

Sol-Gel Heterogeneization of an Ir(III) Complex for Sustainable Visible-Light Redox Photocatalysis

 , and
, and
Abstract

1. Introduction
2. Results
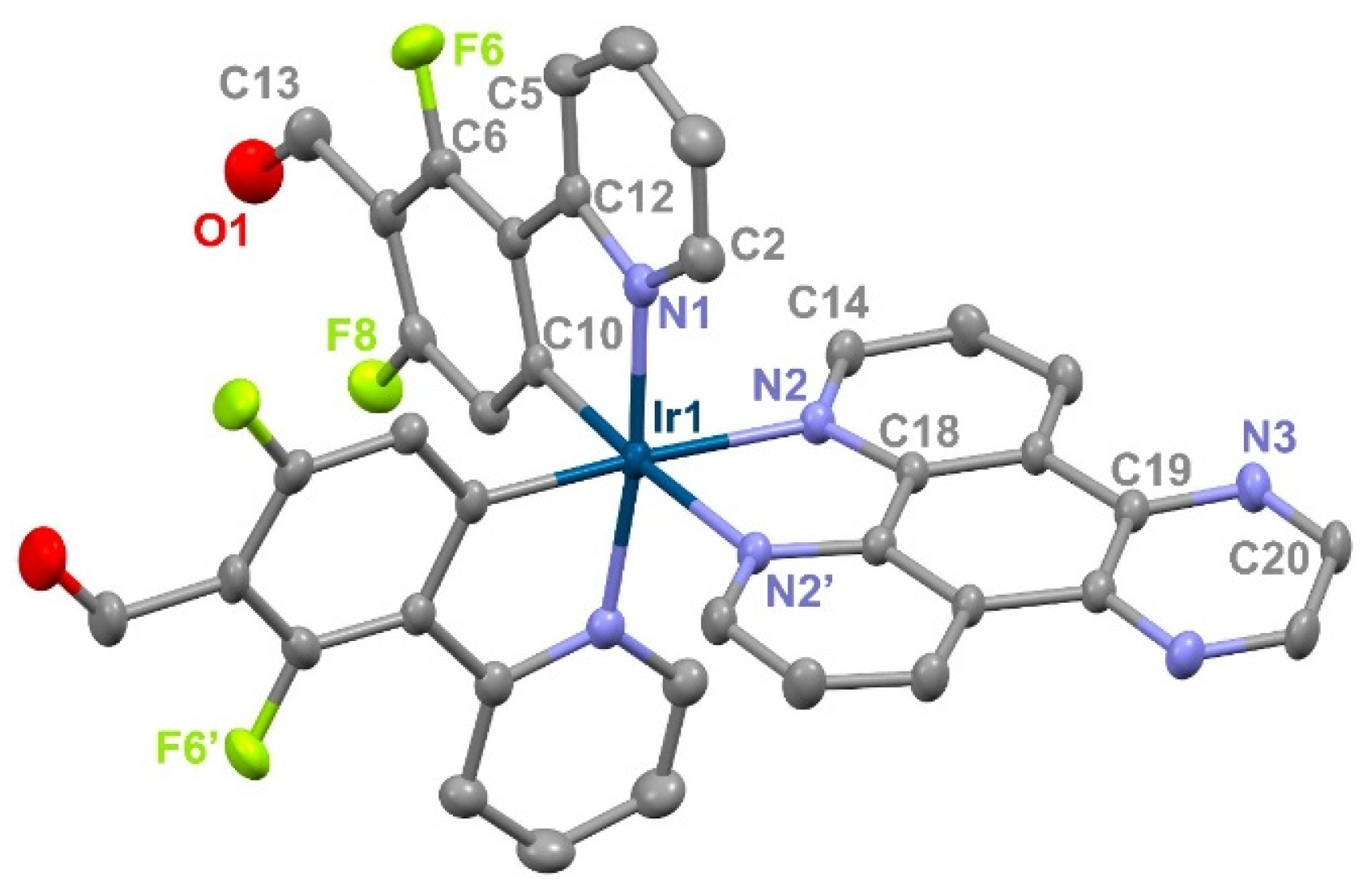
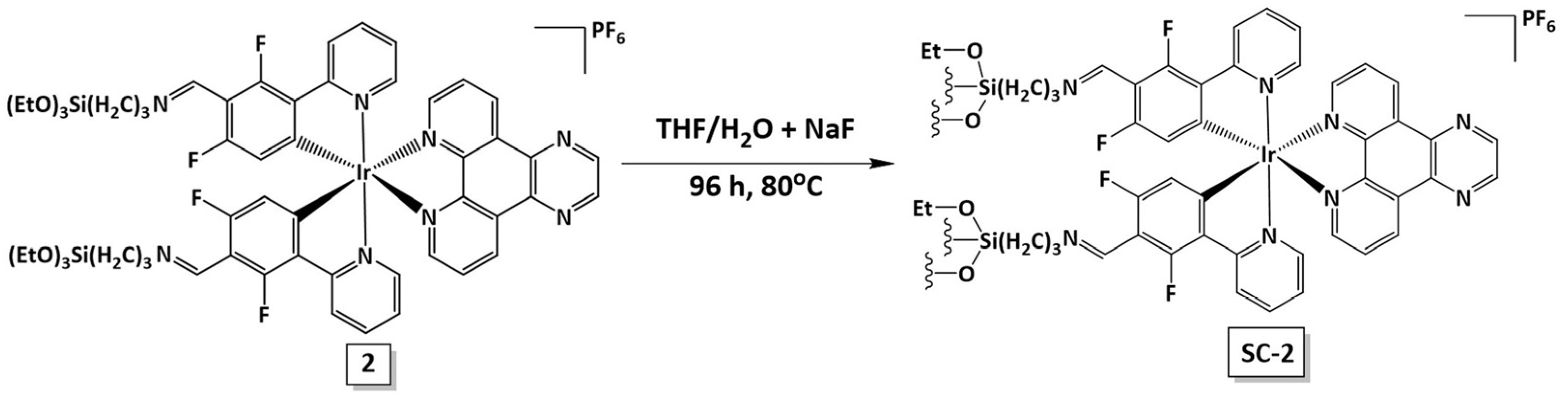
2.1. Synthesis and Characterization of Complexes 1 and 2 and the Self-Condensed Ionosilica SC-2
2.2. Photophysical and Electronic Characterization


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Medium | λem/nm (λexc/nm) | τ/µs 1 | Φ/% |
|---|---|---|---|---|
| [Ir(dfbzapy)2(pirafen)]PF6 (1) | Solid | 560 (440) | 13.24 | 1 |
| MeCN | 510 (360) | 12.62 | 7 | |
| [Ir(Si-dfppy)2(pirafen)]PF6 (2) | MeCN | 500 (375) | 20.19 | 1.2 |
| SC-2 | Solid | 660 (475) | 10.73 | 1 |
| Suspension MeCN | 510 (360) | 13.79 | 2.4 |
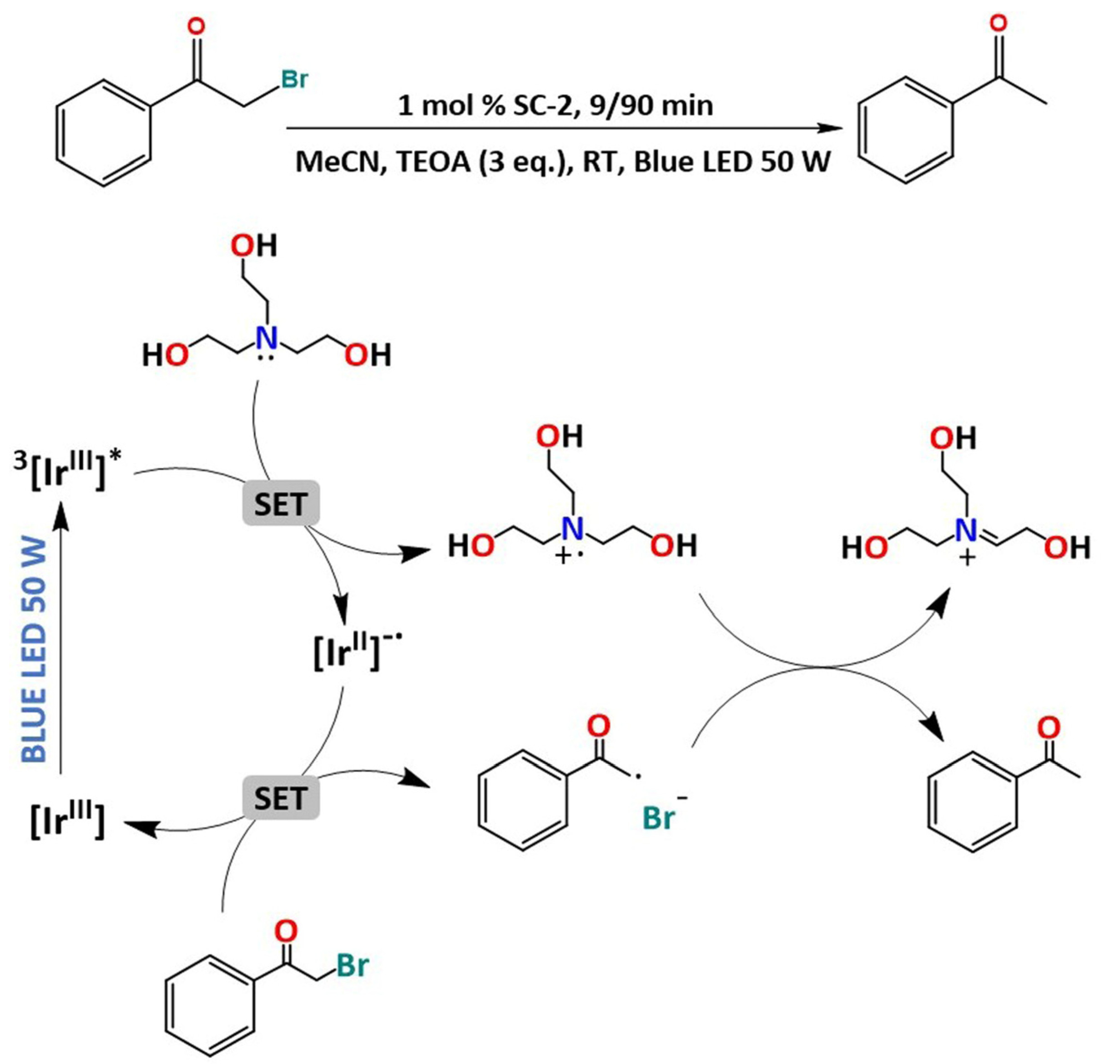
2.3. Photocatalytic Studies
3. Materials and Methods
3.1. General Methods
3.2. Synthesis of [Ir(Dfbzapy)2(pyraphen)]PF6 (1)
3.3. Synthesis of [Ir(Si-Dfbzapy)2(pyraphen)]PF6 (2)
3.4. Synthesis of the Self-Condensed Material (SC-2)
3.5. X-Ray Crystallographic Analysis of Complex 1
3.6. Theoretical Calculations
3.7. Photocatalytic Methods for Dehalogenation of 2-Bromoacetophenone
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kou, J.; Lu, C.; Wang, J.; Chen, Y.; Xu, Z.; Varma, R.S. Selectivity Enhancement in Heterogeneous Photocatalytic Transformations. Chem. Rev. 2017, 117, 1445–1514. [Google Scholar] [CrossRef]
- Ham, R.; Nielsen, C.J.; Pullen, S.; Reek, J.N.H. Supramolecular Coordination Cages for Artificial Photosynthesis and Synthetic Photocatalysis. Chem. Rev. 2023, 123, 5225–5261. [Google Scholar] [CrossRef] [PubMed]
- Chandra, P.; Choudhary, N.; Mobin, S.M. The game between molecular photoredox catalysis and hydrogen: The golden age of hydrogen budge. Mol. Catal. 2023, 537, 112921. [Google Scholar] [CrossRef]
- Millward, F.; Zysman-Colman, E. Alchemy reimagined: Photocatalysis using anthropogenic waste materials. Trends Chem. 2023, 5, 584–587. [Google Scholar] [CrossRef]
- Ciamician, G.; Silber, P. Chemische Lichtwirkungen. Ber. Dtsch. Chem. Ges. 1908, 41, 1928–1935. [Google Scholar] [CrossRef]
- Heindel, N.D.; Pfau, M.A. A profitable partnership: Giacomo Ciamician and Paul Silber. J. Chem. Educ. 1965, 42, 383. [Google Scholar] [CrossRef]
- Yakushev, A.A.; Abel, A.S.; Averin, A.D.; Beletskaya, I.P.; Cheprakov, A.V.; Ziankou, I.S.; Bonneviot, L.; Bessmertnykh-Lemeune, A. Visible-light photocatalysis promoted by solid- and liquid-phase immobilized transition metal complexes in organic synthesis. Coord. Chem. Rev. 2022, 458, 214331. [Google Scholar] [CrossRef]
- Marzo, L.; Pagire, S.K.; Reiser, O.; König, B. Visible-Light Photocatalysis: Does It Make a Difference in Organic Synthesis? Angew. Chem. Int. Ed. 2018, 57, 10034–10072. [Google Scholar] [CrossRef]
- König, B. Photocatalysis in Organic Synthesis—Past, Present, and Future. Eur. J. Org. Chem. 2017, 2017, 1979–1981. [Google Scholar] [CrossRef]
- Lindroth, R.; Materna, K.L.; Hammarström, L.; Wallentin, C.-J. Sustainable Ir-Photoredox Catalysis by Means of Heterogenization. ACS Org. Inorg. Au 2022, 2, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.Y.; Perry, I.B.; Bissonnette, N.B.; Buksh, B.F.; Edwards, G.A.; Frye, L.I.; Garry, O.L.; Lavagnino, M.N.; Li, B.X.; Liang, Y.; et al. Metallaphotoredox: The Merger of Photoredox and Transition Metal Catalysis. Chem. Rev. 2022, 122, 1485–1542. [Google Scholar] [CrossRef] [PubMed]
- Prier, C.K.; Rankic, D.A.; MacMillan, D.W.C. Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis. Chem. Rev. 2013, 113, 5322–5363. [Google Scholar] [CrossRef] [PubMed]
- Bevernaegie, R.; Wehlin, S.A.M.; Elias, B.; Troian-Gautier, L. A Roadmap Towards Visible Light Mediated Electron Transfer Chemistry with Iridium(III) Complexes. ChemPhotoChem 2021, 5, 217–234. [Google Scholar] [CrossRef]
- Bell, J.D.; Murphy, J.A. Recent advances in visible light-activated radical coupling reactions triggered by (i) ruthenium, (ii) iridium and (iii) organic photoredox agents. Chem. Soc. Rev. 2021, 50, 9540–9685. [Google Scholar] [CrossRef]
- Glaser, F.; Wenger, O.S. Recent progress in the development of transition-metal based photoredox catalysts. Coord. Chem. Rev. 2020, 405, 213129. [Google Scholar] [CrossRef]
- Singh, A.; Teegardin, K.; Kelly, M.; Prasad, K.S.; Krishnan, S.; Weaver, J.D. Facile synthesis and complete characterization of homoleptic and heteroleptic cyclometalated Iridium(III) complexes for photocatalysis. J. Organomet. Chem. 2015, 776, 51–59. [Google Scholar] [CrossRef]
- Tritton, D.N.; Tang, F.-K.; Bodedla, G.B.; Lee, F.-W.; Kwan, C.-S.; Leung, K.C.-F.; Zhu, X.; Wong, W.-Y. Development and advancement of iridium(III)-based complexes for photocatalytic hydrogen evolution. Coord. Chem. Rev. 2022, 459, 214390. [Google Scholar] [CrossRef]
- Sakai, K.; Ozawa, H. Homogeneous catalysis of platinum(II) complexes in photochemical hydrogen production from water. Coord. Chem. Rev. 2007, 251, 2753–2766. [Google Scholar] [CrossRef]
- Shi, J.; Su, Z.; Li, X.; Feng, J.; Men, C. Impacts of host–guest assembly on the photophysical and photocatalytic properties of heterogenized molecular photosensitizer and catalysts. J. Mater. Chem. A 2023, 11, 6646–6658. [Google Scholar] [CrossRef]
- Kunzmann, A.; Valero, S.; Sepúlveda, Á.E.; Rico-Santacruz, M.; Lalinde, E.; Berenguer, J.R.; García-Martínez, J.; Guldi, D.M.; Serrano, E.; Costa, R.D. Hybrid Dye-Titania Nanoparticles for Superior Low-Temperature Dye-Sensitized Solar Cells. Adv. Energy Mater. 2018, 8, 1702583. [Google Scholar] [CrossRef]
- Wang, Z.; Li, C.; Domen, K. Recent developments in heterogeneous photocatalysts for solar-driven overall water splitting. Chem. Soc. Rev. 2019, 48, 2109–2125. [Google Scholar] [CrossRef] [PubMed]
- Luo, T.; Gilmanova, L.; Kaskel, S. Advances of MOFs and COFs for photocatalytic CO2 reduction, H2 evolution and organic redox transformations. Coord. Chem. Rev. 2023, 490, 215210. [Google Scholar] [CrossRef]
- Li, Y.-L.; Li, A.-J.; Huang, S.-L.; Vittal, J.J.; Yang, G.-Y. Polypyridyl Ru(ii) or cyclometalated Ir(iii) functionalized architectures for photocatalysis. Chem. Soc. Rev. 2023, 52, 4725–4754. [Google Scholar] [CrossRef]
- Hao, Y.; Lu, Y.-L.; Jiao, Z.; Su, C.-Y. Photocatalysis Meets Confinement: An Emerging Opportunity for Photoinduced Organic Transformations. Angew. Chem. Int. Ed. 2024, 63, e202317808. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Aguirre, M.; Serrano, E.; Ezquerro, C.; Lalinde, E.; Berenguer, J.R.; García-Martínez, J.; Rodríguez, M.A. Hybrid organometallo-silica catalysts for sustainable visible-light promoted olefin isomerization. Catal. Today 2023, 422, 114213. [Google Scholar] [CrossRef]
- Martínez-Aguirre, M.; Herce, J.; Serrano, E.; García-Martínez, J.; Rodríguez, M.A.; Berenguer, J.R. Self-condensed organometallo Ir(III) ionosilica for sustainable visible-light promoted electron-transfer photocatalysis. J. Catal. 2025, 443, 115946. [Google Scholar] [CrossRef]
- Tran Duy, T.; Duarte Rodrigues, A.; Vo-Thanh, G.; Hesemann, P. Dialkyl imidazolium acetate ionosilica as efficient and recyclable organocatalyst for cyanosilylation reactions of ketones. Green Energy Environ. 2020, 5, 130–137. [Google Scholar] [CrossRef]
- Wu, H.; Hesemann, P.; Trens, P.; Silly, G.; Salles, F.; Zajac, J. Ionosilica-based anion exchangers for low-temperature thermochemical storage of energy under mild conditions of adsorbent regeneration and saturation. Chem. Eng. J. 2020, 398, 125634. [Google Scholar] [CrossRef]
- Hesemann, P. Applications of ionosilicas in heterogeneous catalysis: Opportunities for the elaboration of new functional catalytic phases. Curr. Opin. Green Sustain. Chem. 2018, 10, 21–26. [Google Scholar] [CrossRef]
- Materna, K.L.; Hammarström, L. Photoredox Catalysis Using Heterogenized Iridium Complexes. Chem. Eur. J. 2021, 27, 16966–16977. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, D.; Xie, Q.; Xu, C.; Kuang, G.; Tang, J.; Pan, C.; Yu, G. Enhanced Reducibility via Altering Exciton Binding Energy of Conjugated Microporous Polymers for Photocatalytic Reduction. Macromolecules 2023, 56, 4022–4029. [Google Scholar] [CrossRef]
- Devika, S.; Palanichamy, M.; Murugesan, V. Selective oxidation of ethylbenzene over CeAlPO-5. Appl. Catal. A 2011, 407, 76–84. [Google Scholar] [CrossRef]
- Okamura, N.; Nakamura, T.; Yagi, S.; Maeda, T.; Nakazumi, H.; Fujiwara, H.; Koseki, S. Novel bis- and tris-cyclometalated iridium (iii) complexes bearing a benzoyl group on each fluorinated 2-phenylpyridinate ligand aimed at development of blue phosphorescent materials for OLED. RSC Adv. 2016, 6, 51435–51445. [Google Scholar] [CrossRef]
- Millán, G.; Nieddu, M.; López, I.P.; Ezquerro, C.; Berenguer, J.R.; Larráyoz, I.M.; Pichel, J.G.; Lalinde, E. A new family of luminescent iridium complexes: Synthesis, optical, and cytotoxic studies. Dalton Trans. 2023, 52, 6360–6374. [Google Scholar] [CrossRef]
- Ezquerro, C.; Sepulveda, A.E.; Grau-Atienza, A.; Serrano, E.; Lalinde, E.; Berenguer, J.R.; Garcia-Martinez, J. Organometallic phosphors as building blocks in sol-gel chemistry: Luminescent organometallo-silica materials. J. Mater. Chem. C 2017, 5, 9721–9732. [Google Scholar] [CrossRef]
- Ezquerro, C.; López, I.; Serrano, E.; Alfaro-Arnedo, E.; Lalinde Peña, E.; Larráyoz, I.; Pichel, J.G.; García-Martinez, J.; Berenguer, J.R. Highly Emissive Hybrid Mesoporous Organometallo-Silica Nanoparticles for Bioimaging. Mater. Adv. 2022, 3, 3582–3592. [Google Scholar] [CrossRef]
- Bryden, M.A.; Millward, F.; Lee, O.S.; Cork, L.; Gather, M.C.; Steffen, A.; Zysman-Colman, E. Lessons learnt in photocatalysis—The influence of solvent polarity and the photostability of the photocatalyst. Chem. Sci. 2024, 15, 3741–3757. [Google Scholar] [CrossRef]
- Bruker AXS Inc. APEX4, SAINT, and SADABS; Bruker AXS Inc.: Fitchburg, WI, USA, 2021. [Google Scholar]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX suite for small-molecule single-crystal crystallography. J. Appl. Crystallogr. 1999, 32, 837–838. [Google Scholar] [CrossRef]
- Spek, A.L. Single-crystal structure validation with the program PLATON. J. Appl. Crystallogr. 2003, 36, 7–13. [Google Scholar] [CrossRef]
- Spek, A.L. PLATON SQUEEZE: A tool for the calculation of the disordered solvent contribution to the calculated structure factors. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 9–18. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H. Gaussian 16. 3; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Wadt, W.R.; Hay, P.J. Ab initio effective core potentials for molecular calculations. Potentials for main group elements Na to Bi. J. Chem. Phys. 1985, 82, 284–298. [Google Scholar] [CrossRef]
- Barone, V.; Cossi, M. Quantum Calculation of Molecular Energies and Energy Gradients in Solution by a Conductor Solvent Model. J. Phys. Chem. A 1998, 102, 1995–2001. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Tenderholt, A.L.; Langner, K.M. cclib: A library for package-independent computational chemistry algorithms. J. Comput. Chem. 2008, 29, 839–845. [Google Scholar] [CrossRef]


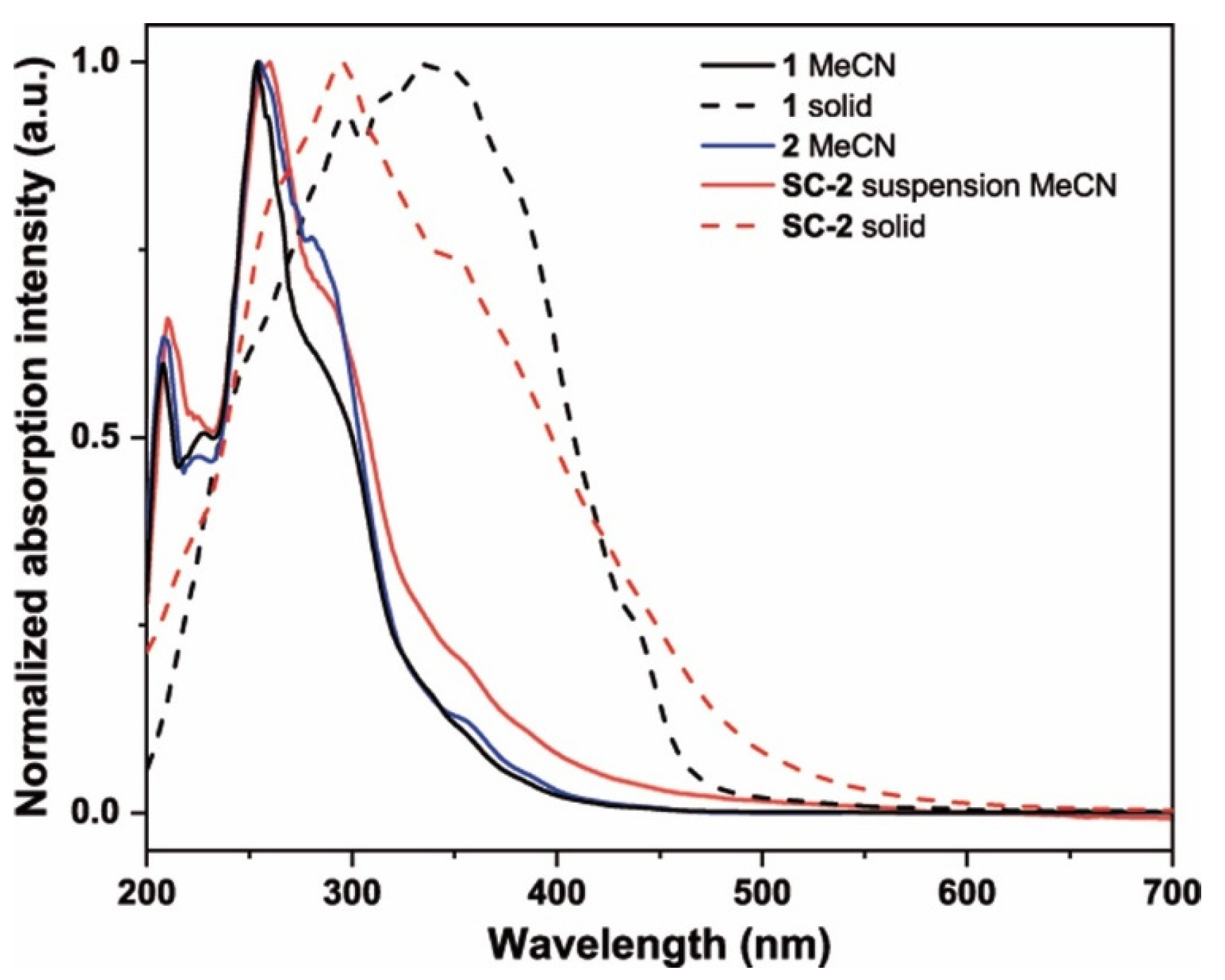

| Sample | Medium | λabs/nm (ε·10−3/M−1·cm−1) |
|---|---|---|
| [Ir(dfbzapy)2(pyraphen)]PF6 (1) | Solid | 245, 290, 341, 380, 440 |
| MeCN 5 × 10−5 M | 255 (20.0), 296 (11.0), 355 (2.2), 385 (0.9), 442 (0.1) | |
| [Ir(Si-dfppy)2(pyraphen)]PF6 (2) | MeCN 5 × 10−5 M | 256 (20.0), 283 (15.4), 360 (2.3), 390 (0.9), 442 (0.1) |
| SC-2 | Solid | 256, 295, 350, 375, 420, 445 |
| Entry | Time (min) | Yield (%) |
|---|---|---|
| 1 | 9 | 93 (±4) 1 |
| 2 2 | 20 | - |
| 3 3 | 20 | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Herce, J.; Martínez-Aguirre, M.; Gómez-Benito, J.; Rodríguez, M.A.; Berenguer, J.R. Sol-Gel Heterogeneization of an Ir(III) Complex for Sustainable Visible-Light Redox Photocatalysis. Molecules 2025, 30, 1680. https://doi.org/10.3390/molecules30081680
Herce J, Martínez-Aguirre M, Gómez-Benito J, Rodríguez MA, Berenguer JR. Sol-Gel Heterogeneization of an Ir(III) Complex for Sustainable Visible-Light Redox Photocatalysis. Molecules. 2025; 30(8):1680. https://doi.org/10.3390/molecules30081680
Chicago/Turabian StyleHerce, Janira, Mónica Martínez-Aguirre, Javier Gómez-Benito, Miguel A. Rodríguez, and Jesús R. Berenguer. 2025. "Sol-Gel Heterogeneization of an Ir(III) Complex for Sustainable Visible-Light Redox Photocatalysis" Molecules 30, no. 8: 1680. https://doi.org/10.3390/molecules30081680
APA StyleHerce, J., Martínez-Aguirre, M., Gómez-Benito, J., Rodríguez, M. A., & Berenguer, J. R. (2025). Sol-Gel Heterogeneization of an Ir(III) Complex for Sustainable Visible-Light Redox Photocatalysis. Molecules, 30(8), 1680. https://doi.org/10.3390/molecules30081680