
Spectroscopic and Microscopic Analysis of Apple Pectins





Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Pectins
2.3. Instrumental Analysis
2.4. Chemical Shift Calculation
3. Results and Discussion
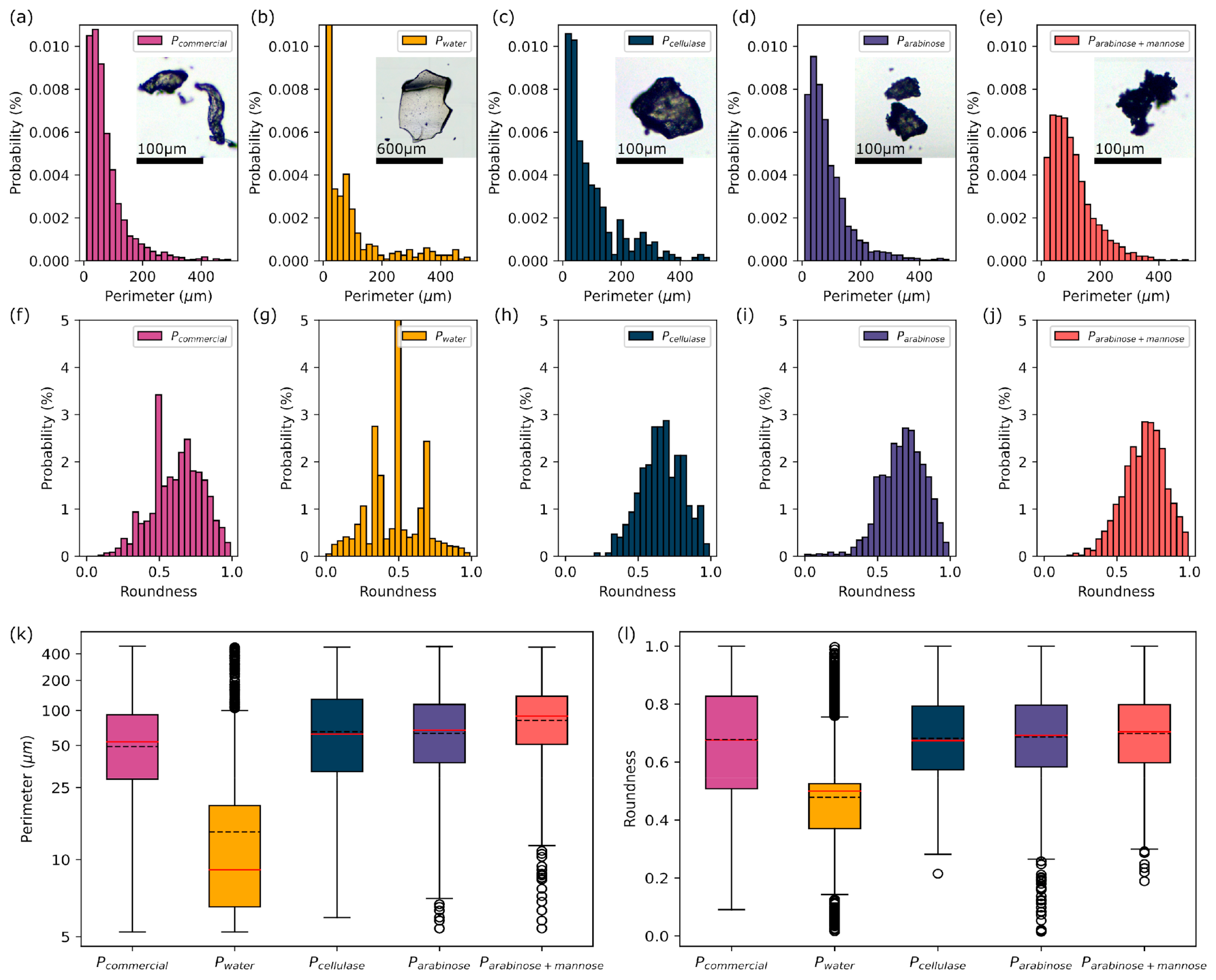
3.1. Morphology Analysis
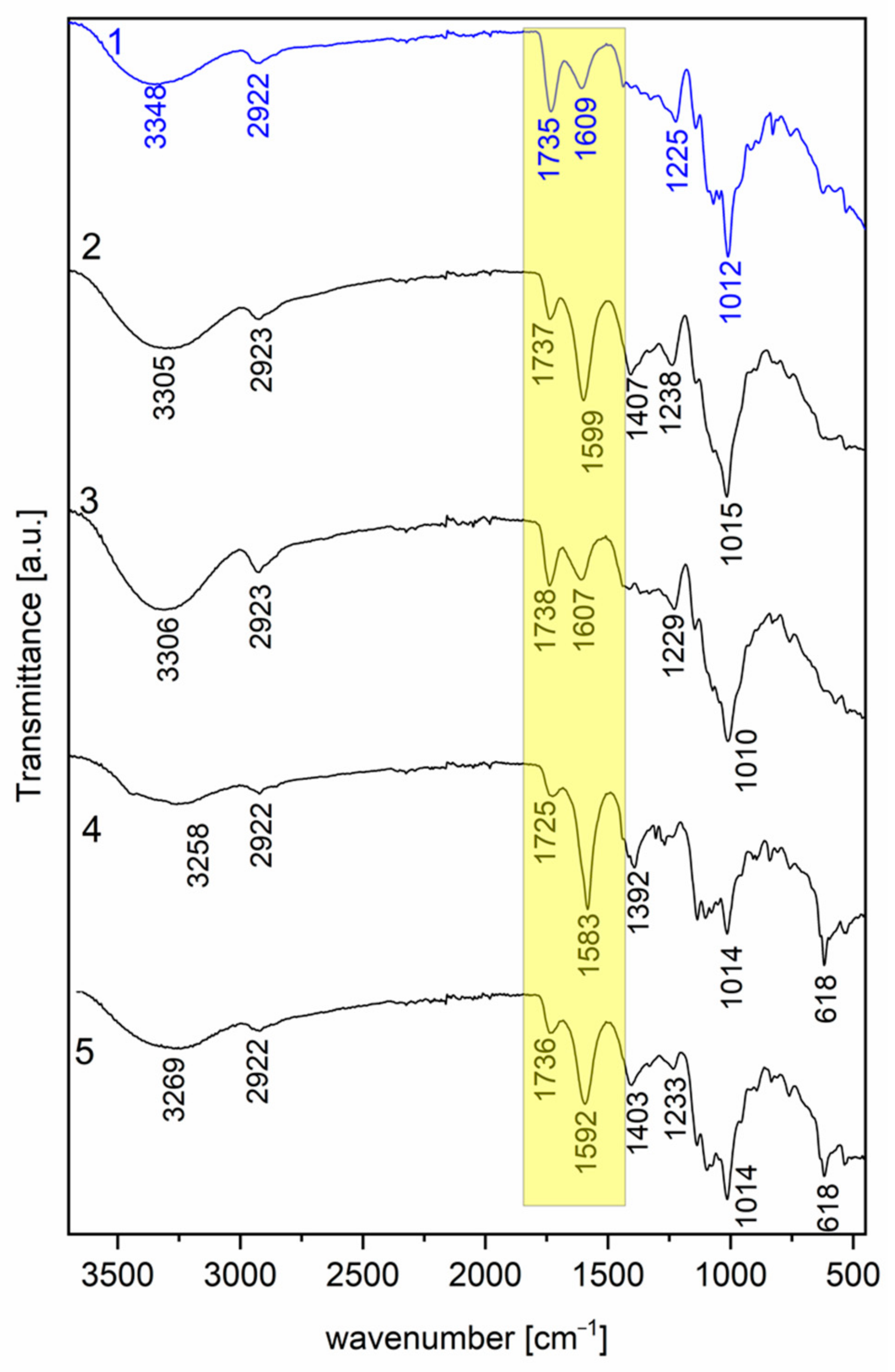
3.2. Characterization of Pectin via FT-IR Spectroscopy
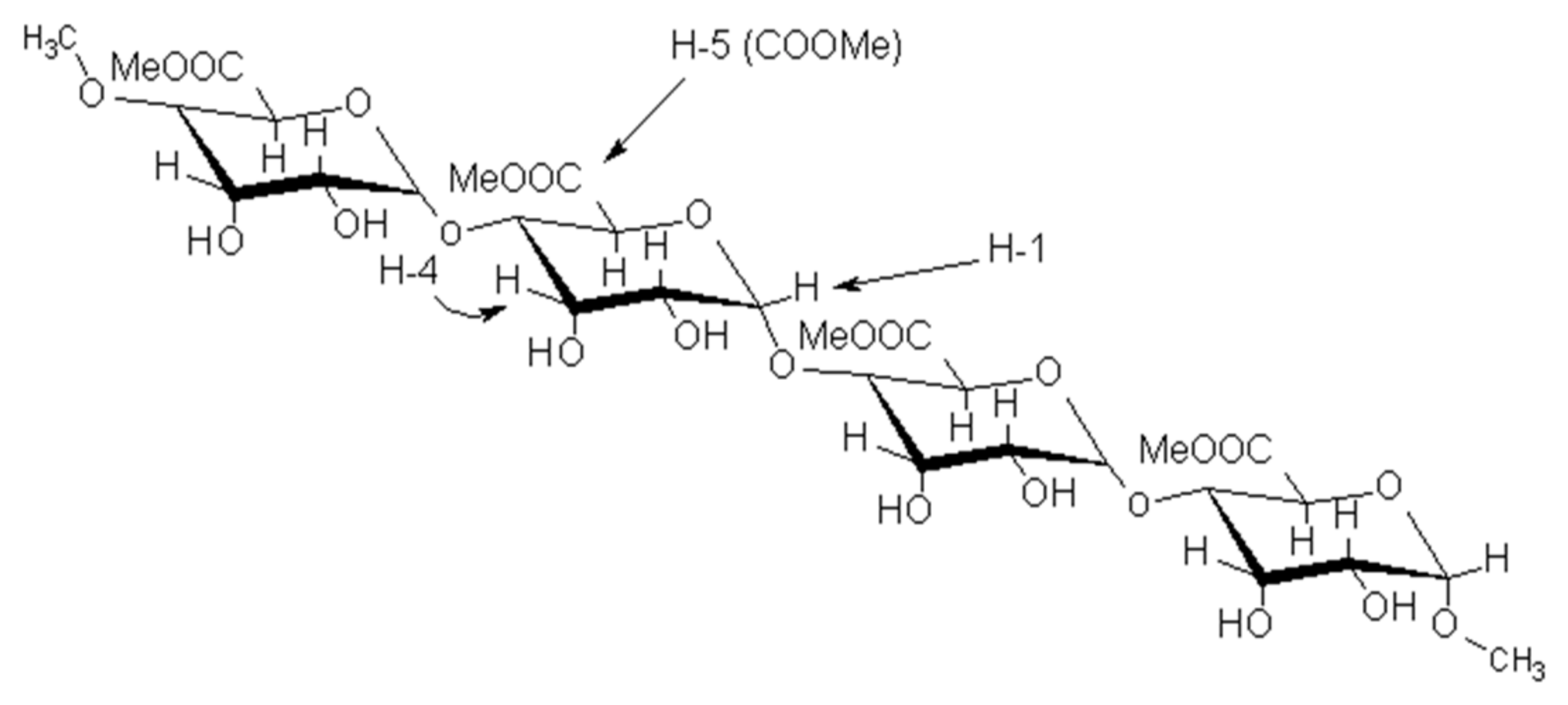
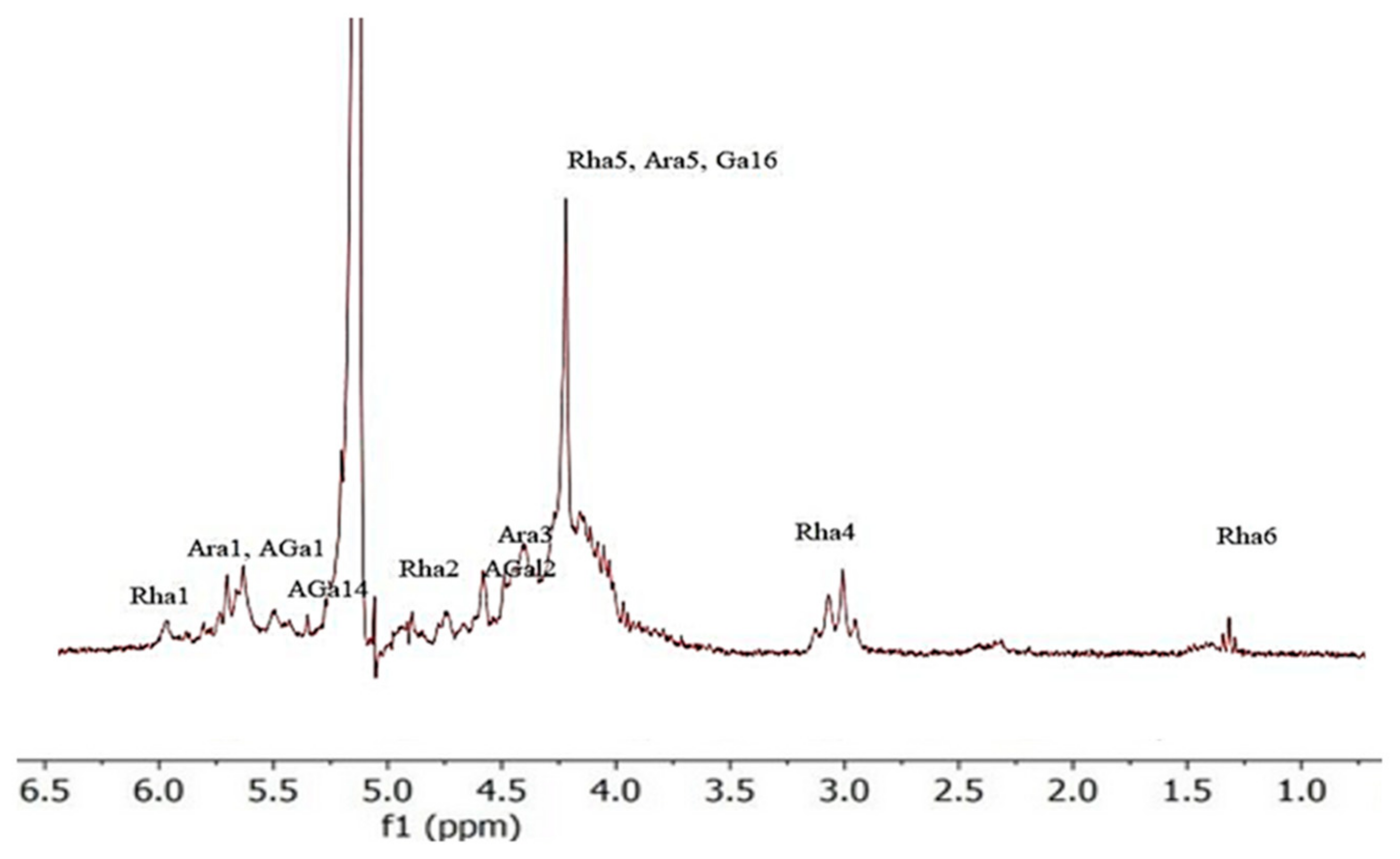
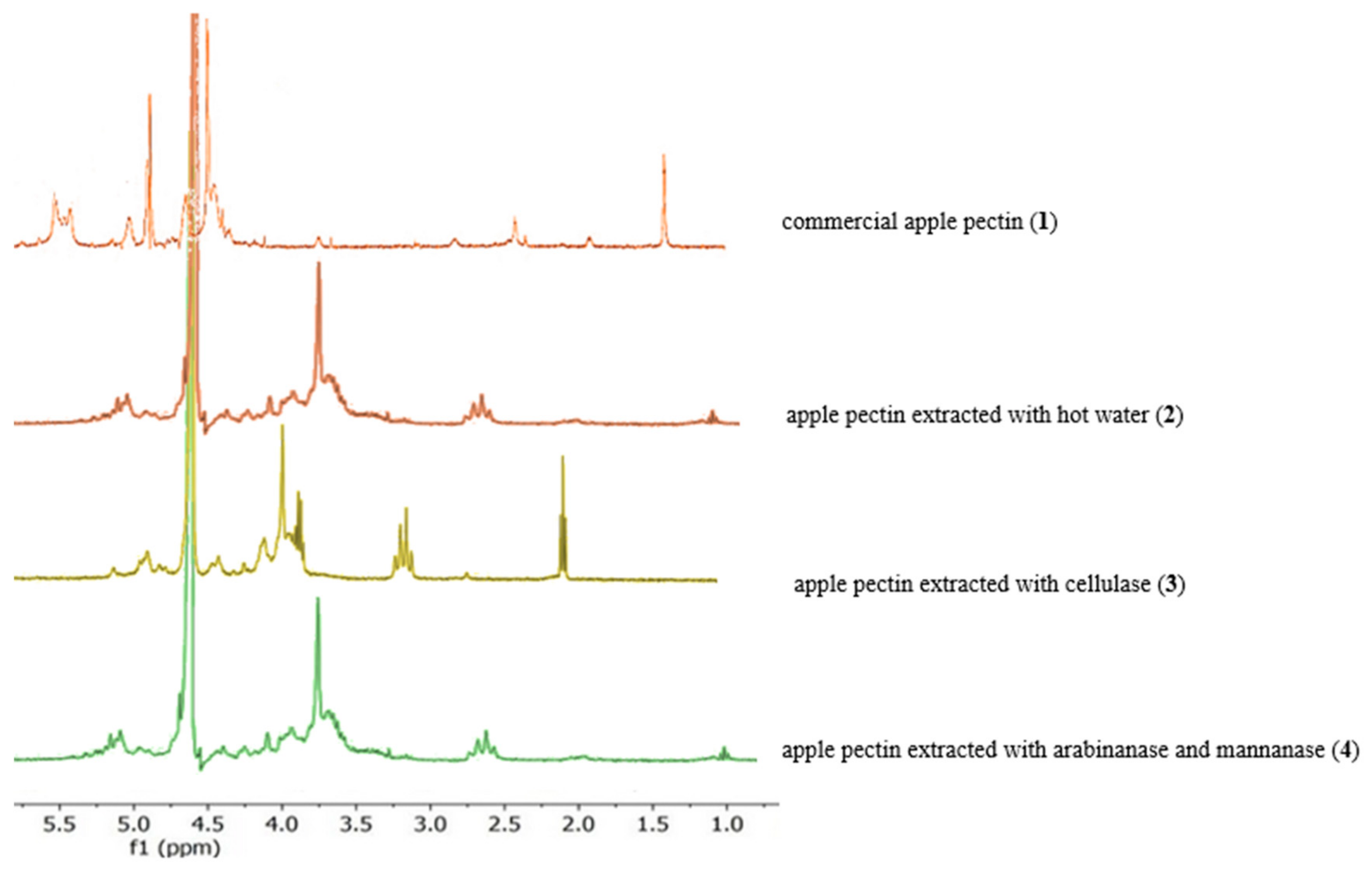
3.3. Characterization of Pectin via 1H NMR Spectroscopy
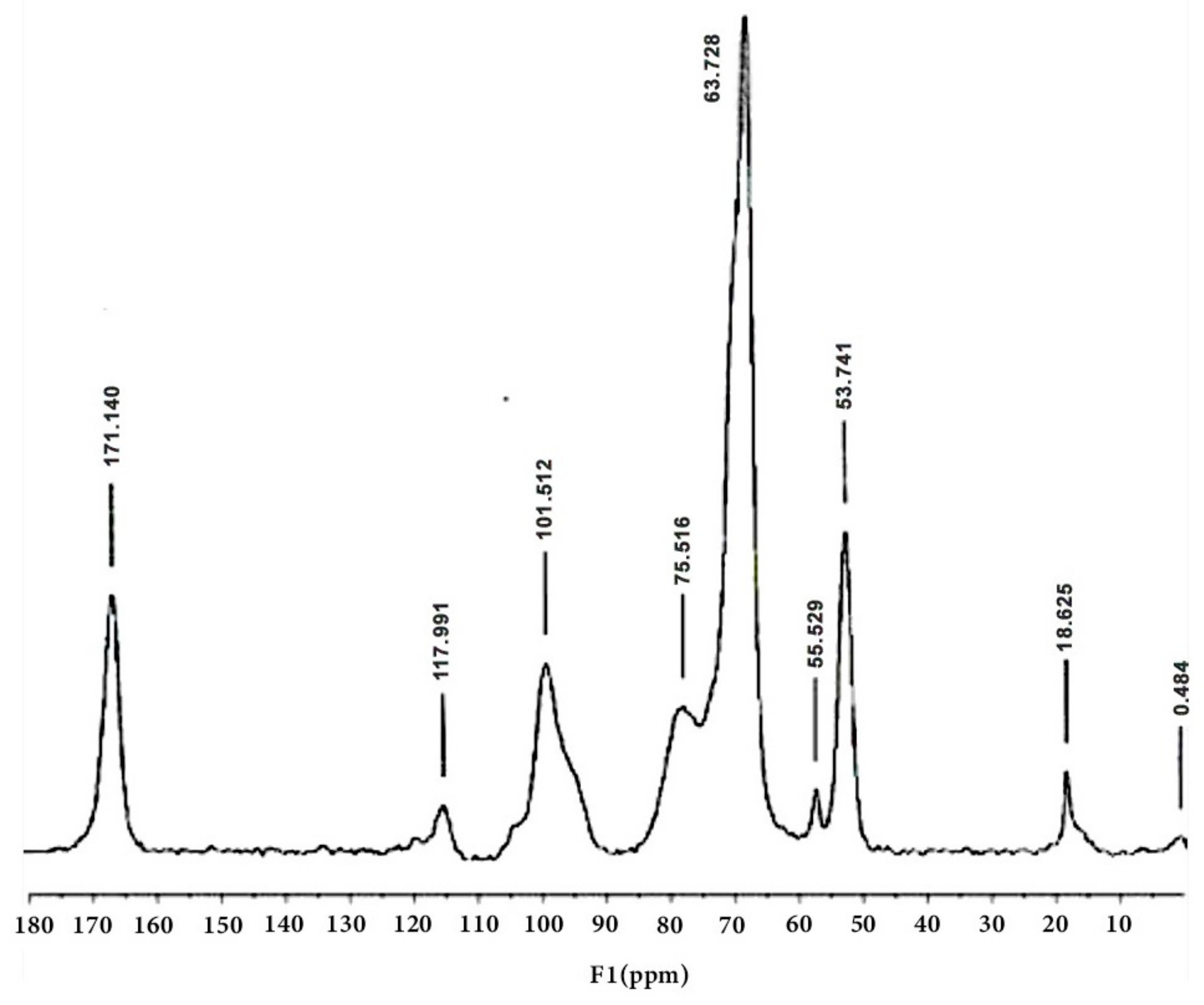
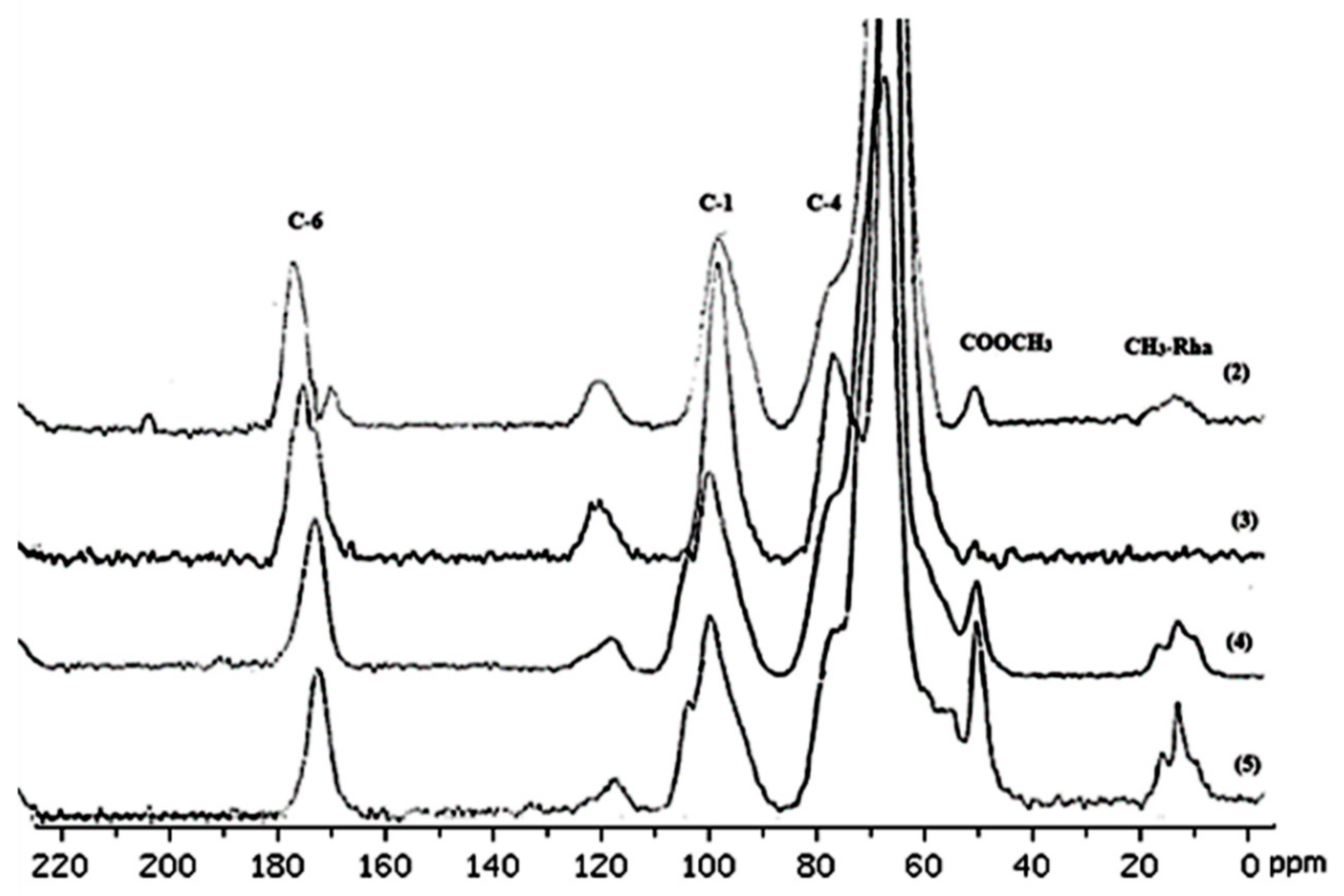
3.4. Characterization of Pectin via 13C NMR Spectroscopy
4. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sirisomboon, P.; Tanaka, M.; Fujita, S.; Akinaga, T.; Kojima, T. A simplified method for the determination of total oxalate-soluble pectin content in Japanese pear. J. Food Comp. Anal. 2001, 14, 83–91. [Google Scholar]
- Srivastava, P.; Malviya, R. Sources of pectin, extraction and its applications in pharmaceutical industry—An overview. Indian J. Nat. Prod. Resour. 2011, 2, 10–18. [Google Scholar]
- Narasimman, P.; Sethuraman, P. An overview on the fundamentals of pectin. Int. J. Adv. Res. 2016, 4, 1855–1860. [Google Scholar] [CrossRef] [PubMed]
- Chandel, V.; Biswas, D.; Roy, S.; Vaidya, D.; Verma, A.; Gupta, A. Current Advancements in Pectin: Extraction, Properties and Multifunctional Applications. Foods 2022, 11, 2683. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Alimardani-Theuil, P.; Gainvors-Claise, A.; Duchiron, F. Yeasts: An attractive source of pectinases—From gene expression to potential applications: A review. Process Biochem. 2011, 46, 1525–1537. [Google Scholar] [CrossRef]
- Leclere, L.; Cutsem, P.V.; Michiels, C. Anti-Cancer Activities of pH- or Heat-Modified Pectin. Front. Pharmacol. 2013, 4, 128. [Google Scholar] [CrossRef]
- Wikiera, A.; Mika, M.; Starzyńska-Janiszewska, A.; Stodolak, B. Application of Celluclast 1.5 L in pectin extraction. Carbohyd. Polym. 2015, 134, 251–257. [Google Scholar]
- Lee, J.H.; Shim, J.S.; Lee, J.S.; Kim, M.K.; Chung, M.S.; Kim, K.H. Pectin-Like Acidic Polysaccharide from Panax Ginseng with Selective Antiadhesive Activity Against Pathogenic Bacteria. Carbohyd. Res. 2006, 341, 1154–1163. [Google Scholar] [CrossRef]
- Khotimchenko, M.; Kovalev, V.; Kolenchenko, E.; Khotimchenko, Y. Acidic Method for the Low Molecular Pectin Preparation. Int. J. Pharm. Pharm. Sci. 2012, 4, 279–283. [Google Scholar]
- Wikiera, A.; Grabacka, M.; Byczyński, Ł.; Stodolak, B.; Mika, M. Enzymatically Extracted Apple Pectin Possesses Antioxidant and Antitumor Activity. Molecules 2021, 26, 1434. [Google Scholar] [CrossRef]
- Marić, M.; Grassino, A.N.; Zhu, Z.; Barba, F.J.; Brnčić, M.; Rimac Brnčić, S. An overview of the traditional and innovative approaches for pectin extraction from plant food wastes and by-products: Ultrasound-, microwaves-, and enzyme-assisted extraction. Trends Food Sci. Technol. 2018, 76, 28–37. [Google Scholar] [CrossRef]
- Kozioł, A.; Środa-Pomianek, K.; Górniak, A.; Wikiera, A.; Cyprych, K.; Malik, M. Structural Determination of Pectins by Spectroscopy Methods. Coatings 2022, 12, 546. [Google Scholar] [CrossRef]
- Li, D.Q.; Du, G.M.; Jing, W.W.; Li, J.F.; Yan, J.Y.; Liu, Z.Y. Combined effects of independent variables on yield and protein content of pectin extracted from sugar beet pulp by citric acid. Carbohydr. Polym. 2015, 129, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Gnanasambandam, R.; Proctor, A. Determination of pectin degree of esterification by diffuse reflectance Fourier transform infrared spectroscopy. Food Chem. 2000, 68, 327–332. [Google Scholar] [CrossRef]
- Zouambia, Y.; Moulai-Mostefa, N.; Krea, M. Structural characterization and surface activity of hydrophobically functionalized extracted pectins. Carbohydr. Polym. 2009, 78, 841–846. [Google Scholar] [CrossRef]
- Almohammed, F.; Koubaa, M.; Khelfa, A.; Nakaya, M.; Mhemdi, H.; Vorobiev, E. Pectin recovery from sugar beet pulp enhanced by high-voltage electrical discharges. Food Bioprod. Process. 2017, 103, 95–103. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, L.; Chen, F.; Lai, S.; Yang, H. Effects of vacuum impregnation with calcium ascorbate and disodium stannous citrate on Chinese red bayberry. Food Bioprocess. Technol. 2018, 11, 1300–1316. [Google Scholar] [CrossRef]
- Synytsya, A.; Čopı́ková, J.; Matějka, P.; Machovič, V. Fourier transform Raman and infrared spectroscopy of pectins. Carbohydr. Polym. 2003, 54, 64–73. [Google Scholar] [CrossRef]
- Shivamathi, C.S.; Gunaseelan, S.; Soosai, M.R.; Vignesh, N.S.; Varalakshmi, P.; Kumar, R.S.; Karthikumar, S.; Kumar, R.V.; Baskar, R.; Rigby, S.P.; et al. Process optimization and characterization of pectin derived from underexploited pineapple peel biowaste as a value-added product. Food Hydrocoll. 2022, 123, 10714. [Google Scholar] [CrossRef]
- Santos, E.E.; Amaro, R.C.; Bustamante, C.C.C.; Guerra, M.H.A.; Soares, L.C.; Froes, R.E.S. Extraction of pectin from agroindustrial residue with an ecofriendly solvent: Use of FTIR and chemometrics to differentiate pectins according to degree of methyl esterification. Food Hydrocoll. 2020, 107, 105921. [Google Scholar] [CrossRef]
- Lara-Espinoza, C.; Carvajal-Millán, E.; Balandrán-Quintana, R.; López-Franco, Y.; Rascón-Chu, A. Pectin and Pectin-Based Composite Materials: Beyond Food Texture. Molecules 2018, 23, 942. [Google Scholar] [CrossRef] [PubMed]
- Golovchenko, V.V.; Khlopin, V.A.; Patova, O.A.; Feltsinger, L.S.; Bilan, M.I.; Dmitrenok, A.S.; Shashkov, A.S. Pectin from leaves of birch (Betula pendula Roth.): Results of NMR experiments and hypothesis of the RG-I structure. Carbohydr. Polym. 2022, 284, 119186. [Google Scholar] [CrossRef] [PubMed]
- Voragen, A.G.J.; Pilnik, W.; Thibault, J.-F.; Axelos, M.A.V.; Renard, C.M.G.C. Pectins. In Food Polysaccharides and their Applications; Stephen, A.M., Ed.; Marcel Dekker: New York, NY, USA, 1995; pp. 287–339. [Google Scholar]
- Vriesmann, L.C.; Petkowicz, C.L. Polysaccharides from the pulp of cupuassu (Theobroma grandiflorum): Structural characterization of a pectic fraction. Carbohydr. Polym. 2009, 77, 72–79. [Google Scholar] [CrossRef]
- Yapo, B.M. Pectin: Structure and applications. In Food Carbohydrates: Chemistry, Physical Properties and Industrial Applications; CRC Press: Boca Raton, FL, USA, 2012; pp. 379–415. [Google Scholar]
- Kacuráková, M.; Smith, A.C.; Gidley, M.J. NMR spectroscopy of pectins from sugar beet and apple: Comparison of structure and gelation properties. Carbohydr. Res. 2007, 342, 11–18. [Google Scholar]
- Voragen, A.G.J.; Coenen, G.-J.; Verhoef, R.P.; Schols, H.A. Pectin, a versatile polysaccharide present in plant cell walls. Struct. Chem. 2009, 20, 263–275. [Google Scholar] [CrossRef]
- Habibi, Y.; Heyraud, A.; Mahrouz, M.; Vignon, M.R. Structural features of pectic polysaccharides from the skin of Opuntia ficus-indica prickly pear fruits. Carbohydr. Res. 2004, 339, 1119–1127. [Google Scholar] [CrossRef]
- Rondeau-Mouro, C.; Bléon, A.; Lahaye, M. Caractérisation par RMN des biopolymères d’origine végétale, de la molécule à l’organisation supramoléculaire. Comptes Rendus Chim. 2008, 11, 370–379. [Google Scholar] [CrossRef]
- Yu, L.; Zhang, X.; Li, S.; Liu, X.; Sun, L.; Liu, H.; Iteku, J.; Zhou, Y.; Tai, G. Rhamnogalacturonan I domains from ginseng pectin. Carbohydr. Polym. 2010, 79, 811–817. [Google Scholar] [CrossRef]
- Tamaki, Y.; Konishi, T.; Fukuta, M.; Tako, M. Isolation and structural characterization of pectin from endocarp of Citrus depressa. Food Chem. 2008, 107, 352–361. [Google Scholar] [CrossRef]
- Wikiera, A.; Kozioł, A.; Mika, M.; Stodolak, B. Structure and bioactivity of apple pectin isolated with arabinanase and mannanase. Food Chem. 2022, 388, 133020. [Google Scholar] [CrossRef]
- Schneider, C.; Rasband, W.; Eliceiri, K. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Wikiera, A.; Mika, M.; Starzyńska-Janiszewska, A.; Stodolak, B. Endo-xylanase and endo-cellulase-assisted extraction of pectin from apple pomace. Carbohydr. Polym. 2016, 142, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Chen, J.; Yin, Y.; Xiao, S.; Zhang, R.; Yang, Y.; Li, L.; Xu, H.; Zhang, X.; Hu, P. Chemical structure elucidation and functional activities comparison of two polysaccharides purified from Citrus reticulata Blanco peels. Chem. Biol. Technol. Agric. 2024, 11, 37. [Google Scholar]
- Renard, C.; Jarvis, M.C. Acetylation and methylation of homogalacturonan 2: Effect on ion--binding properties and conformations. Carbohyd. Polym. 1999, 39, 209–216. [Google Scholar] [CrossRef]
- Yang, M.; Liu, Z.; Zhang, J.; Zhu, X.; Xie, W.; Lan, H.; Huang, Y.; Ye, X.; Yang, J. Simultaneous quantification of cellulose and pectin in tobacco using a robust solid-state NMR method. Carbohydr. Res. 2022, 521, 108676. [Google Scholar] [CrossRef]
- Sinitsya, A.; Čopikova, J.; Pavlikova, H.; Skoblya, S.; Machovic, V. Amidation of highly methoxy-lated citrus pectin with primary amines. Carbohyd. Polym. 2000, 42, 359–368. [Google Scholar]
- Freitas, C.M.P.; Sousa, R.C.S.; Dias, M.M.S.; Coimbra, J.S.R. Extraction of pectin from passion fruit peel. Food Eng. Rev. 2020, 12, 460–472. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mw (kDa) | GalA (%) | DM (%) | NS * (%) | Protein (%) | Reactive with F–C Reagent (%) |
|---|---|---|---|---|---|
| 378 ± 45 | 80.9 ± 2.4 | 56.9 ± 2.1 | 14.3 ± 0.7 | 0.78 ± 0.12 | 0.49 ± 0.08 |
| Pectin | Yield (% Wet Basis) | Mw (kDa) | Source |
|---|---|---|---|
| Pcommercial | - | 378 ± 45 | [10] |
| Pcellulase | 15.20 | 589 ± 66 | [10,35] |
| Parabinanase | 16.34 | 452 ± 8 | [35] |
| Parabinanase+mannanase | 13.51 | 341 ± 6 | [31] |
| Pacid | 13.33 | 312 ± 5 | [31] |
| FT-IR Band (cm⁻1) | Assignment | Integration Area/Peak Height | Application in DM Calculation | References |
|---|---|---|---|---|
| ~1740–1750 | Carboxyl esters (C=O) | Integration area or peak height | Proportional to the content of the methoxyl groups | [12,14,18] |
| ~1630–1600 | Free carboxyl groups (COO⁻) | Integration area or peak height | Proportional to the content of free galacturonic acid groups | [12,14,18] |
| Ratio of 1740/1630 | Ratio of peak height or area integral | Ratio of esterified carboxyl groups compared to free carboxyl groups | The ratio of the peak area or height of these two bands is used to calculate the degree of methoxylation (DM) | [12,14] |
| PECTIN | %DM * | |
|---|---|---|
| Pcommercial | (1) | 57% ± 0.5% |
| Pcellulase | (2) | 58% ± 0.3% |
| Pwater | (3) | 51% ± 0.5% |
| Parabinose+mannose | (4) | 30% ± 0.34% |
| Parabinose | (5) | 36% ± 0.4% |
| Feature | Apple Pectins | Apple Pectin from Literature 1 [21] | Apple Pectin from Literature 2 [22] |
|---|---|---|---|
| Galacturonic Residue Distribution | Random or block | Block | Random or block |
| Presence of Acetyl Groups | Yes | Yes | Yes |
| Presence of Other Sugars | Rhamnose, arabinose, galactose | Rhamnose, arabinose, galactose | Rhamnose, arabinose, galactose, xylose |
| Region 3.0–4.0 ppm (Protons in the Galacturonic Ring) | Broad signals | Broad signals | Broad signals |
| Region 4.8–5.2 ppm (Anomeric Protons) | Multiple signals | Multiple signals | Multiple signals |
| Region 3.7–3.8 ppm (Methoxy Protons) | Singlet | Singlet | Singlet |
| Region 2.0–2.2 ppm (Acetyl Protons) | Singlet | Singlet | Singlet |
| Region 1.2–1.3 ppm (Rhamnose Protons) | Doublet | Doublet | Doublet |
| Other Significant Signals | - | - | - |
| Chemical Shift Assignment (ppm) | Structure/Functional Group | Justification from the Literature | References |
|---|---|---|---|
| 170–175 | Carboxyl groups (C=O) in galacturonic acid | Characteristic shift of carboxyl groups in uronic acids. | [12,25,26] |
| 100–105 | Anomeric carbons (C-1) | Characteristic shift of anomeric carbons in polysaccharides. | [12,26] |
| 70–85 | Galacturonic ring carbons (C-2, C-3, C-4, C-5) | Shifts in this range correspond to the carbons in the sugar ring. | [25,27] |
| 52–55 | Methoxy groups (OCH3) | Characteristic shift of methoxy groups. | [12,26] |
| 20–22 | Acetyl groups (COCH3) | Characteristic shift of acetyl groups. | [25,26] |
| 17–18 | Rhamnose carbons (C-6) | Characteristic shift of methyl groups in rhamnose. | [12] |
| Polymer | Carbon | Shift (ppm) | Sample | C-1 | C-2,C-3,C-5 | COOCH3 | OCOCH3 | |
|---|---|---|---|---|---|---|---|---|
| Galacturonan | C-6 | 173.31 | 1 | 98.9 | 65.7 | 54.2 | 21.9 | |
| Galacturonan | C-6 | 171.27 | 2 | 101.6 | 66.1 | 54.4 | 21.2 | |
| Arabinan | C-1 | 105.6 | 13C NMR | 3 | 101.9 | 66.1 | 54.6 | 19.6 |
| Galacturonan | C-1 | 100.56 | chemical shifts | 4 | 99.6 | 65.5 | 54.2 | 21.0 |
| Galactan | C-4 | 78.84 | for the | 5 | 101.0 | 66.1 | 54.5 | 19.9 |
| Galacturonan | C-3 | 71.23 | samples of | |||||
| Galacturonan | C-2 | 69.12 | pectins. | |||||
| Arabinan | C-5 | 68.92 | ||||||
| Galacturonan | OCH3 | 53.54 | ||||||
| Rhamnose | CH3 | 17.22 | ||||||
| Area, 106 | |||||
|---|---|---|---|---|---|
| Sample | A1COOCH3 34 | A2COOCH3 (C-6) | AOCOCH3 | DM (%) | DAc (%) |
| 1 | 25.80 | 45.90 | 19.92 | 58.38 | 0.43 |
| 2 | 37.84 | 58.23 | 78.79 | 64.98 | 1.35 |
| 3 | 17.34 | 52.54 | 77.79 | 33.00 | 1.48 |
| 4 | 37.43 | 51.33 | 78.79 | 72.93 | 1.53 |
| 5 | 33.88 | 51.44 | 77.79 | 65.87 | 1.51 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Serrafi, A.; Wikiera, A.; Cyprych, K.; Malik, M. Spectroscopic and Microscopic Analysis of Apple Pectins. Molecules 2025, 30, 1633. https://doi.org/10.3390/molecules30071633
Serrafi A, Wikiera A, Cyprych K, Malik M. Spectroscopic and Microscopic Analysis of Apple Pectins. Molecules. 2025; 30(7):1633. https://doi.org/10.3390/molecules30071633
Chicago/Turabian StyleSerrafi, Agata, Agnieszka Wikiera, Konrad Cyprych, and Magdalena Malik. 2025. "Spectroscopic and Microscopic Analysis of Apple Pectins" Molecules 30, no. 7: 1633. https://doi.org/10.3390/molecules30071633
APA StyleSerrafi, A., Wikiera, A., Cyprych, K., & Malik, M. (2025). Spectroscopic and Microscopic Analysis of Apple Pectins. Molecules, 30(7), 1633. https://doi.org/10.3390/molecules30071633