu, K.; Chae, J.I.; Shim, J.H. Targeted inhibition of c-MET by podophyllotoxin promotes caspase-dependent apoptosis and suppresses cell growth in gefitinib-resistant non-small cell lung cancer cells. Phytomed. Int. J. Phytother. Phytopharm. 2021, 80, 153355. [Google Scholar] [CrossRef]
- Yu, X.; Gao, F.; Li, W.; Zhou, L.; Liu, W.; Li, M. Formononetin inhibits tumor growth by suppression of EGFR-Akt-Mcl-1 axis in non-small cell lung cancer. J. Exp. Clin. Cancer Res. CR 2020, 39, 62. [Google Scholar] [CrossRef]
- Yin, R.; Gao, J.; Liu, Y. Mechanisms analysis for Formononetin counteracted-Osimertinib resistance in non-small cell lung cancer cells: From the insight into the gene transcriptional level. Chem. Biol. Drug Des. 2024, 103, e14435. [Google Scholar] [CrossRef]
- Yang, C.; Xie, Q.; Zeng, X.; Tao, N.; Xu, Y.; Chen, Y.; Wang, J.; Zhang, L. Novel hybrids of podophyllotoxin and formononetin inhibit the growth, migration and invasion of lung cancer cells. Bioorganic Chem. 2019, 85, 445–454. [Google Scholar] [CrossRef]
- Xue, X.; Lindstrom, A.; Li, Y. Porphyrin-Based Nanomedicines for Cancer Treatment. Bioconjugate Chem. 2019, 30, 1585–1603. [Google Scholar] [CrossRef]
- Alzeibak, R.; Mishchenko, T.A.; Shilyagina, N.Y.; Balalaeva, I.V.; Vedunova, M.V.; Krysko, D.V. Targeting immunogenic cancer cell death by photodynamic therapy: Past, present and future. J. Immunother. Cancer 2021, 9, e001926. [Google Scholar] [CrossRef]
- Ling, Y.; Chen, Y.; Chen, P.; Hui, H.; Song, X.; Lu, Z.; Li, C.; Lu, N.; Guo, Q. Baicalein potently suppresses angiogenesis induced by vascular endothelial growth factor through the p53/Rb signaling pathway leading to G1/s cell cycle arrest. Exp. Biol. Med. 2011, 236, 851–858. [Google Scholar] [CrossRef]
- Zhang, S.; Wang, Y.; Shan, J.; Qi, X.; Liu, Q. Improved Bioavailability and Hepatoprotective Activity of Baicalein Via a Self-assembled Solutol HS15 Micelles System. Curr. Drug Deliv. 2024, 21, 461–472. [Google Scholar] [CrossRef]
- Thorat, N.M.; Sarkate, A.P.; Lokwani, D.K.; Tiwari, S.V.; Azad, R.; Thopate, S.R. N-Benzylation of 6-aminoflavone by reductive amination and efficient access to some novel anticancer agents via topoisomerase II inhibition. Mol. Divers. 2021, 25, 937–948. [Google Scholar] [CrossRef] [PubMed]
- Tangeti, V.S.; Vasundhara, D.; Satyanarayana, K.V.V.V.; Kumar, K.S.P. Synthesis and antiproliferative activity of some dihydro-1H-furo [2,3-c]pyrazole-flavone hybrids. Asian J. Chem. 2017, 29, 1525–1532. [Google Scholar] [CrossRef]
- Fernandes, A.P.; Gandin, V. Selenium compounds as therapeutic agents in cancer. Biochim. Et Biophys. Acta 2015, 1850, 1642–1660. [Google Scholar] [CrossRef] [PubMed]
- Wallenberg, M.; Misra, S.; Björnstedt, M. Selenium cytotoxicity in cancer. Basic. Clin. Pharmacol. Toxicol. 2014, 114, 377–386. [Google Scholar] [CrossRef]
- Fonseca, S.F.; Padilha, N.B.; Thurow, S.; Roehrs, J.A.; Savegnago, L.; de Souza, M.N.; Fronza, M.G.; Collares, T.; Buss, J.; Seixas, F.K.; et al. Ultrasound-promoted copper-catalyzed synthesis of bis-arylselanyl chrysin derivatives with boosted antioxidant and anticancer activities. Ultrason. Sonochem. 2017, 39, 827–836. [Google Scholar] [CrossRef]
- Arya, G.C.; Kaur, K.; Jaitak, V. Isoxazole derivatives as anticancer agent: A review on synthetic strategies, mechanism of action and SAR studies. Eur. J. Med. Chem. 2021, 221, 113511. [Google Scholar] [CrossRef]
- Arya, J.S.; Joseph, M.M.; Sherin, D.R.; Nair, J.B.; Manojkumar, T.K.; Maiti, K.K. Exploring Mitochondria-Mediated Intrinsic Apoptosis by New Phytochemical Entities: An Explicit Observation of Cytochrome c Dynamics on Lung and Melanoma Cancer Cells. J. Med. Chem. 2019, 62, 8311–8329. [Google Scholar] [CrossRef]
- Mobbili, G.; Romaldi, B.; Sabbatini, G.; Amici, A.; Marcaccio, M.; Galeazzi, R.; Laudadio, E.; Armeni, T.; Minnelli, C. Identification of Flavone Derivative Displaying a 4′-Aminophenoxy Moiety as Potential Selective Anticancer Agent in NSCLC Tumor Cells. Molecules 2023, 28, 3239. [Google Scholar] [CrossRef]
- Chang, H.L.; Su, J.H.; Yeh, Y.T.; Lee, Y.C.; Chen, H.M.; Wu, Y.C.; Yuan, S.S. Protoapigenone, a novel flavonoid, inhibits ovarian cancer cell growth in vitro and in vivo. Cancer Lett. 2008, 267, 85–95. [Google Scholar] [CrossRef]
- Wang, H.C.; Lee, A.Y.; Chou, W.C.; Wu, C.C.; Tseng, C.N.; Liu, K.Y.; Lin, W.L.; Chang, F.R.; Chuang, D.W.; Hunyadi, A.; et al. Inhibition of ATR-dependent signaling by protoapigenone and its derivative sensitizes cancer cells to interstrand cross-link-generating agents in vitro and in vivo. Mol. Cancer Ther. 2012, 11, 1443–1453. [Google Scholar] [CrossRef]
- Danko, B.; Martins, A.; Chuang, D.W.; Wang, H.C.; Amaral, L.; Molnar, J.; Chang, F.R.; Wu, Y.C.; Hunyadi, A. In vitro cytotoxic activity of novel protoflavone analogs—Selectivity towards a multidrug resistant cancer cell line. Anticancer. Res. 2012, 32, 2863–2870. [Google Scholar] [PubMed]
- Stompor, M.; Świtalska, M.; Wietrzyk, J. Synthesis and biological evaluation of acyl derivatives of hydroxyflavones as potent antiproliferative agents against drug resistance cell lines. Z. Fur Naturforschung. C J. Biosci. 2018, 73, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Stompor, M.; Switalska, M.; Podgorski, R.; Uram, L.; Aebisher, D.; Wietrzyk, J. Synthesis and biological evaluation of 4′-O-acetylisoxanthohumol and its analogues as antioxidant and antiproliferative agents. Acta Biochim. Pol. 2017, 64, 577–583. [Google Scholar] [CrossRef]
- Margiotta, E.; van der Lubbe, S.C.C.; de Azevedo Santos, L.; Paragi, G.; Moro, S.; Bickelhaupt, F.M.; Fonseca Guerra, C. Halogen Bonds in Ligand-Protein Systems: Molecular Orbital Theory for Drug Design. J. Chem. Inf. Model. 2020, 60, 1317–1328. [Google Scholar] [CrossRef]
- Oselusi, S.O.; Dube, P.; Odugbemi, A.I.; Akinyede, K.A.; Ilori, T.L.; Egieyeh, E.; Sibuyi, N.R.; Meyer, M.; Madiehe, A.M.; Wyckoff, G.J.; et al. The role and potential of computer-aided drug discovery strategies in the discovery of novel antimicrobials. Comput. Biol. Med. 2024, 169, 107927. [Google Scholar] [CrossRef]
- Vemula, D.; Jayasurya, P.; Sushmitha, V.; Kumar, Y.N.; Bhandari, V. CADD, AI and ML in drug discovery: A comprehensive review. Eur. J. Pharm. Sci. Off. J. Eur. Fed. Pharm. Sci. 2023, 181, 106324. [Google Scholar] [CrossRef]
- Zhang, B.; Wang, M.; Sun, L.; Liu, J.; Yin, L.; Xia, M.; Zhang, L.; Liu, X.; Cheng, Y. Recent Advances in Targeted Cancer Therapy: Are PDCs the Next Generation of ADCs? J. Med. Chem. 2024, 67, 11469–11487. [Google Scholar] [CrossRef]
- Cooper, B.M.; Iegre, J.; DH, O.D.; Ölwegård Halvarsson, M.; Spring, D.R. Peptides as a platform for targeted therapeutics for cancer: Peptide-drug conjugates (PDCs). Chem. Soc. Rev. 2021, 50, 1480–1494. [Google Scholar] [CrossRef]
- Sartor, O.; de Bono, J.; Chi, K.N.; Fizazi, K.; Herrmann, K.; Rahbar, K.; Tagawa, S.T.; Nordquist, L.T.; Vaishampayan, N.; El-Haddad, G.; et al. Lutetium-177-PSMA-617 for Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2021, 385, 1091–1103. [Google Scholar] [CrossRef]
- Hennrich, U.; Eder, M. [(177)Lu]Lu-PSMA-617 (Pluvicto(TM)): The First FDA-Approved Radiotherapeutical for Treatment of Prostate Cancer. Pharmaceuticals 2022, 15, 1292. [Google Scholar] [CrossRef]
- Fu, C.; Yu, L.; Miao, Y.; Liu, X.; Yu, Z.; Wei, M. Peptide-drug conjugates (PDCs): A novel trend of research and development on targeted therapy, hype or hope? Acta Pharm. Sin. B 2023, 13, 498–516. [Google Scholar] [CrossRef] [PubMed]
- Shano, L.B.; Karthikeyan, S.; Kennedy, L.J.; Chinnathambi, S.; Pandian, G.N. MOFs for next-generation cancer therapeutics through a biophysical approach-a review. Front. Bioeng. Biotechnol. 2024, 12, 1397804. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.; Hmar, E.B.L.; Pathak, H.; Sharma, H.K. 6-An overview on nanocarriers. In Nanocarriers for Drug-Targeting Brain Tumors; Kumar, L., Pathak, Y.Y., Eds.; Elsevier: Amsterdam, The Netherlands, 2022; pp. 145–204. [Google Scholar] [CrossRef]
- Abed, S.N.; Deb, P.K.; Surchi, H.S.; Kokaz, S.F.; Jamal, S.M.; Bandopadhyay, S.; Tekade, R.K. Chapter 17—Nanocarriers in Different Preclinical and Clinical Stages. In Basic Fundamentals of Drug Delivery; Tekade, R.K., Ed.; Academic Press: Cambridge, MA, USA, 2019; pp. 685–731. [Google Scholar] [CrossRef]
- Cunha, C.; Daniel-da-Silva, A.L.; Oliveira, H. Drug Delivery Systems and Flavonoids: Current Knowledge in Melanoma Treatment and Future Perspectives. Micromachines 2022, 13, 1838. [Google Scholar] [CrossRef] [PubMed]
- Yi, P.; Wu, Y.; Wang, J.; Liu, Q.; Xing, Y.; Lu, Y.; Ma, C.; Duan, L.; Zhao, J.; Meng, Q. Photocatalytic acceptorless dehydrogenation of flavanones by cationic Eosin Y as a bifunctional catalyst. Org. Biomol. Chem. 2025, 23, 1574–1580. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Modification | Compound ID | Activity | Cells Line | Reference |
|---|---|---|---|---|
| C-7 substituted | 18c | 4.1 ± 0.1 μM | NCI-H460 | [54] |
| 19a | 3.9 ± 0.1 μM | |||
| 19b | 2.7 ± 0.8 μM | |||
| 20a | 0.24 μM | NCI-H522 | [55] | |
| 23a | 4.76 ± 0.96 μM | A549 | [61] | |
| 26a | 0.753 μM | A549 | [66] | |
| C-6 substituted | 28h | 4.73 μM | A549 | [47] |
| Other modifications | 33 | 1.2 μM | A549 | [77] |
| 34 | 0.76 μM | |||
| 35 | 0.4 μM 2.3 ± 0.2 μM | A549 | [78] | |
| H1975 | ||||
| 37c | 2 μM | A549 | [81] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yao, J.; Zhu, F.; Feng, Y.; Gu, C.; Wang, T.; Li, X.; Yang, H.; Hu, X.; Bonnet, P.-A.; Meng, X. Research Progress on the Structure–activity Relationship and Mechanism of Flavonoid Derivatives in the Treatment of Lung Cancer. Molecules 2025, 30, 1827. https://doi.org/10.3390/molecules30081827
Yao J, Zhu F, Feng Y, Gu C, Wang T, Li X, Yang H, Hu X, Bonnet P-A, Meng X. Research Progress on the Structure–activity Relationship and Mechanism of Flavonoid Derivatives in the Treatment of Lung Cancer. Molecules. 2025; 30(8):1827. https://doi.org/10.3390/molecules30081827
Chicago/Turabian StyleYao, Jiacheng, Feng Zhu, Yikun Feng, Chen Gu, Tianyu Wang, Xinyu Li, Hao Yang, Xiamin Hu, Pierre-Antoine Bonnet, and Xiangguo Meng. 2025. "Research Progress on the Structure–activity Relationship and Mechanism of Flavonoid Derivatives in the Treatment of Lung Cancer" Molecules 30, no. 8: 1827. https://doi.org/10.3390/molecules30081827
APA StyleYao, J., Zhu, F., Feng, Y., Gu, C., Wang, T., Li, X., Yang, H., Hu, X., Bonnet, P.-A., & Meng, X. (2025). Research Progress on the Structure–activity Relationship and Mechanism of Flavonoid Derivatives in the Treatment of Lung Cancer. Molecules, 30(8), 1827. https://doi.org/10.3390/molecules30081827