
N,N- and N,O-Bidentate-Chelation-Assisted Alkenyl C–H Functionalization

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
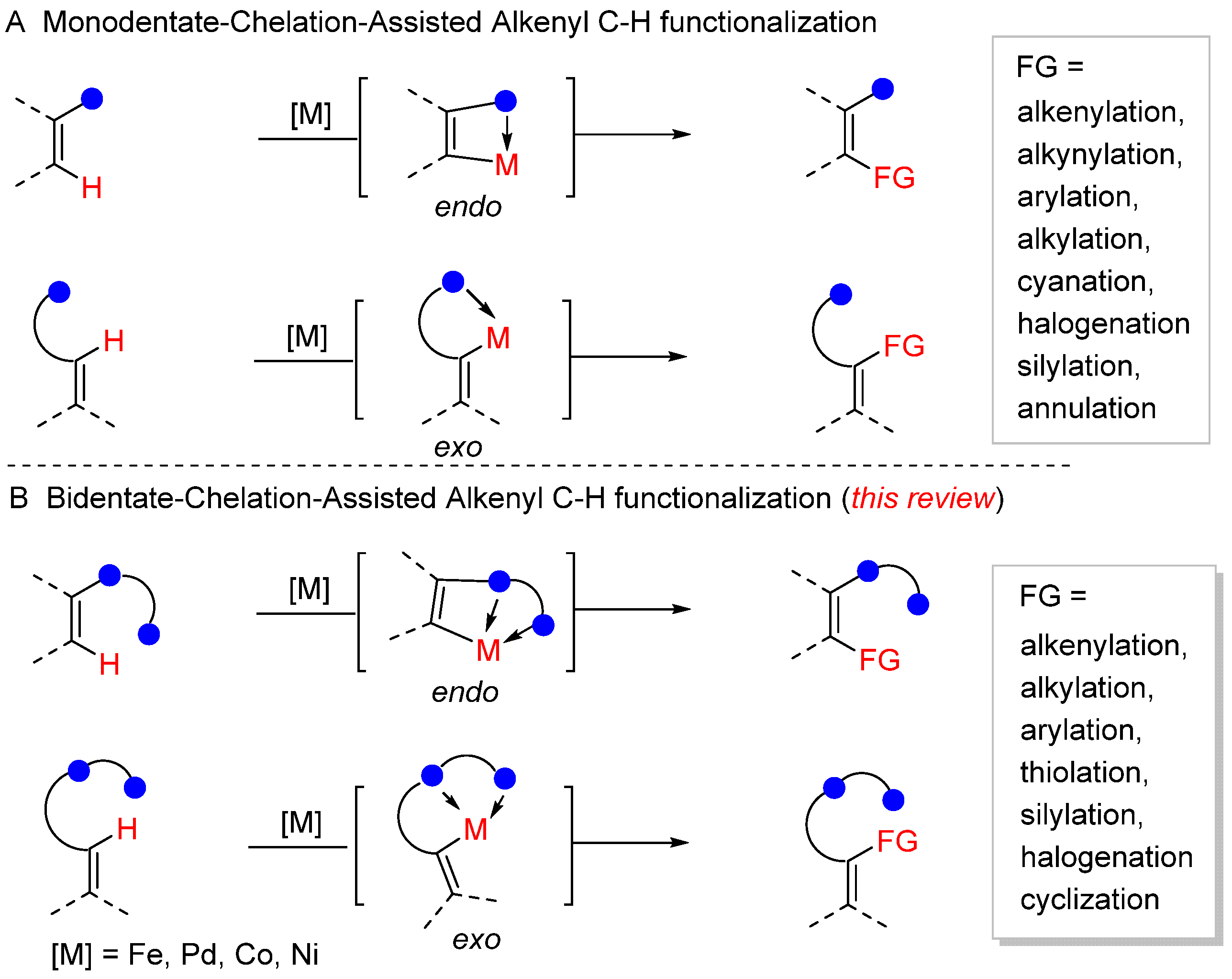
1. Introduction
2. Bidentate-Chelation-Assisted Alkenyl C–H Functionalization
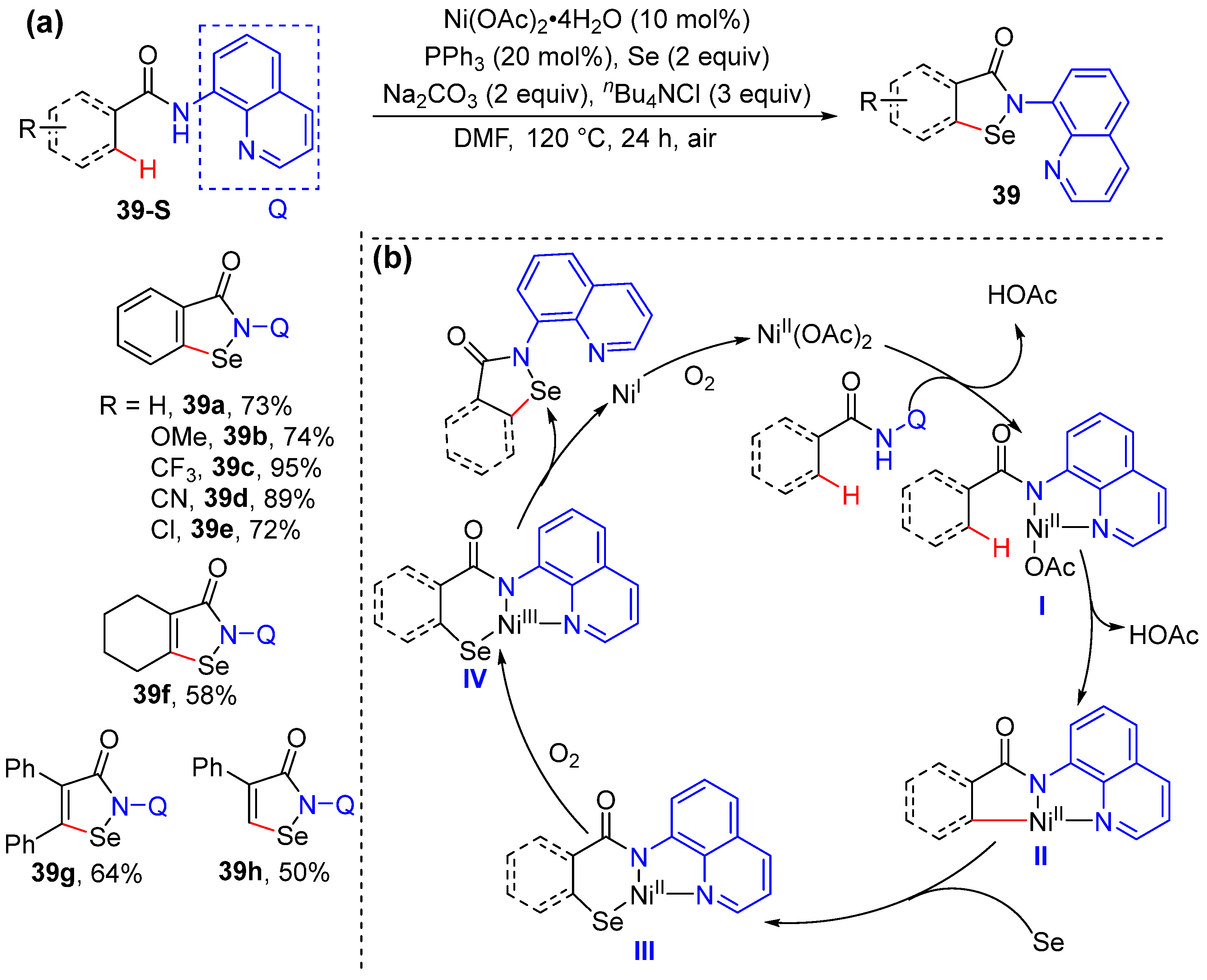
2.1. N,N-Bidentate-Chelation-Assisted Alkenyl C–H Functionalization
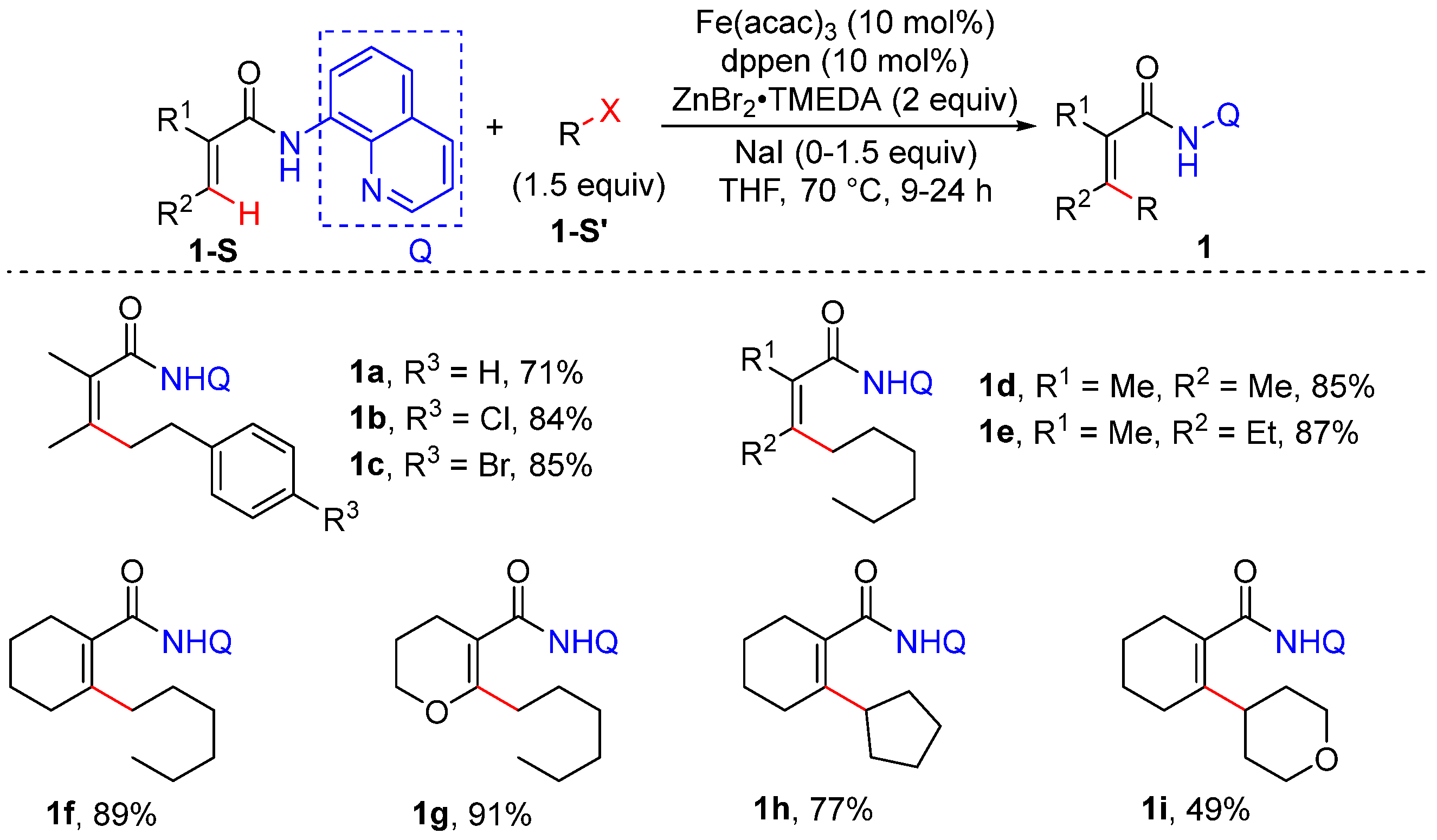
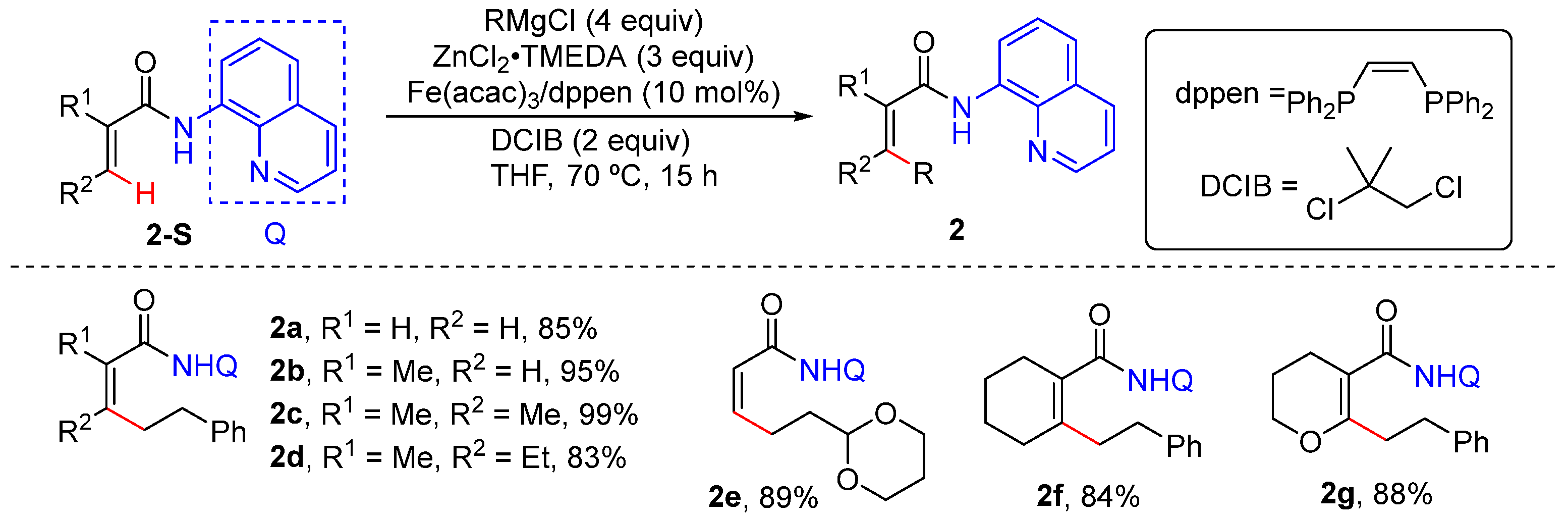
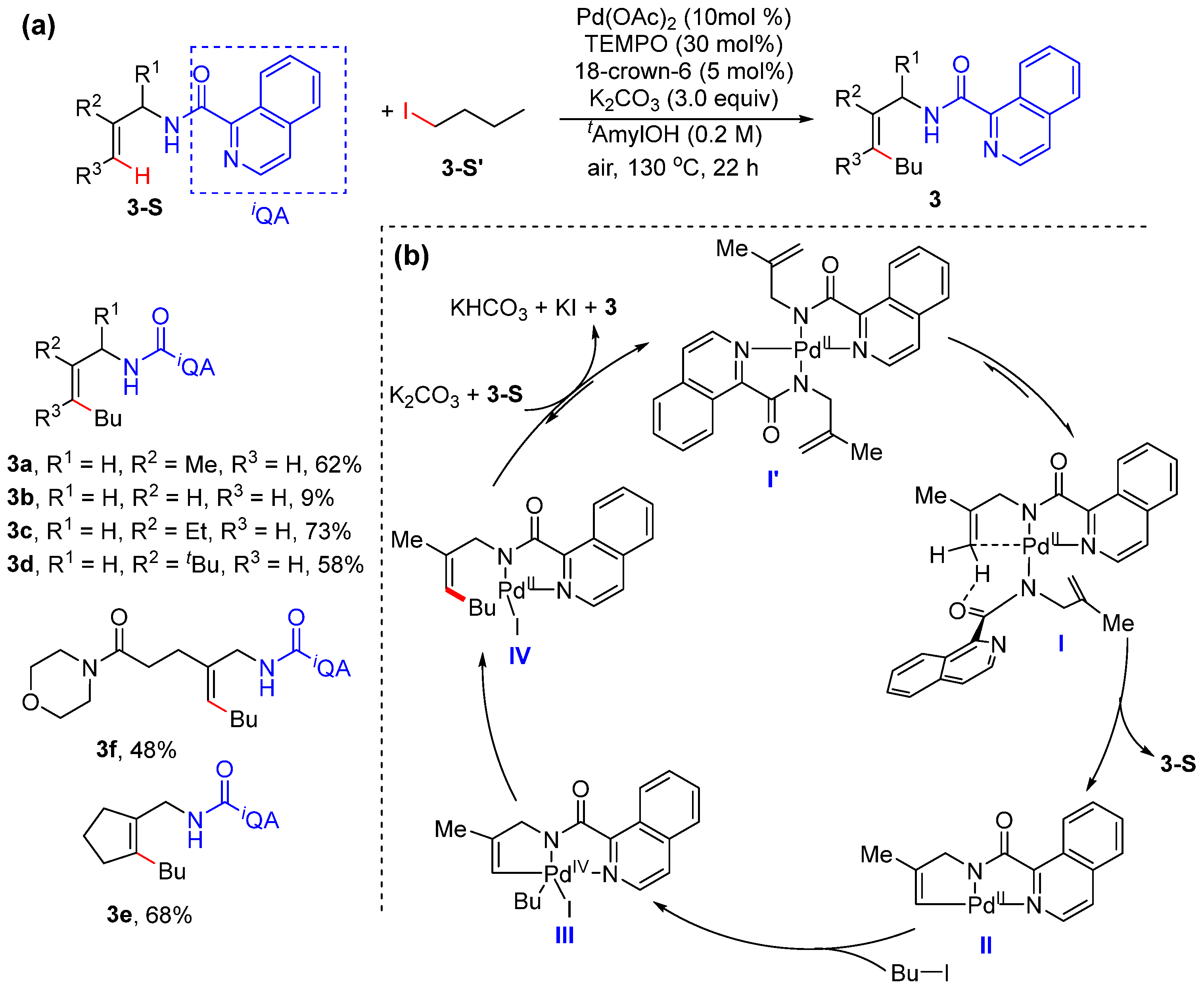
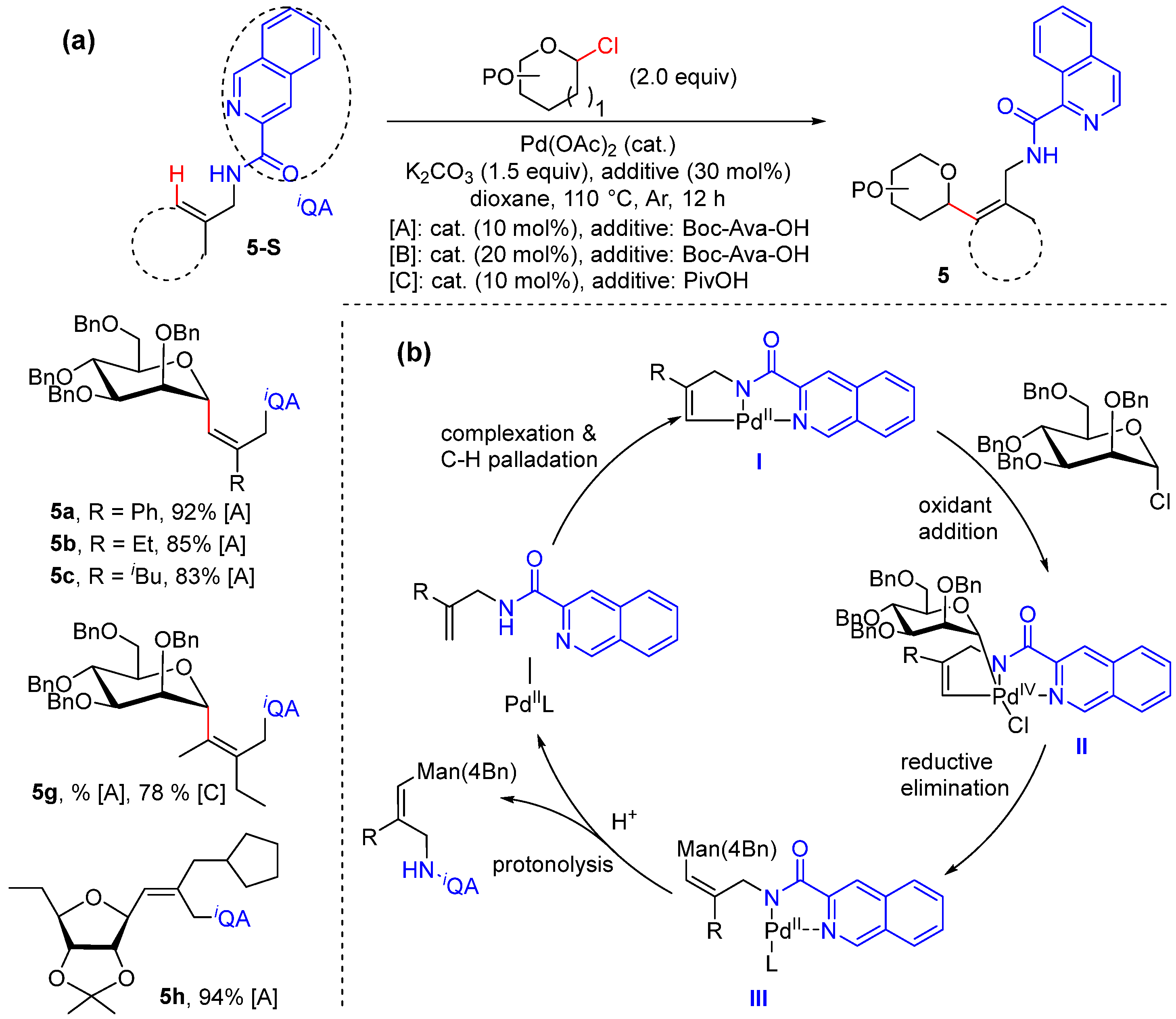
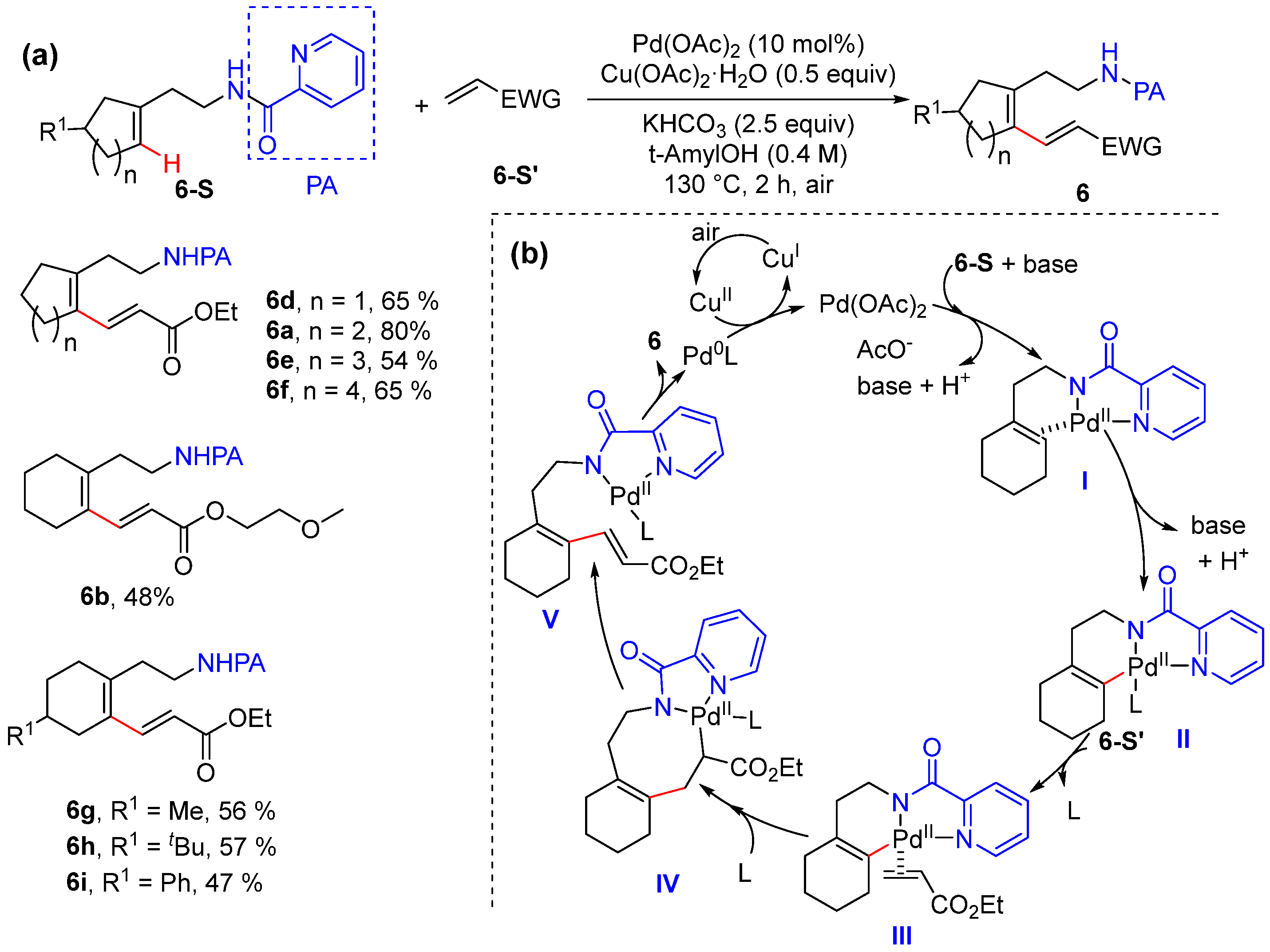
Alkenyl C–H Alkylation
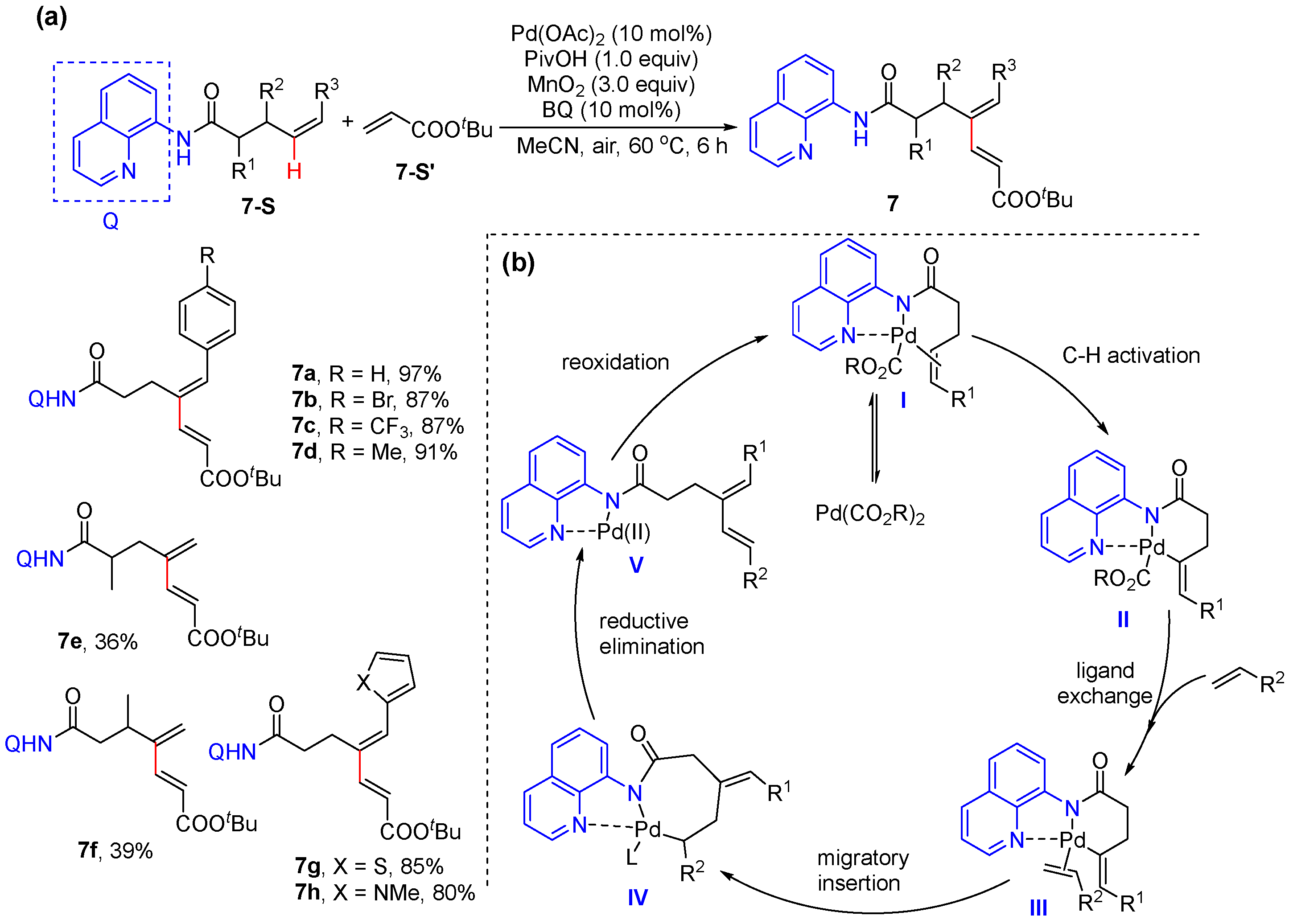
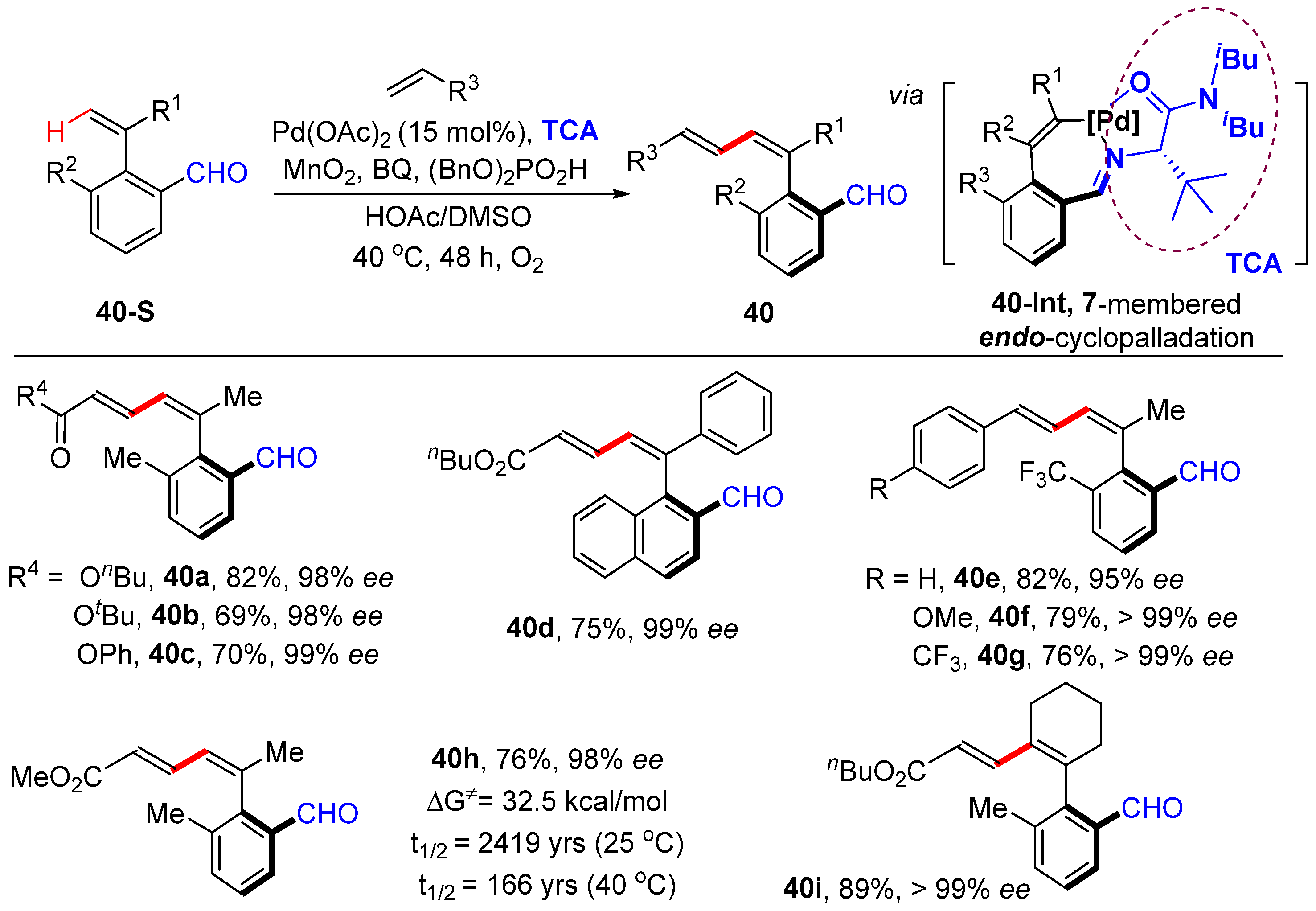
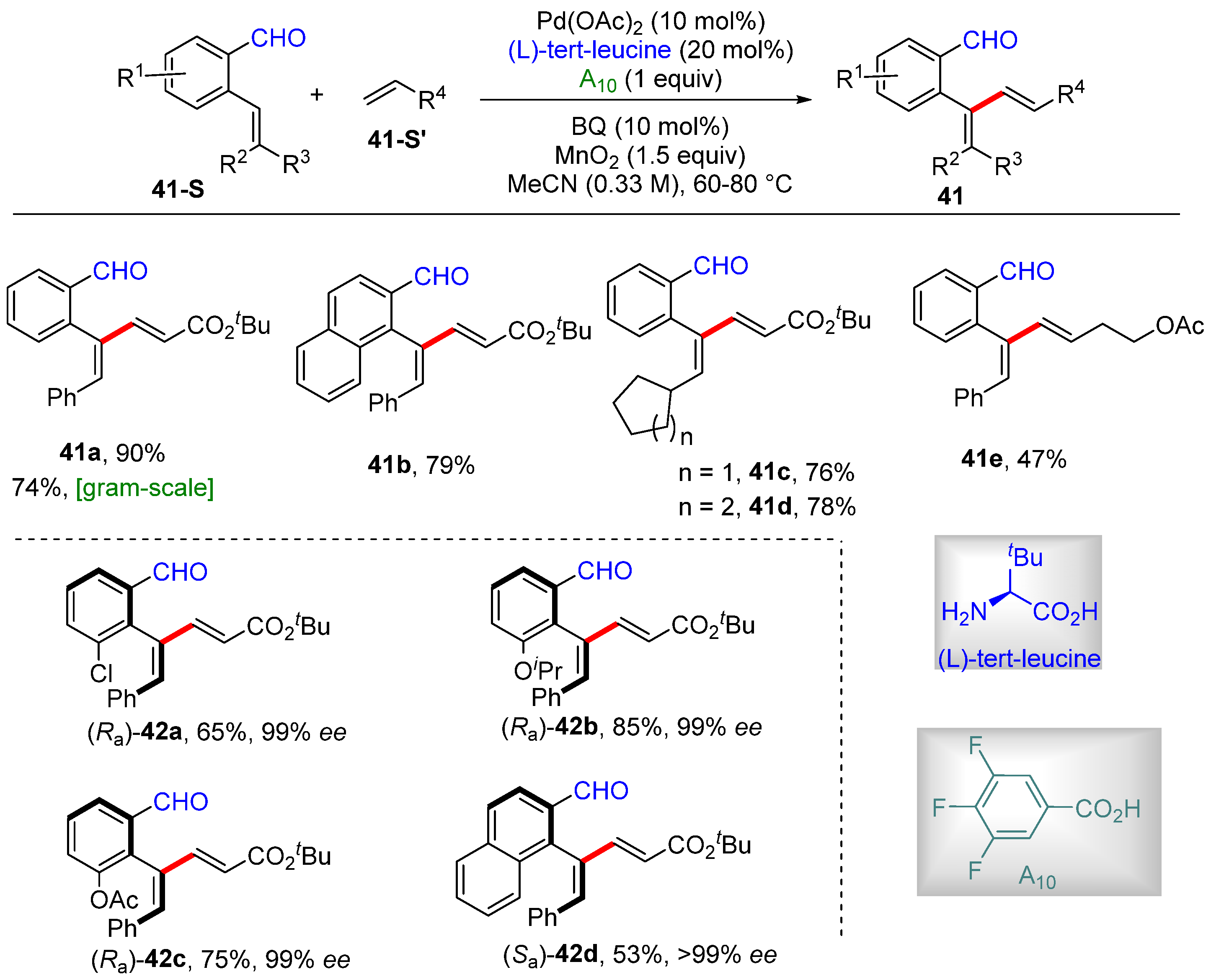
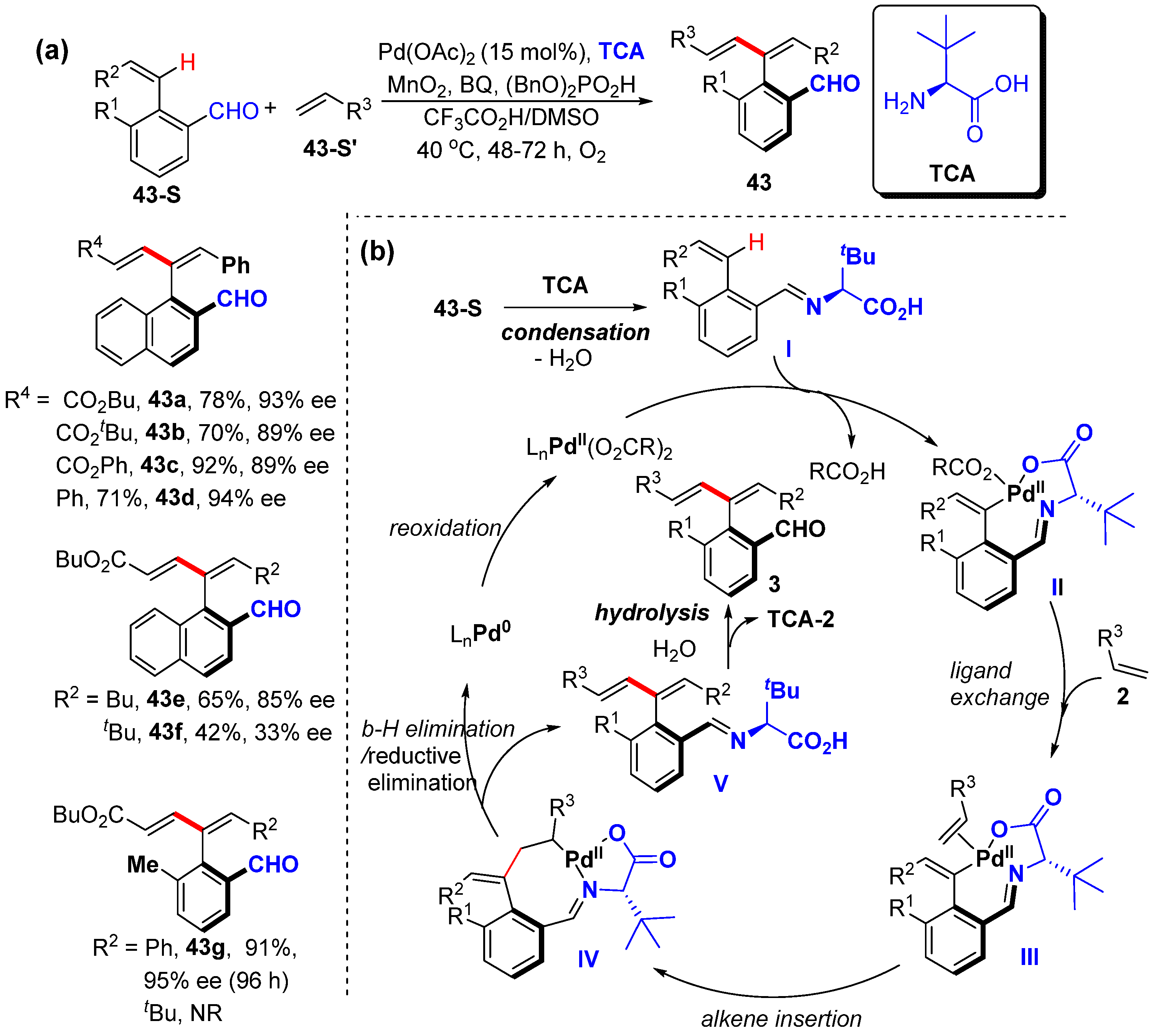
2.2. Alkenyl C–H Alkenylation
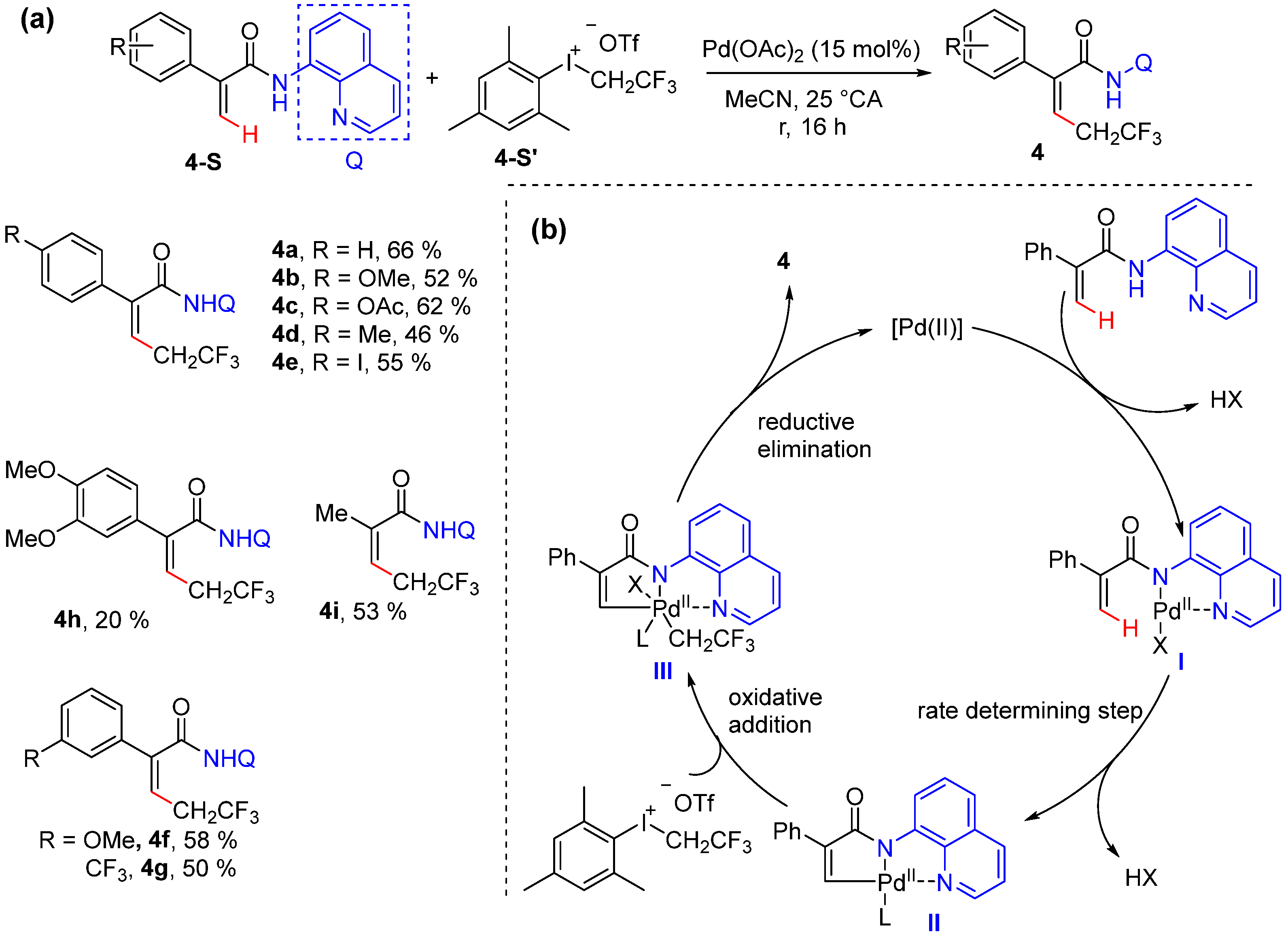
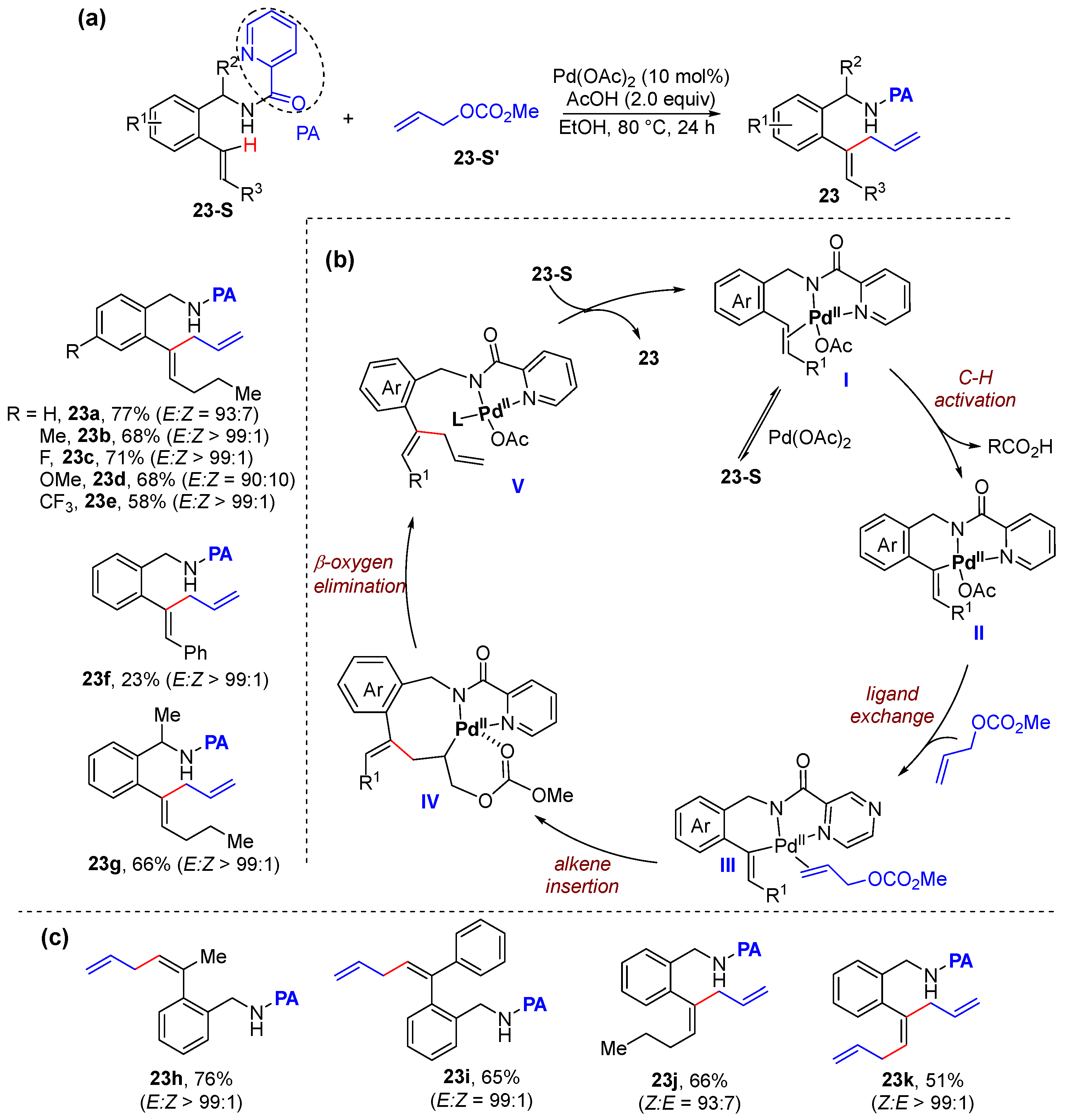
2.3. Alkenyl C–H Allylation
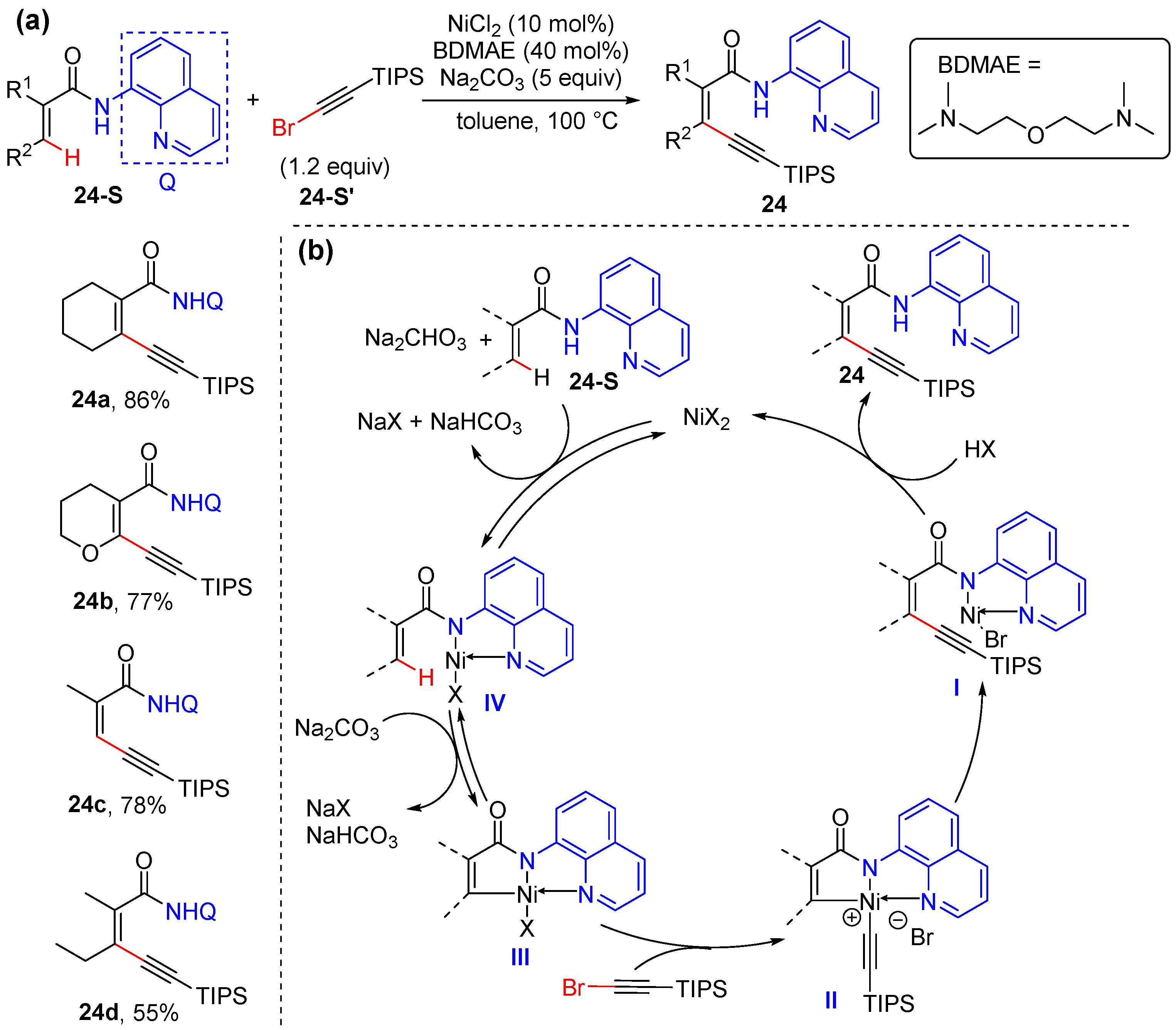
2.4. Alkenyl C–H Alkynylation
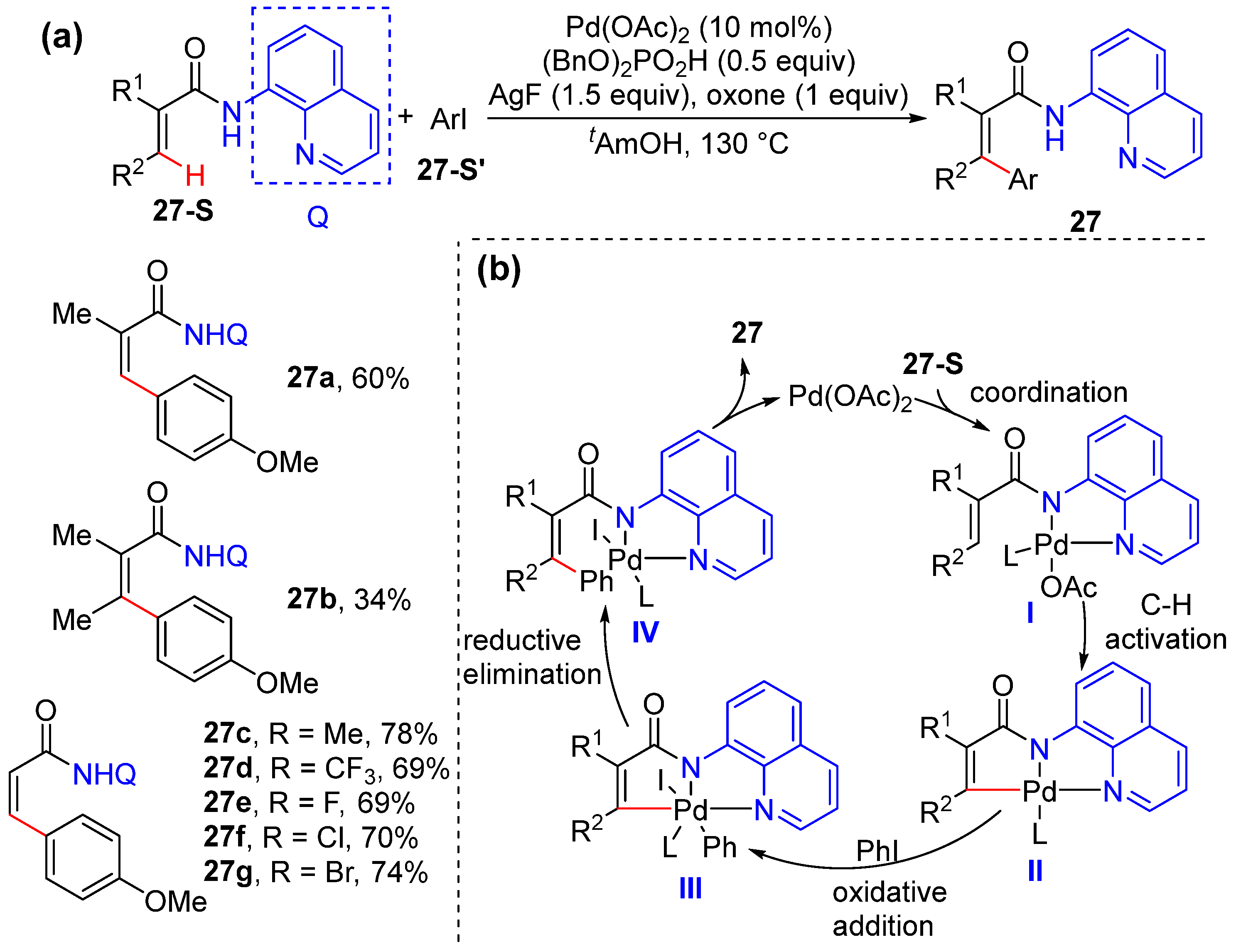
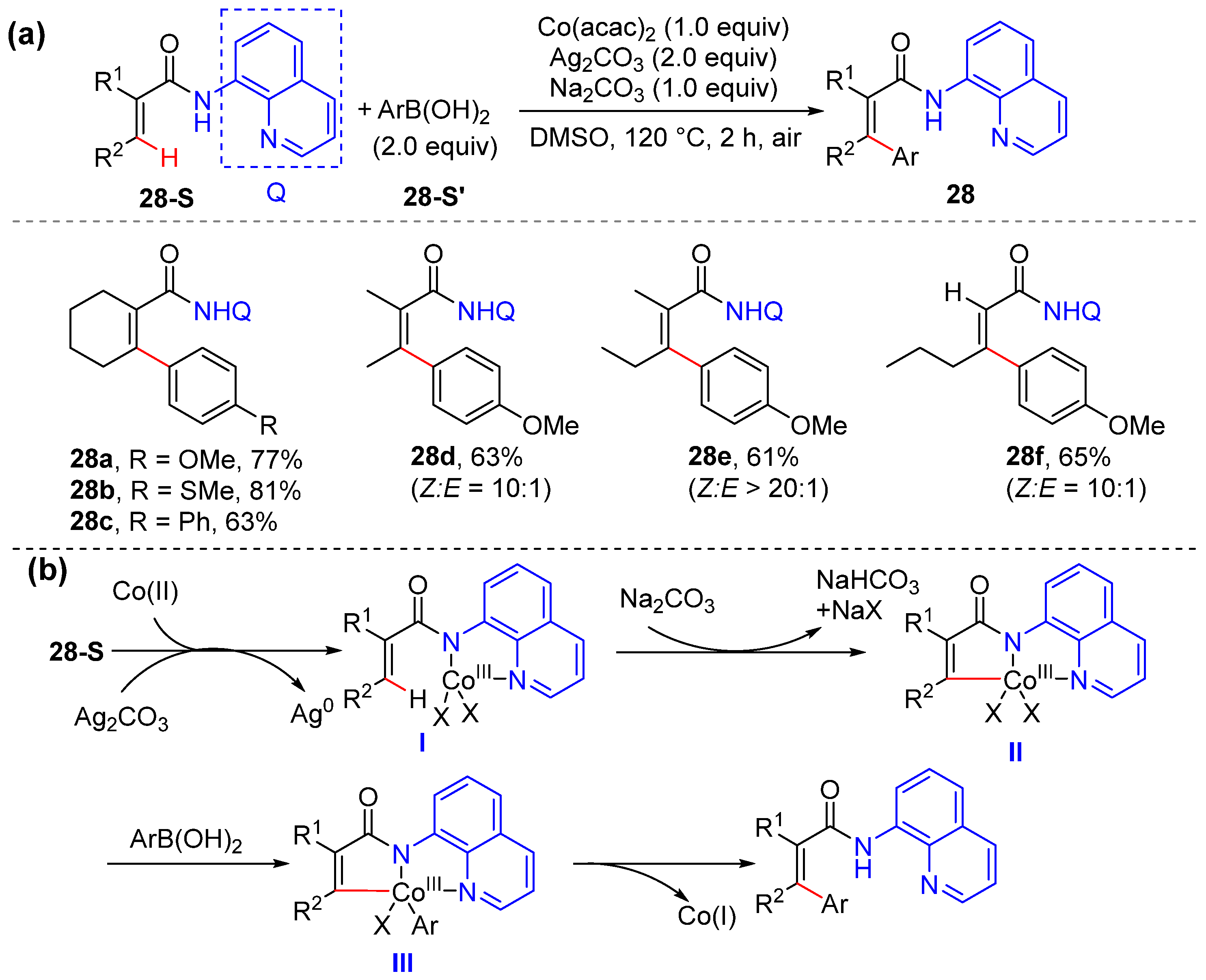
2.5. Alkenyl C–H Arylation
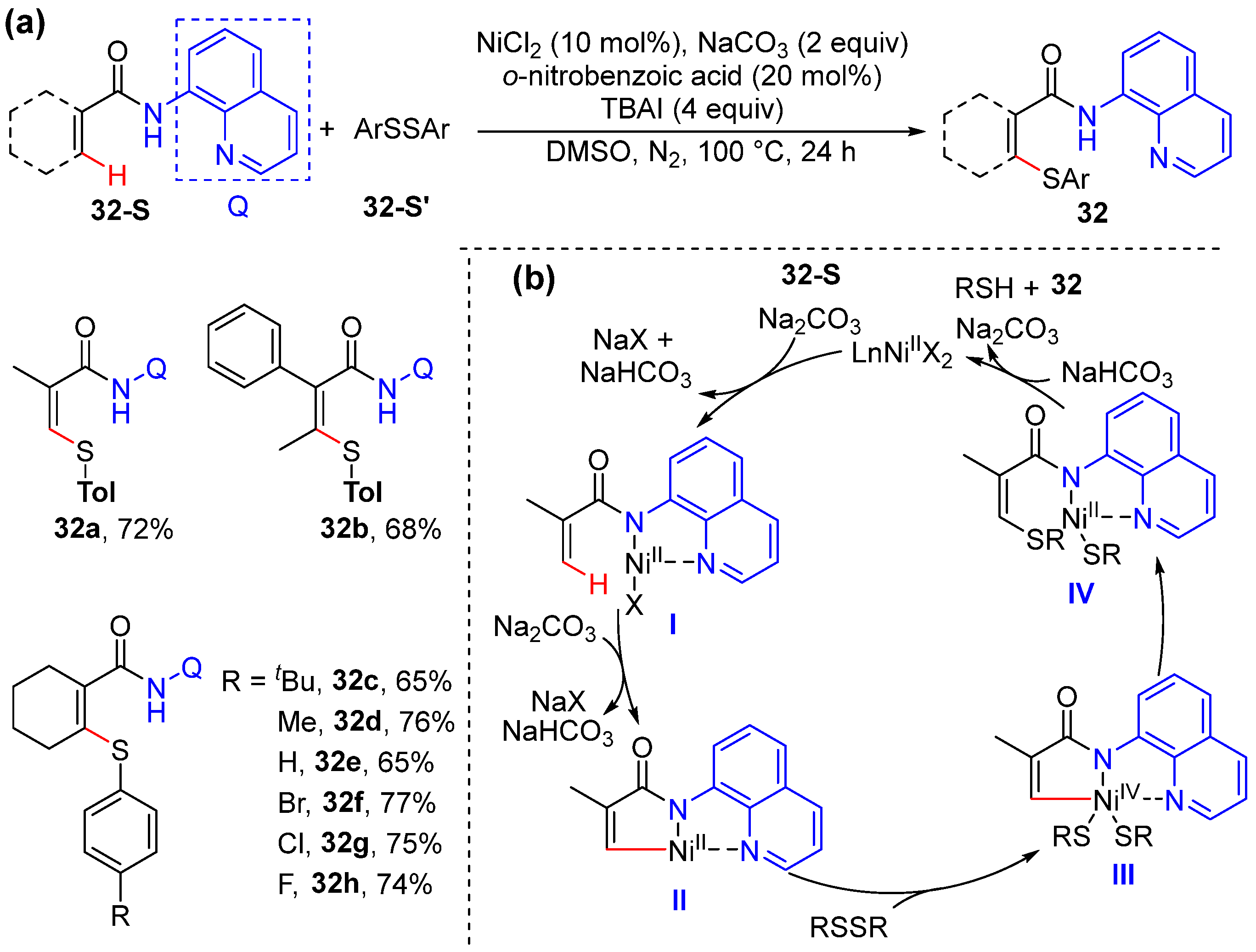
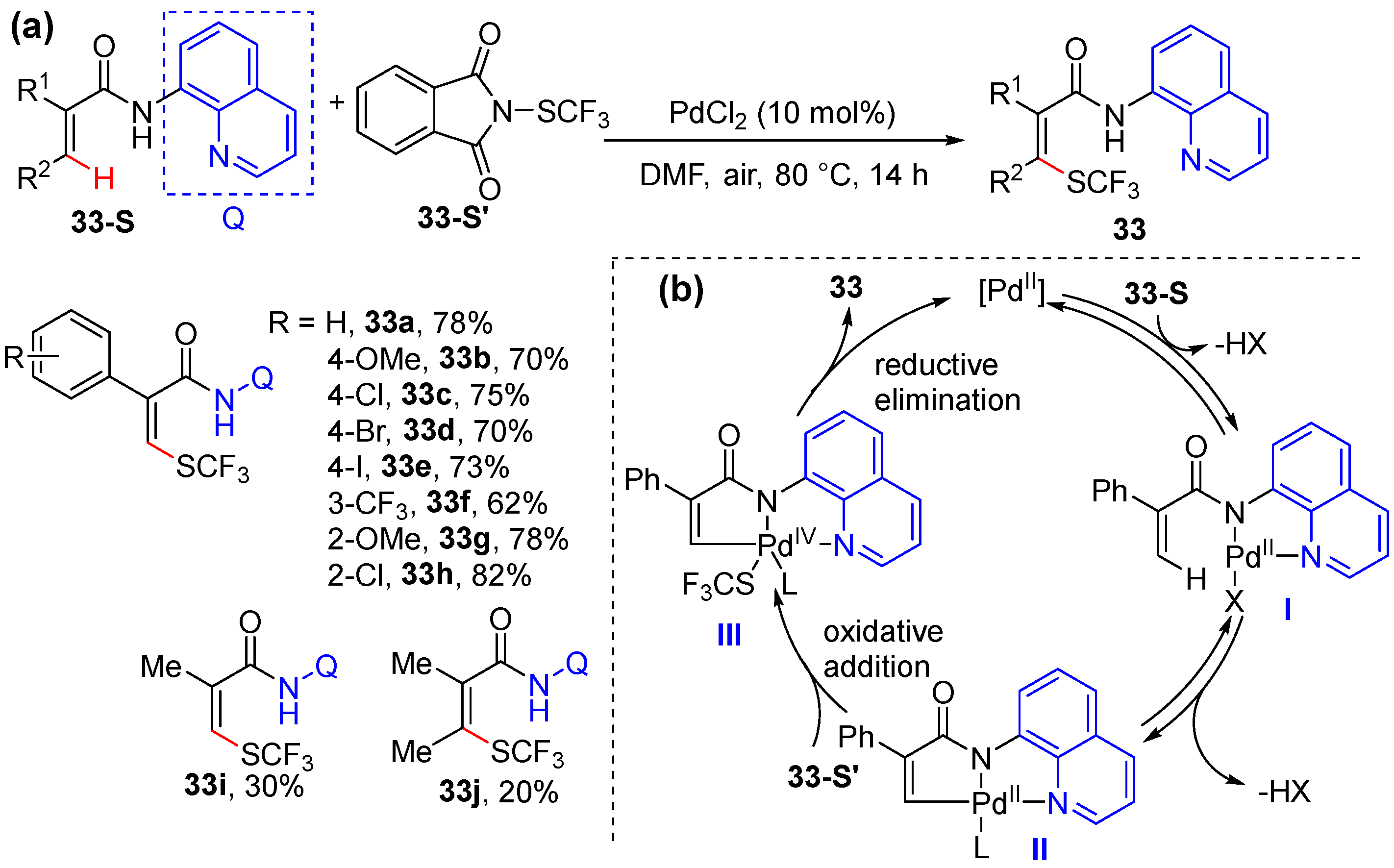
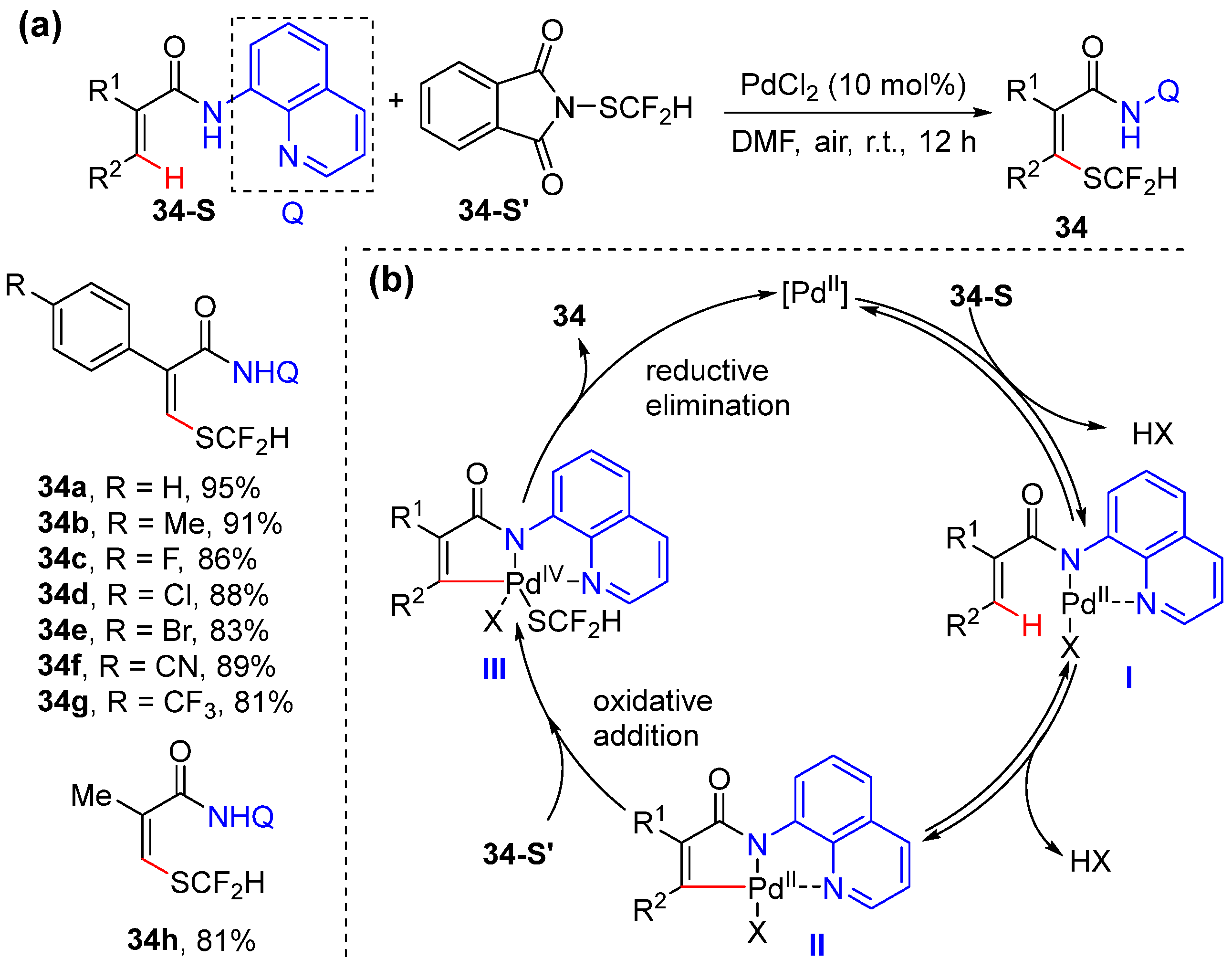
2.6. Alkenyl C–H Thiolation
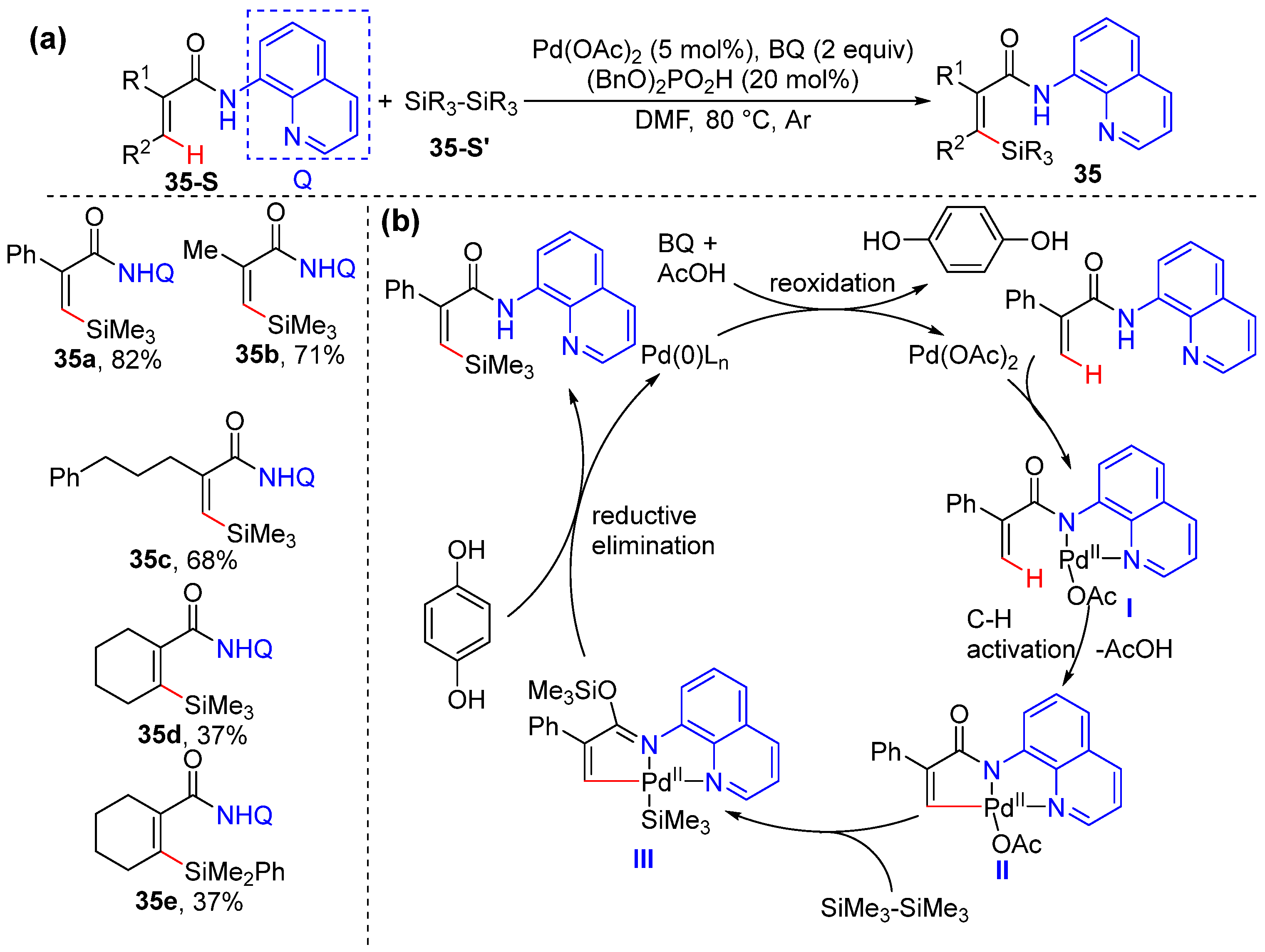
2.7. Alkenyl C–H Silylation
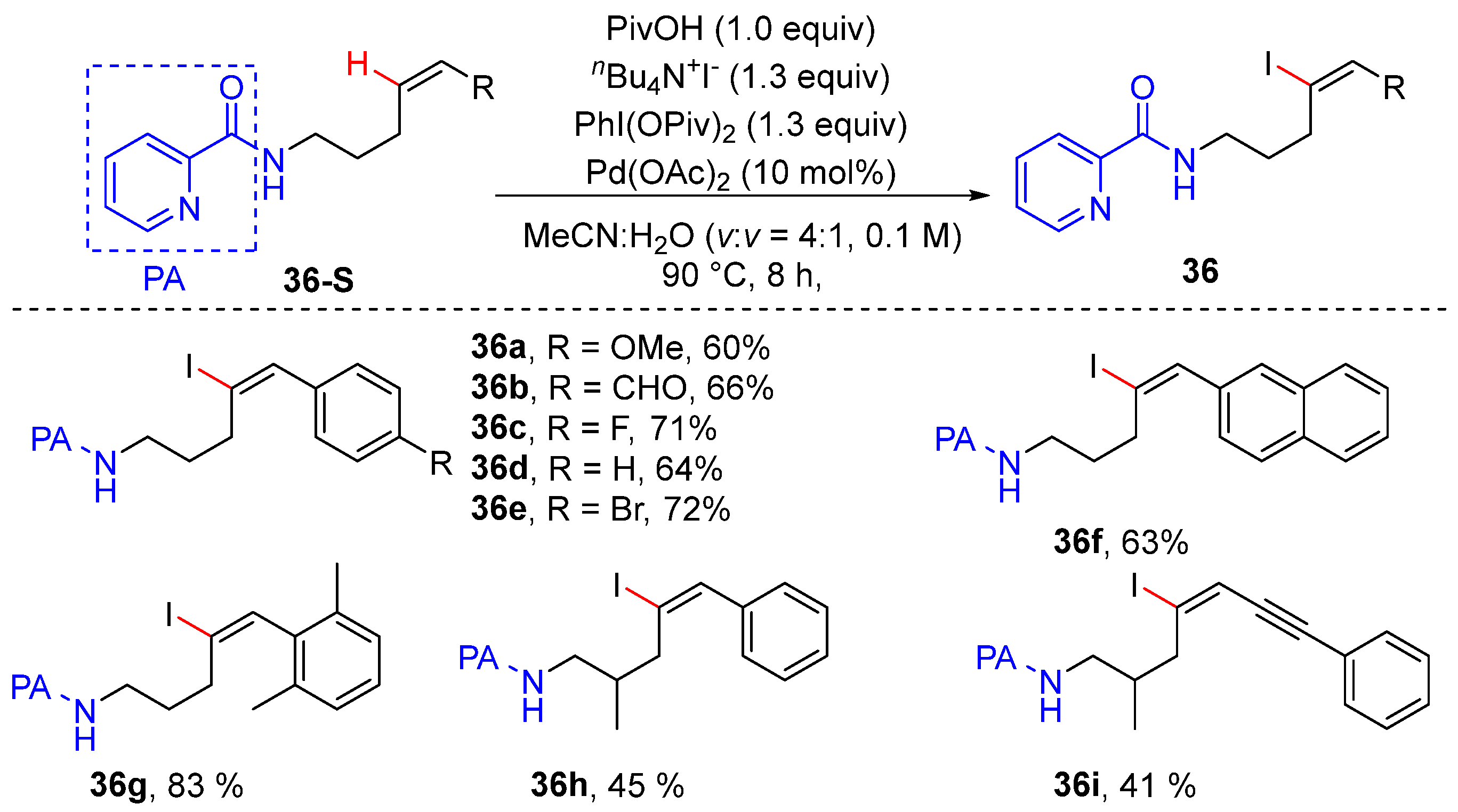
2.8. Alkenyl C–H Halogenation
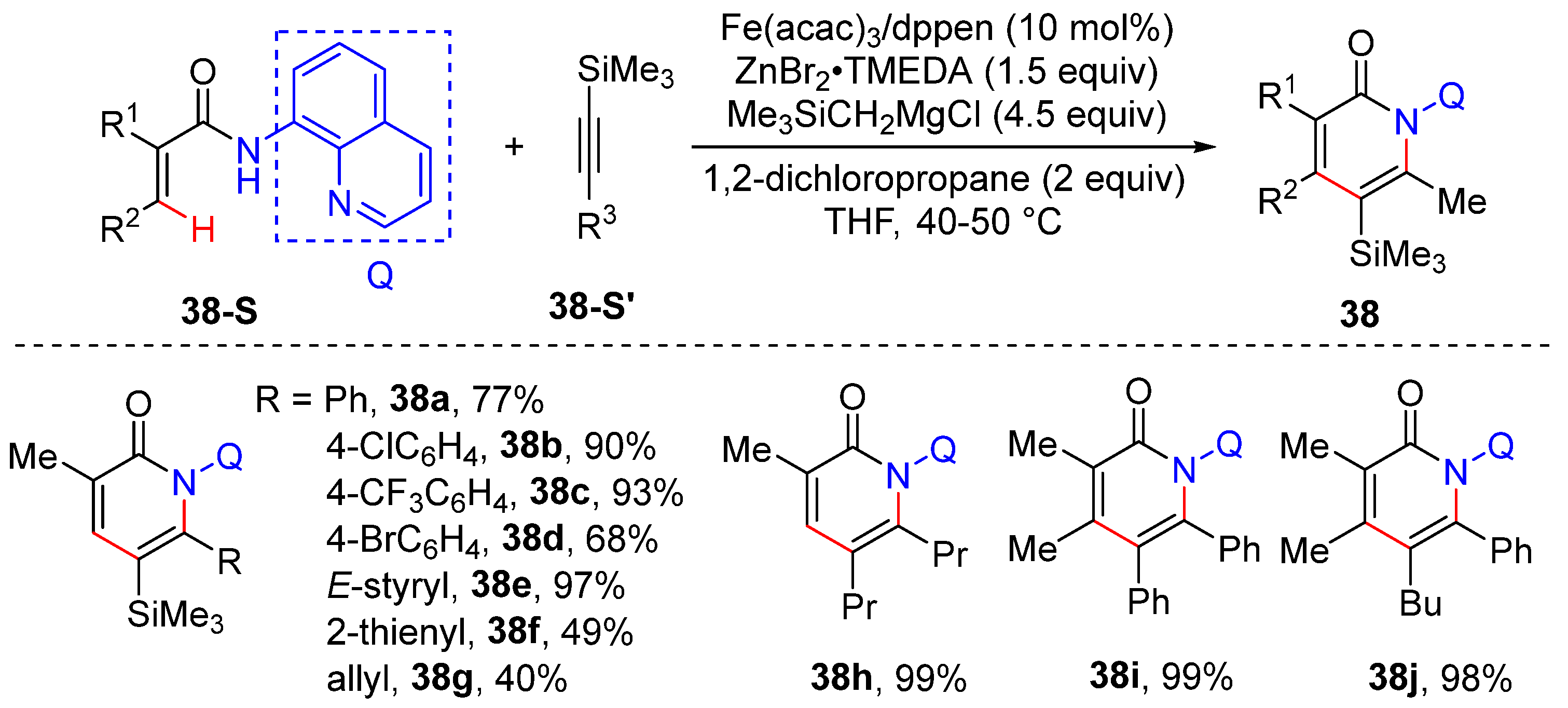
2.9. Cyclization by Alkenyl C–H Functionalization
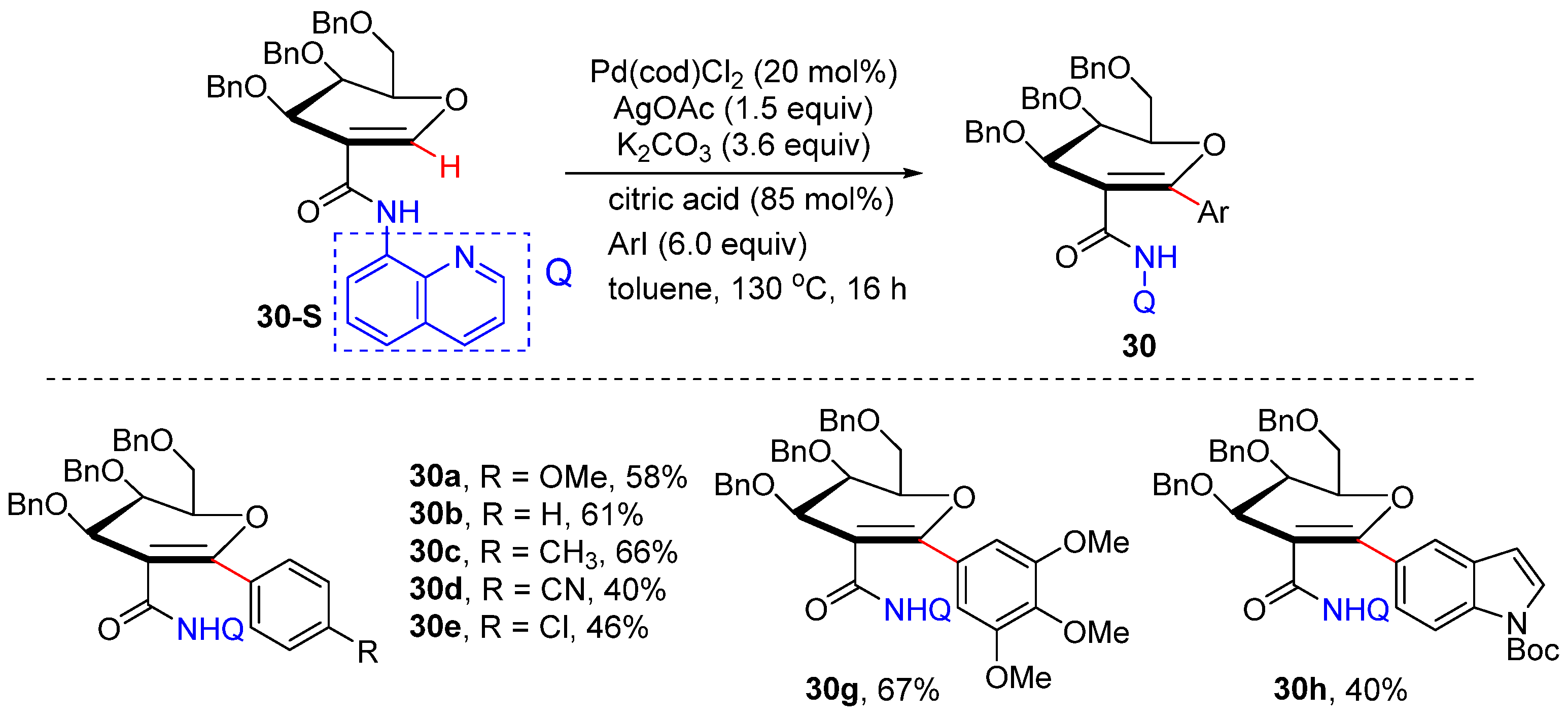
2.10. N,O-Bidentate-Chelation-Assisted Alkenyl C–H Functionalization
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Newton, C.G.; Wang, S.-G.; Oliveira, C.C.; Cramer, N. Catalytic Enantioselective Transformations Involving C–H Bond Cleavage by Transition-Metal Complexes. Chem. Rev. 2017, 117, 8908–8976. [Google Scholar] [CrossRef] [PubMed]
- Engle, K.M.; Mei, T.-S.; Wasa, M.; Yu, J.-Q. Weak Coordination as a Powerful Means for Developing Broadly Useful C–H Functionalization Reactions. Acc. Chem. Res. 2012, 45, 788–802. [Google Scholar] [CrossRef]
- Colby, D.A.; Bergman, R.G.; Ellman, J.A. Rhodium-Catalyzed C−C Bond Formation via Heteroatom-Directed C–H Bond Activation. Chem. Rev. 2010, 110, 624–655. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, L. Carboxylate-Assisted Transition-Metal-Catalyzed C–H Bond Functionalizations: Mechanism and Scope. Chem. Rev. 2011, 111, 1315–1345. [Google Scholar] [CrossRef] [PubMed]
- Arockiam, P.B.; Bruneau, C.; Dixneuf, P.H. Ruthenium(II)-Catalyzed C–H Bond Activation and Functionalization. Chem. Rev. 2012, 112, 5879–5918. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.K.; Wang, B.J.; Zhang, J.T.; Yu, W.L.; Liu, Z.X.; Zhang, Y.H. Transition metal-catalyzed C–H bond functionalizations by the use of diverse directing groups. Org. Chem. Front. 2015, 2, 1107–1295. [Google Scholar] [CrossRef]
- Daugulis, O.; Roane, J.; Tran, L.D. Bidentate, Monoanionic Auxiliary-Directed Functionalization of Carbon–Hydrogen Bonds. Acc. Chem. Res. 2015, 48, 1053–1064. [Google Scholar] [CrossRef] [PubMed]
- Rej, S.; Chatani, N. Rhodium-Catalyzed C(sp2)- or C(sp3)−H Bond Functionalization Assisted by Removable Directing Groups. Angew. Chem. Int. Ed. 2019, 58, 8304–8329. [Google Scholar] [CrossRef]
- Gandeepan, P.; Müller, T.; Zell, D.; Cera, G.; Warratz, S.; Ackermann, L. 3d Transition Metals for C–H Activation. Chem. Rev. 2019, 119, 2192–2452. [Google Scholar] [CrossRef] [PubMed]
- Mandal, R.; Garai, B.; Sundararaju, B. Weak-Coordination in C–H Bond Functionalizations Catalyzed by 3d Metals. ACS Catal. 2022, 12, 3452–3506. [Google Scholar] [CrossRef]
- Lyons, T.W.; Sanford, M.S. Palladium-Catalyzed Ligand-Directed C–H Functionalization Reactions. Chem. Rev. 2010, 110, 1147–1169. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Spring, D.R. Arene C–H functionalisation using a removable/modifiable or a traceless directing group strategy. Chem. Soc. Rev. 2014, 43, 6906–6919. [Google Scholar] [CrossRef] [PubMed]
- Yadav, M.R.; Rit, R.K.; Shankar, M.; Sahoo, A.K. Reusable and Removable Directing Groups for C(sp2)−H Bond Functionalization of Arenes. Asian J. Org. Chem. 2015, 4, 846–864. [Google Scholar] [CrossRef]
- Dey, A.; Sinha, S.K.; Achar, T.K.; Maiti, D. Accessing Remote meta- and para-C(sp2)−H Bonds with Covalently Attached Directing Groups. Angew. Chem. Int. Ed. 2019, 58, 10820–10843. [Google Scholar] [CrossRef]
- He, J.; Wasa, M.; Chan, K.S.L.; Shao, Q.; Yu, J.-Q. Palladium-Catalyzed Transformations of Alkyl C–H Bonds. Chem. Rev. 2017, 117, 8754–8786. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Romine, A.M.; Rubel, C.Z.; Engle, K.M.; Shi, B.-F. Transition-Metal-Catalyzed, Coordination-Assisted Functionalization of Nonactivated C(sp3)–H Bonds. Chem. Rev. 2021, 121, 14957–15074. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Hu, F.D.; Zhang, Y.; Wang, J.B. Directing group-assisted transition-metal-catalyzed vinylic C-H bond functionalization. Sci. China Chem. 2015, 58, 1252–1265. [Google Scholar] [CrossRef]
- Zhang, J.; Lu, X.N.; Shen, C.; Xu, L.Y.; Ding, L.Y.; Zhong, G.F. Recent advances in chelation-assisted site- and stereoselective alkenyl C–H functionalization. Chem. Soc. Rev. 2021, 50, 3263–3314. [Google Scholar] [CrossRef] [PubMed]
- Yazhou Wang, Y.Z. Lixia Xu, Rui He, Jian Zhang, Recent Advances in Geminal-Group-Directed Alkenyl C—H Functionalization. Chin. J. Org. Chem. 2022, 42, 2000–2014. [Google Scholar] [CrossRef]
- Liu, B.; Yang, L.; Li, P.; Wang, F.; Li, X. Recent advances in transition metal-catalyzed olefinic C–H functionalization. Org. Chem. Front. 2021, 8, 1085–1101. [Google Scholar] [CrossRef]
- Sambiagio, C.; Schönbauer, D.; Blieck, R.; Dao-Huy, T.; Pototschnig, G.; Schaaf, P.; Wiesinger, T.; Zia, M.F.; Wencel-Delord, J.; Besset, T.; et al. A comprehensive overview of directing groups applied in metal-catalysed C–H functionalisation chemistry. Chem. Soc. Rev. 2018, 47, 6603–6743. [Google Scholar] [PubMed]
- Zhu, Y.H.; Liao, Y.L.; Jin, S.Q.; Ding, L.Y.; Zhong, G.F.; Zhang, J. Functionality-Directed Regio- and Enantio-Selective Olefinic C–H Functionalization of Aryl Alkenes. Chem. Rec. 2023, 23, e202300012. [Google Scholar] [CrossRef]
- Lu, X.; Wang, Y.N.; Xie, K.L.; Zhang, J. Cross-Coupling Reactions between Alkenes by C–H Cyclometalation. Synlett 2023, 34, 2262–2292. [Google Scholar]
- Kodama, H.; Katsuhira, T.; Nishida, T.; Hino, T.; Tsubata, K. Process for the Preparation of 2-Halobenzoic Acids. Worldwide Patent WO 2001083421 A1, 2001, 27 April 2001. [Google Scholar]
- Zaitsev, V.G.; Shabashov, D.; Daugulis, O. Highly Regioselective Arylation of sp3 C–H Bonds Catalyzed by Palladium Acetate. J. Am. Chem. Soc. 2005, 127, 13154–13155. [Google Scholar] [CrossRef]
- Rej, S.; Ano, Y.; Chatani, N. Bidentate Directing Groups: An Efficient Tool in C–H Bond Functionalization Chemistry for the Expedient Construction of C–C Bonds. Chem. Rev. 2020, 120, 1788–1887. [Google Scholar] [CrossRef] [PubMed]
- Lapuh, M.I.; Mazeh, S.; Besset, T. Chiral Transient Directing Groups in Transition-Metal-Catalyzed Enantioselective C–H Bond Functionalization. ACS Catalysis 2020, 10, 12898–12919. [Google Scholar] [CrossRef]
- Bag, D.; Verma, P.K.; Sawant, S.D. Chiral Transient Directing Group Strategies in Asymmetric Synthesis. Chem. Asian J. 2020, 15, 3225–3238. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.L.; Hong, K.; Li, T.J.; Park, H.; Yu, J.Q. Functionalization of C(sp3)–H bonds using a transient directing group. Science 2016, 351, 252–256. [Google Scholar] [CrossRef]
- Ilies, L.; Matsubara, T.; Ichikawa, S.; Asako, S.; Nakamura, E. Iron-Catalyzed Directed Alkylation of Aromatic and Olefinic Carboxamides with Primary and Secondary Alkyl Tosylates, Mesylates, and Halides. J. Am. Chem. Soc. 2014, 136, 13126–13129. [Google Scholar] [CrossRef] [PubMed]
- Ilies, L.; Ichikawa, S.; Asako, S.; Matsubara, T.; Nakamura, E. Iron-Catalyzed Directed Alkylation of Alkenes and Arenes with Alkylzinc Halides. Adv. Synth. Catal. 2015, 357, 2175–2179. [Google Scholar] [CrossRef]
- Luo, Y.C.; Yang, C.; Qiu, S.Q.; Liang, Q.J.; Xu, Y.H.; Loh, T.P. Palladium(II)-Catalyzed Stereospecific Alkenyl C–H Bond Alkylation of Allylamines with Alkyl Iodides. ACS Catal. 2019, 9, 4271–4276. [Google Scholar] [CrossRef]
- Ruyet, L.; Lapuh, M.I.; Koshti, V.S.; Földesi, T.; Jubault, P.; Poisson, T.; Novák, Z.; Besset, T. Z-Selective Pd-catalyzed 2,2,2-trifluoroethylation of acrylamides at room temperature. Chem. Commun. 2021, 57, 6241–6244. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Zhang, H.; Wang, Q.; Qiao, T.; He, G.; Chen, G. Stereoselective Synthesis of C-Vinyl Glycosides via Palladium-Catalyzed C–H Glycosylation of Alkenes. Angew. Chem. Int. Ed. 2021, 60, 19620–19625. [Google Scholar] [CrossRef] [PubMed]
- Mao, C.L.; Zhao, S.; Zang, Z.L.; Xiao, L.; Zhou, C.H.; He, Y.; Cai, G.X. Pd-Catalyzed Remote Site-Selective and Stereoselective C(Alkenyl)–H Alkenylation of Unactivated Cycloalkenes. J. Org. Chem. 2020, 85, 774–787. [Google Scholar] [CrossRef]
- Liu, M.; Yang, P.; Karunananda, M.K.; Wang, Y.; Liu, P.; Engle, K.M. C(alkenyl)–H Activation via Six-Membered Palladacycles: Catalytic 1,3-Diene Synthesis. J. Am. Chem. Soc. 2018, 140, 5805–5813. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Hirano, K.; Miura, M. Pd-Catalyzed Regioselective C–H Alkenylation and Alkynylation of Allylic Alcohols with the Assistance of a Bidentate Phenanthroline Auxiliary. Org. Lett. 2020, 22, 9059–9064. [Google Scholar] [CrossRef] [PubMed]
- Schreib, B.S.; Son, M.; Aouane, F.A.; Baik, M.-H.; Carreira, E.M. Allene C(sp2)–H Activation and Alkenylation Catalyzed by Palladium. J. Am. Chem. Soc. 2021, 143, 21705–21712. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; Zhu, Y.H.; Jin, S.Q.; Xu, K.J.; Luo, S.X.; Xu, L.X.; Zhong, G.F.; Zhong, L.J.; Zhang, J. Regio- and stereo-selective olefinic C–H functionalization of aryl alkenes in ethanol. Org. Chem. Front. 2022, 9, 989–994. [Google Scholar] [CrossRef]
- Zhu, Y.H.; Wang, Y.N.; Shen, W.Z.; Chen, X.Y.; Liu, Q.H.; Yang, L.M.; Zhong, G.F.; Zhang, J. Stereoselective Synthesis of Complex Polyenes through Sequential α-/β-C–H Functionalization of trans-Styrenes. Angew. Chem. Int. Ed. 2024, 63, e202315273. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.H.; Li, X.L.; Zhang, C.; Liu, X.Y.; Huang, L.Z.; Zhang, Y.B.; Shen, C.; Ding, L.Y.; Zhong, G.F.; Zhang, J. Stereo-selective synthesis of complex dienes and eneynes by sequential hydroarylation and olefinic C–H functionalization. Org. Chem. Front. 2024, 11, 4456–4463. [Google Scholar] [CrossRef]
- Zhang, C.; Lin, Y.; Zhu, Y.H.; Peng, C.X.; Lv, B.B.; Zhao, L.; Zhong, G.F.; Wei, J.F.; Zhang, J. Chelation-assisted α and β C–H functionalization of aryl alkenes with alkynes and alkenes. Org. Chem. Front. 2024, 11, 4862–4868. [Google Scholar] [CrossRef]
- Shen, C.; Lu, X.N.; Zhang, J.; Ding, L.Y.; Sun, Y.L.; Zhong, G.F. Bidentate auxiliary-directed alkenyl C–H allylation via exo-palladacycles: Synthesis of branched 1,4-dienes. Chem. Commun. 2019, 55, 13582–13585. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.L.; Zhang, X.D.; Li, X.L.; Liu, X.Y.; Chen, J.K.; Shen, C.; He, R.; Zhong, G.F.; Zhang, J. Palladium-catalysed α and β C–H allylation of aryl alkenes. Org. Chem. Front. 2024, 11, 3341–3347. [Google Scholar] [CrossRef]
- Yi, J.; Yang, L.; Xia, C.G.; Li, F.W. Nickel-Catalyzed Alkynylation of a C(sp2)–H Bond Directed by an 8-Aminoquinoline Moiety. J. Org. Chem. 2015, 80, 6213–6221. [Google Scholar] [CrossRef] [PubMed]
- Schreib, B.S.; Fadel, M.; Carreira, E.M. Palladium-Catalyzed C–H Alkynylation of Unactivated Alkenes. Angew. Chem. Int. Ed. 2020, 59, 7818–7822. [Google Scholar] [CrossRef] [PubMed]
- Parella, R.; Babu, S.A. Pd(OAc)2-Catalyzed, AgOAc-Promoted Z Selective Directed β-Arylation of Acrylamide Systems and Stereoselective Construction of Z-Cinnamamide Scaffolds. J. Org. Chem. 2015, 80, 12379–12396. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.Z.; Chen, Z.; Gao, Y.D.; Xue, F.T.; Jiang, C. Aminoquinoline-assisted vinylic C–H arylation of unsubstituted acrylamide for the selective synthesis of Z olefins. Org. Biomol. Chem. 2016, 14, 3298–3306. [Google Scholar] [CrossRef]
- Hu, L.; Gui, Q.W.; Chen, X.; Tan, Z.; Zhu, G.G. Cobalt-promoted selective arylation of benzamides and acrylamides with arylboronic acids. Org. Biomol. Chem. 2016, 14, 11070–11075. [Google Scholar] [CrossRef] [PubMed]
- Parella, R.; Babu, S.A. Pd(II)-Catalyzed, Picolinamide-Assisted, Z-Selective γ-Arylation of Allylamines To Construct Z-Cinnamylamines. J. Org. Chem. 2017, 82, 6550–6567. [Google Scholar] [CrossRef]
- de Robichon, M.; Bordessa, A.; Malinowski, M.; Uziel, J.; Lubin-Germain, N.; Ferry, A. Access to C-aryl/alkenylglycosides by directed Pd-catalyzed C–H functionalisation of the anomeric position in glycal-type substrates. Chem. Commun. 2019, 55, 11806–11808. [Google Scholar] [CrossRef] [PubMed]
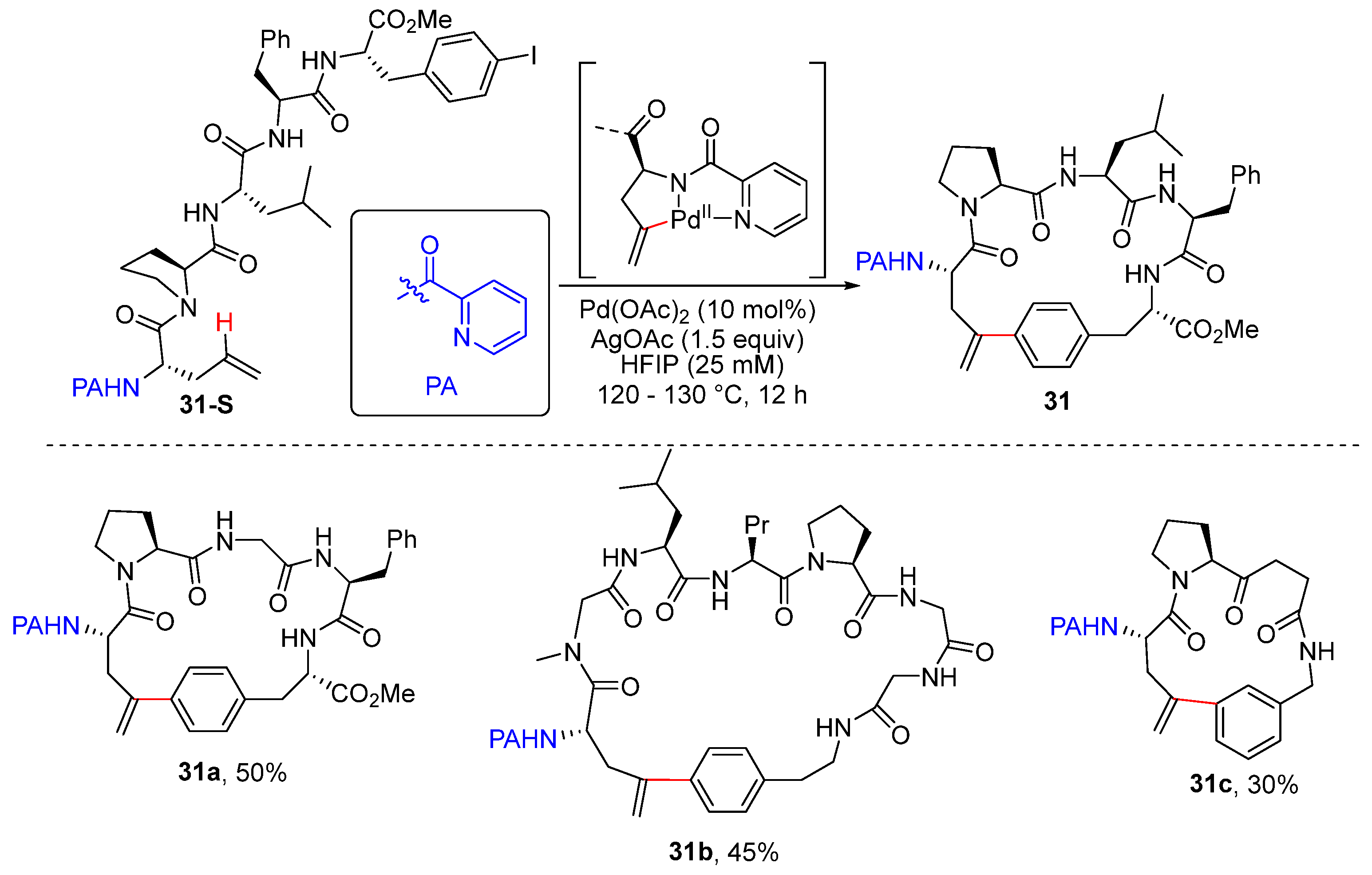
- Han, B.Y.; Li, B.; Qi, L.P.; Yang, P.; He, G.; Chen, G. Construction of Cyclophane-Braced Peptide Macrocycles via Palladium-Catalyzed Picolinamide-Directed Intramolecular C(sp2)–H Arylation. Org. Lett. 2020, 22, 6879–6883. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Li, D.Y.; Wang, B.; Yao, J.Z.; Zhang, Y.H. Direct ortho-Thiolation of Arenes and Alkenes by Nickel Catalysis. Org. Lett. 2015, 17, 1328–1331. [Google Scholar] [CrossRef]
- Zhao, Q.; Poisson, T.; Pannecoucke, X.; Bouillon, J.-P.; Besset, T. Pd-Catalyzed Diastereoselective Trifluoromethylthiolation of Functionalized Acrylamides. Org. Lett. 2017, 19, 5106–5109. [Google Scholar] [CrossRef]
- Xiang, T.X.; Liu, Y.Z.; Xu, Q.L.; He, K.H.; Pan, F. Palladium-Catalyzed Regio- and Diastereoselective Olefinic C–H Difluoromethylthiolation at Room Temperature. J. Org. Chem. 2022, 87, 3135–3144. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.L.; Chen, C.; Ma, Z.G.; Zhou, J.; Wang, L.R.; Zhang, S.Y. Stereoselective Synthesis of Z-Vinylsilanes via Palladium-Catalyzed Direct Intermolecular Silylation of C(sp2)–H Bonds. Org. Lett. 2017, 19, 5216–5219. [Google Scholar] [CrossRef] [PubMed]
- Schreib, B.S.; Carreira, E.M. Palladium-Catalyzed Regioselective C–H Iodination of Unactivated Alkenes. J. Am. Chem. Soc. 2019, 141, 8758–8763. [Google Scholar] [CrossRef] [PubMed]
- Grigorjeva, L.; Daugulis, O. Cobalt-Catalyzed Direct Carbonylation of Aminoquinoline Benzamides. Org. Lett. 2014, 16, 4688–4690. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, T.; Ilies, L.; Nakamura, E. Oxidative C–H Activation Approach to Pyridone and Isoquinolone through an Iron-Catalyzed Coupling of Amides with Alkynes. Chem. Asian J. 2016, 11, 380–384. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, M.; Miki, N.; Tsuchiya, Y.; Nakajima, K.; Nishihara, Y. Synthesis of Benzoisoselenazolone Derivatives by Nickel-Catalyzed Dehydrogenative Direct Selenation of C(sp2)–H Bonds with Elemental Selenium in Air. Org. Lett. 2017, 19, 1092–1095. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; Zhu, Y.H.; Shen, W.Z.; Jin, S.Q.; Zhong, G.F.; Luo, S.X.; Xu, L.X.; Zhong, L.J.; Zhang, J. Access to axially chiral aryl 1,3-dienes by transient group directed asymmetric C–H alkenylations. Org. Chem. Front. 2022, 9, 2109–2115. [Google Scholar] [CrossRef]
- Oxtoby, L.J.; Li, Z.-Q.; Tran, V.T.; Erbay, T.G.; Deng, R.; Liu, P.; Engle, K.M. A Transient-Directing-Group Strategy Enables Enantioselective Reductive Heck Hydroarylation of Alkenes. Angew. Chem. Int. Ed. 2020, 59, 8885–8890. [Google Scholar] [CrossRef]
- Shen, C.; Zhu, Y.H.; Shen, W.Z.; Jin, S.Q.; Zhong, L.J.; Luo, S.X.; Xu, L.X.; Zhong, G.F.; Zhang, J. Construction of axial chirality by asymmetric alpha C–H alkenylation of aryl alkenes. Org. Chem. Front. 2022, 9, 4834–4839. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Peng, C.; Li, X.; Liu, X.; Ding, L.; Zhong, G.; Zhang, J. N,N- and N,O-Bidentate-Chelation-Assisted Alkenyl C–H Functionalization. Molecules 2025, 30, 1669. https://doi.org/10.3390/molecules30081669
Zhang Y, Peng C, Li X, Liu X, Ding L, Zhong G, Zhang J. N,N- and N,O-Bidentate-Chelation-Assisted Alkenyl C–H Functionalization. Molecules. 2025; 30(8):1669. https://doi.org/10.3390/molecules30081669
Chicago/Turabian StyleZhang, Yawei, Chengxing Peng, Xiaoli Li, Xiuying Liu, Liyuan Ding, Guofu Zhong, and Jian Zhang. 2025. "N,N- and N,O-Bidentate-Chelation-Assisted Alkenyl C–H Functionalization" Molecules 30, no. 8: 1669. https://doi.org/10.3390/molecules30081669
APA StyleZhang, Y., Peng, C., Li, X., Liu, X., Ding, L., Zhong, G., & Zhang, J. (2025). N,N- and N,O-Bidentate-Chelation-Assisted Alkenyl C–H Functionalization. Molecules, 30(8), 1669. https://doi.org/10.3390/molecules30081669