
Electrocatalytic CO2 Reduction Coupled with Water Oxidation by bi- and Tetranuclear Copper Complexes Based on di-2-pyridyl Ketone Ligand



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
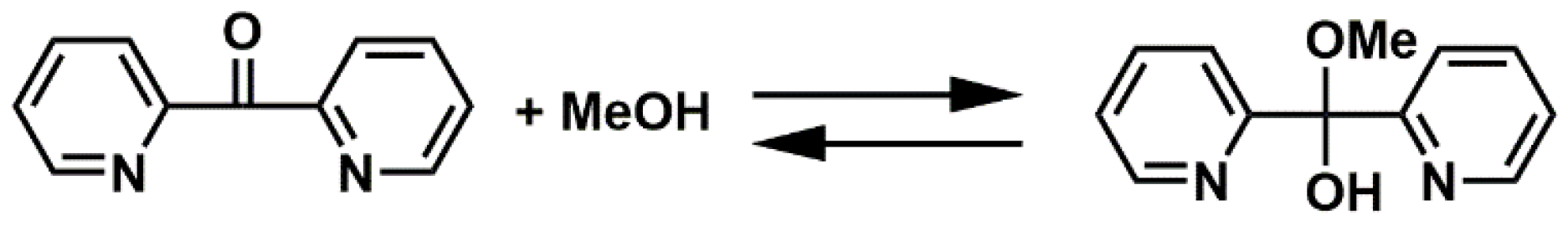
2.1. dpk·MeOH Synthesis
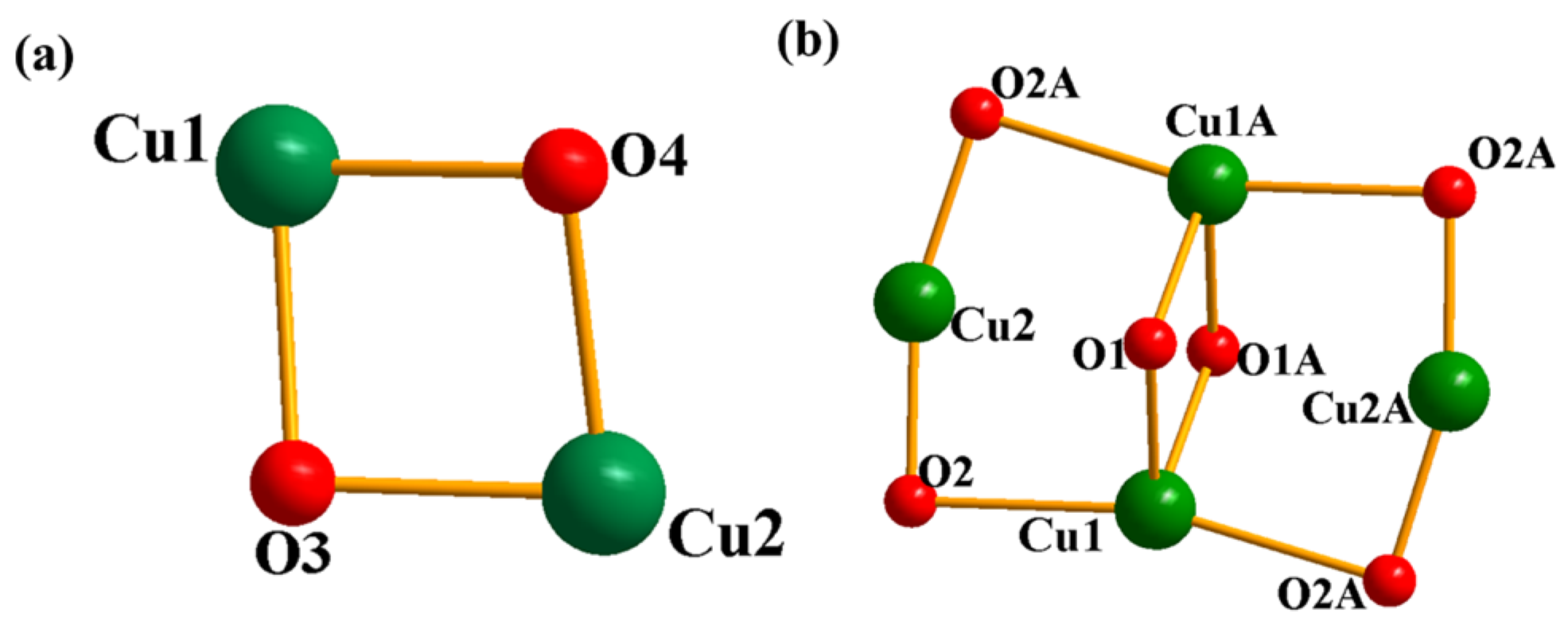
2.2. Analysis of Crystal Structures
2.3. Electrochemical Properties of Complexes 1 and 2
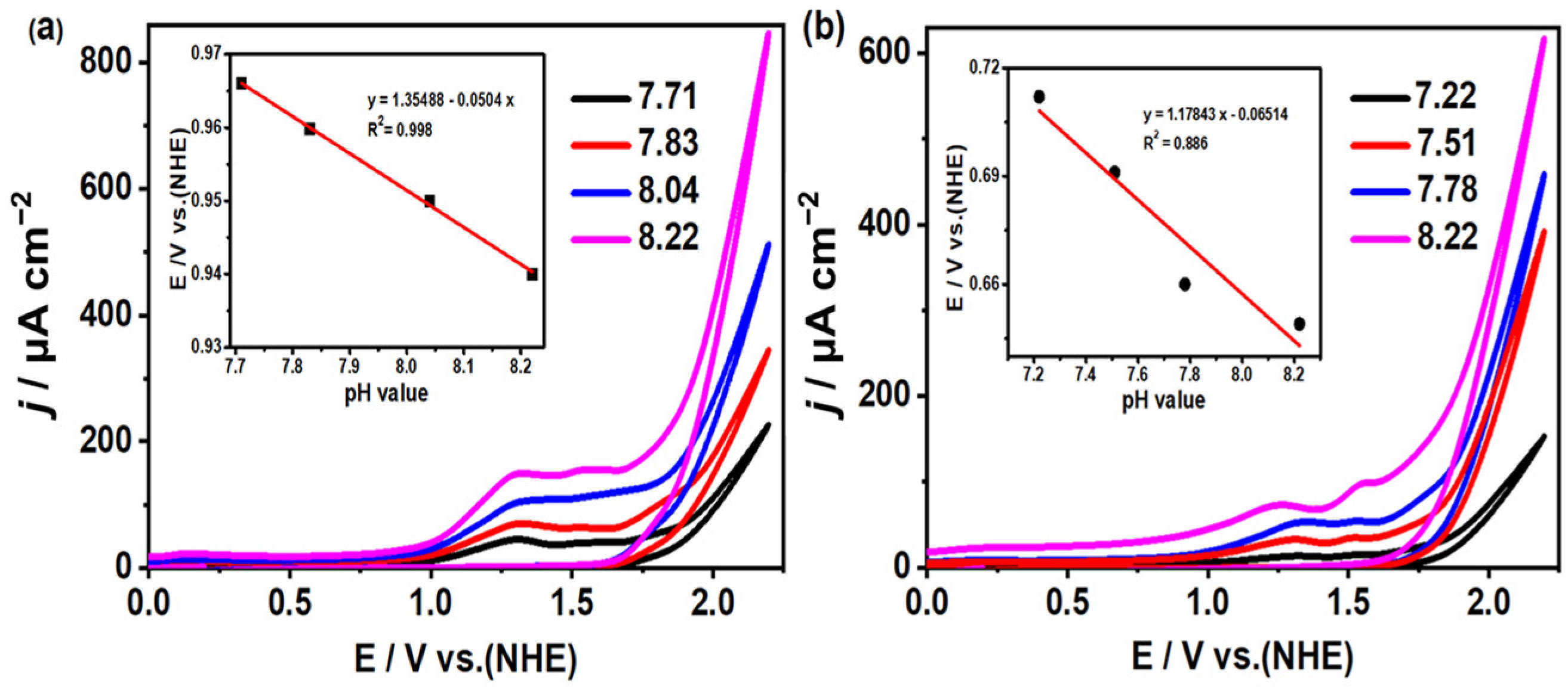
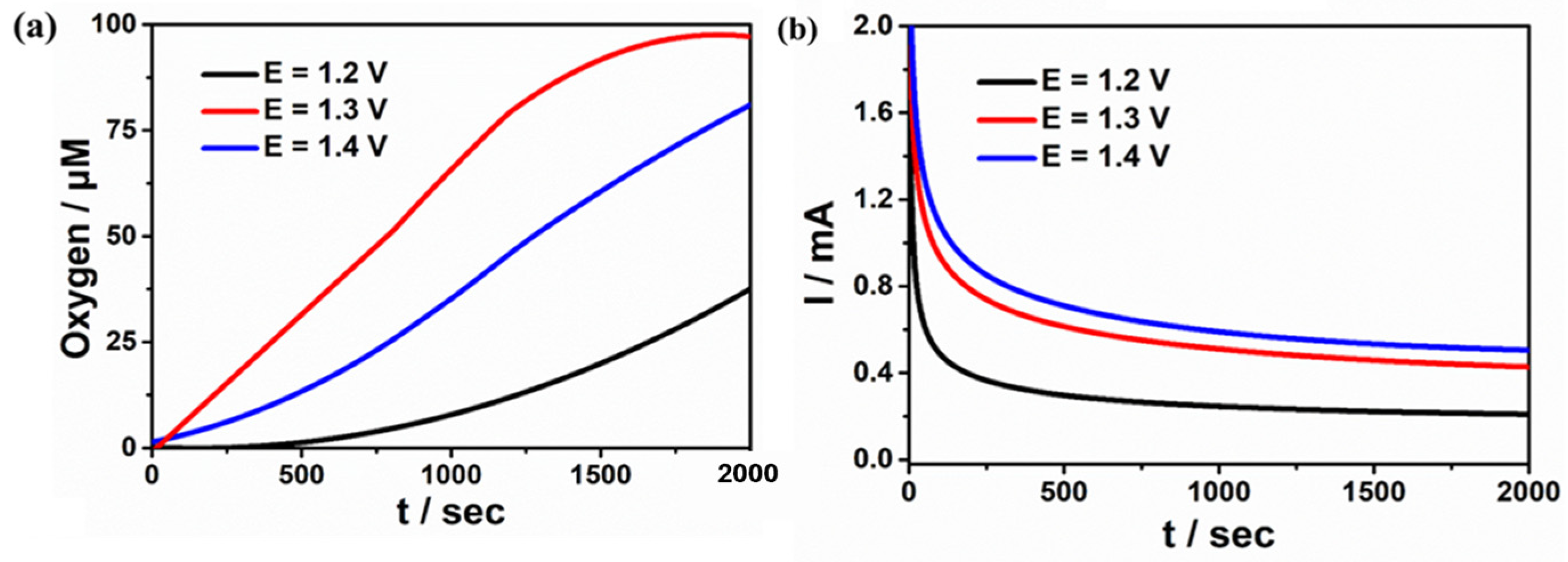
2.4. Study on Electrocatalytic Performance of Water Oxidation of Complexes 1 and 2
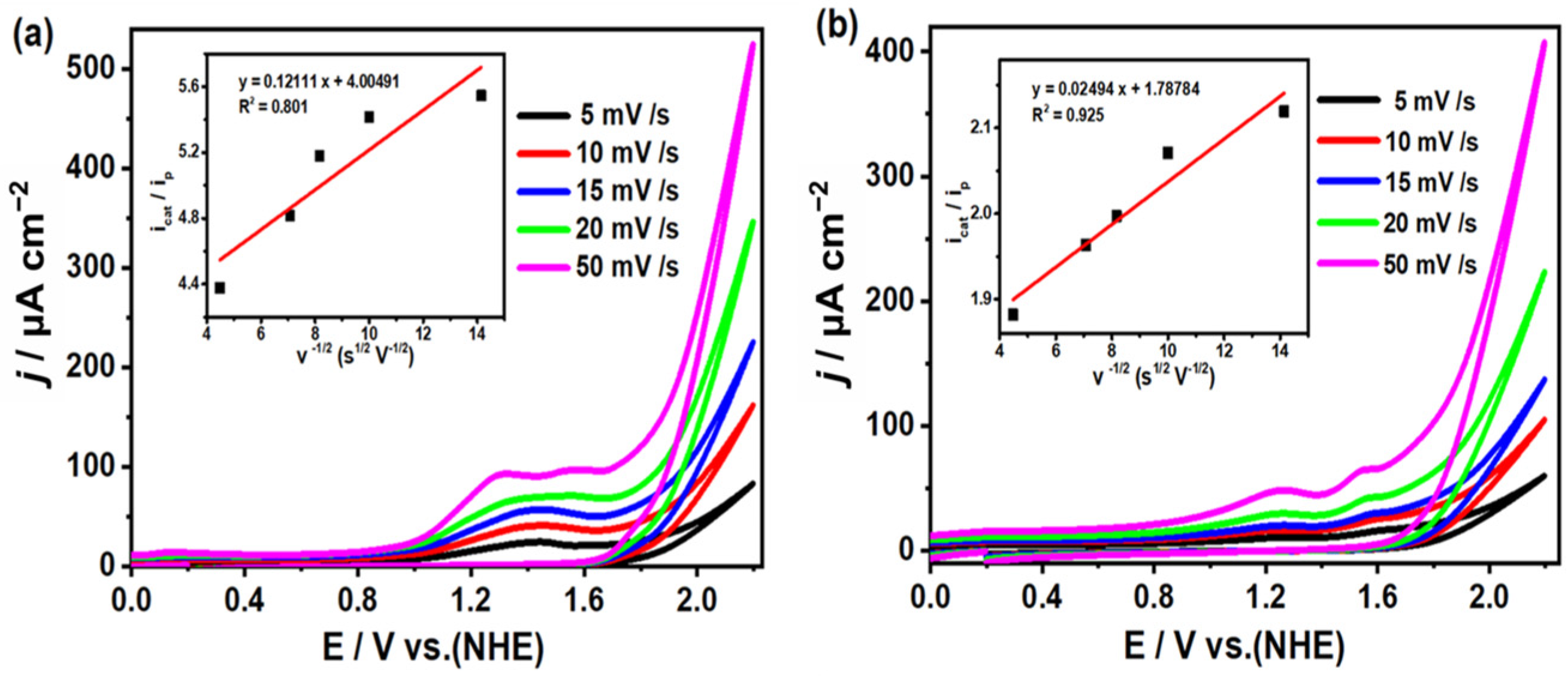
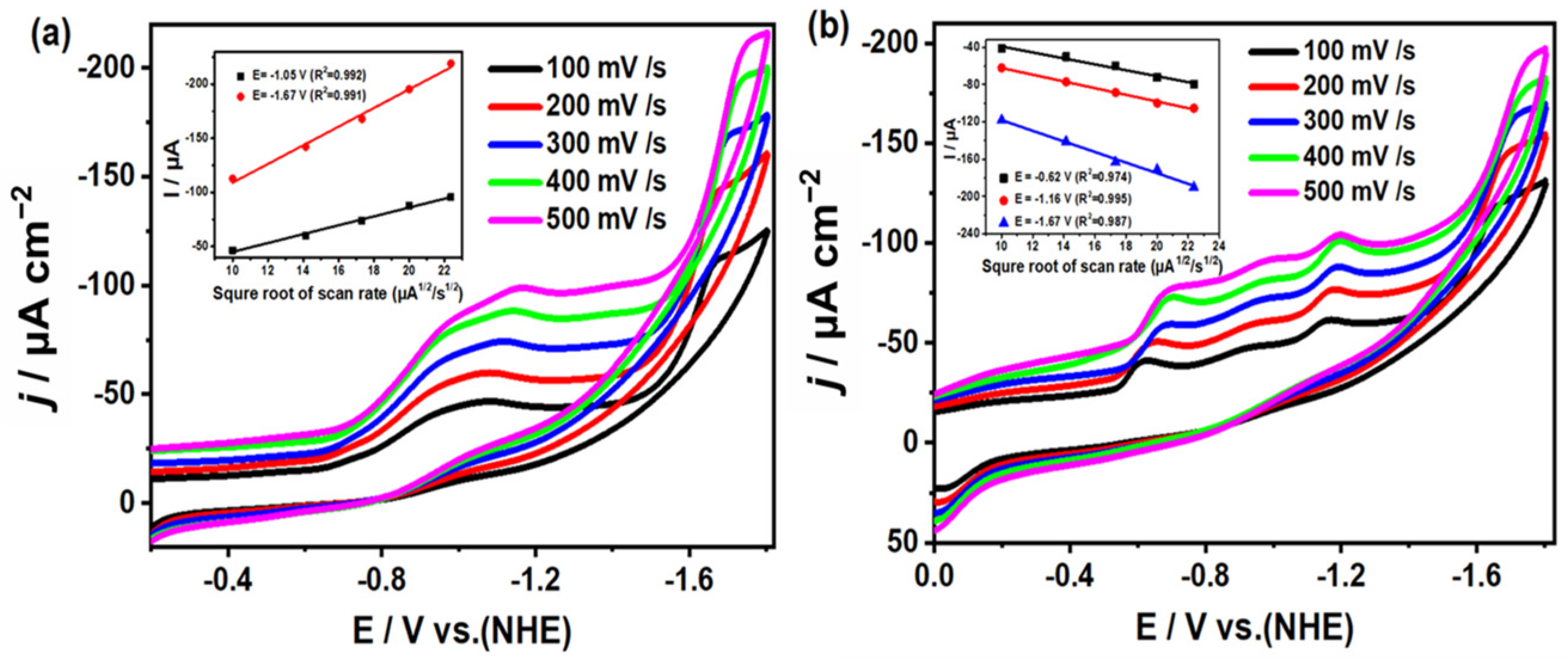
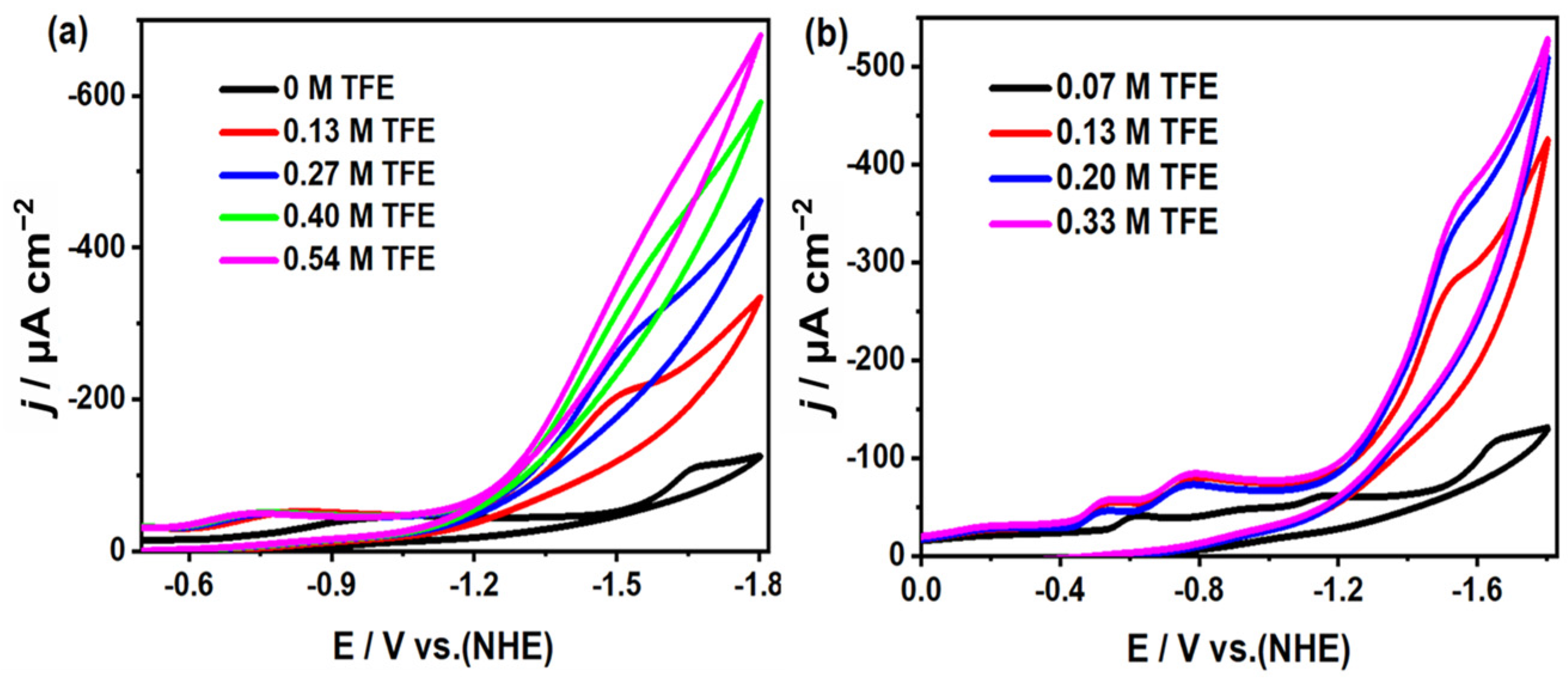
2.5. Study on Electrocatalytic Reduction of CO2 by Complexes 1 and 2
3. Experiments
3.1. General Procedures
3.2. Synthesis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zeng, Z.; Gan, L.Y.; Bin Yang, H.; Su, X.; Gao, J.; Liu, W.; Matsumoto, H.; Gong, J.; Zhang, J.; Cai, W. Orbital coupling of hetero-diatomic nickel-iron site for bifunctional electrocatalysis of CO2 reduction and oxygen evolution. Nat. Commun. 2021, 12, 4088. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.H.; Mathew, S.; Hessels, J.; Reek, J.N.; Yu, F. Homogeneous Catalysts Based on First-Row Transition-Metals for Electrochemical Water Oxidation. ChemSusChem 2021, 14, 234–250. [Google Scholar] [PubMed]
- Bairagi, A.; Pereverzev, A.Y.; Tinnemans, P.; Pidko, E.A.; Roithová, J. Electrocatalytic CO2 reduction: Monitoring of catalytically active, downgraded, and upgraded cobalt complexes. J. Am. Chem. Soc. 2024, 146, 5480–5492. [Google Scholar] [PubMed]
- Barma, A.; Sarkar, A.; Roy, S.; Show, B.; Roy, P. Synthesis and Characterization of a Mononuclear Nickel (II) Complex: Its Dual Electrocatalytic Performance towards Hydrogen Evolution Reaction and Hydrazine Oxidation. ChemistrySelect 2023, 8, e202302725. [Google Scholar] [CrossRef]
- Paul, A.; Adhikary, S.D.; Kapurwan, S.; Konar, S. En route to artificial photosynthesis: The role of polyoxometalate based photocatalysts. J. Mater. Chem. A 2022, 10, 13152–13169. [Google Scholar]
- Yoshino, S.; Takayama, T.; Yamaguchi, Y.; Iwase, A.; Kudo, A. CO2 reduction using water as an electron donor over heterogeneous photocatalysts aiming at artificial photosynthesis. Acc. Chem. Res. 2022, 55, 966–977. [Google Scholar]
- Jiang, S.; Zhang, M.; Xu, C.; Liu, G.; Zhang, K.; Zhang, Z.; Peng, H.-Q.; Liu, B.; Zhang, W. Recent Developments in Nickel-Based Layered Double Hydroxides for Photo (-/) electrocatalytic Water Oxidation. ACS nano 2024, 18, 16413–16449. [Google Scholar] [CrossRef]
- Ruan, G.; Fridman, N.; Maayan, G. Borate Buffer as a Key Player in Cu-Based Homogeneous Electrocatalytic Water Oxidation. Chem. Eur. J. 2022, 28, e202202407. [Google Scholar]
- Gond, M.; Pandey, S.K.; Chaudhari, U.; Sonker, P.; Bharty, M.; Ganesan, V.; Prashanth, B.; Singh, S. Synthesis, crystal structures and electrocatalytic water oxidation by Mn (II), Co (II) and Ni (II) complexes of thiophene-2-carbohydrazide. J. Mol. Struct. 2022, 1270, 133886. [Google Scholar]
- Zhao, X.; Li, J.; Jian, H.; Lu, M.; Wang, M. Two novel schiff base manganese complexes as bifunctional electrocatalysts for CO2 reduction and water oxidation. Molecules 2023, 28, 1074. [Google Scholar] [CrossRef]
- Kamboj, N.; Metre, R.K. Designing a Phenalenyl-Based Dinuclear Ni (II) Complex: An Electrocatalyst with Two Single Ni Sites for the Oxygen Evolution Reaction (OER). Inorg. Chem. 2024, 63, 9771–9785. [Google Scholar] [PubMed]
- Kour, G.; Mao, X.; Du, A. First principles studies of mononuclear and dinuclear Pacman complexes for electrocatalytic reduction of CO2. Catal. Sci. Technol. 2021, 11, 637–645. [Google Scholar]
- Ghosh, T.; Maayan, G. Efficient homogeneous electrocatalytic water oxidation by a manganese cluster with an overpotential of only 74 mV. Angew. Chem. 2019, 131, 2811–2816. [Google Scholar]
- Tamtaji, M.; Goddard, W.A.; Chen, G. High-throughput screening of mechanically interlocked Catenane metal complexes for enhanced electrocatalytic activity. J. Mater. Chem. A 2024, 12, 33948–33957. [Google Scholar]
- Vijayakumar, M.; Yhobu, Z.; Małecki, J.G.; Nagaraju, D.H.; Keri, R.S.; Budagumpi, S. Comprehensive enhancement in electrocatalytic oxygen evolution performance of nickel and cobalt complexes derived from π-conjugated N-heterocyclic carbene ligands through carbon composite strategy. Catal. Sci. Technol. 2024, 14, 2489–2502. [Google Scholar]
- Jiang, X.; Li, J.; Yang, B.; Wei, X.Z.; Dong, B.W.; Kao, Y.; Huang, M.Y.; Tung, C.H.; Wu, L.Z. A Bio-inspired Cu4O4 Cubane: Effective Molecular Catalysts for Electrocatalytic Water Oxidation in Aqueous Solution. Angew. Chem. 2018, 130, 7976–7980. [Google Scholar]
- Jiao, F.; Zhang, X.; Zhang, T.; Hu, Y.; Lu, R.; Ma, G.; Chen, T.; Guo, H.; Li, D.; Pan, Y. Insights into carbon-neutral treatment of rural wastewater by constructed wetlands: A review of current development and future direction. Environ. Res. 2024, 262, 119796. [Google Scholar]
- Caiardi, F.; Azzaro-Pantel, C.; Le-Boulch, D. Exploring carbon neutrality scenarios through the life cycle assessment lens: A review of literature and methodological challenges. Environ. Dev. Sustain. 2024, 1–24. [Google Scholar] [CrossRef]
- Zhang, X.; Huang, W.; Yu, L.; García-Melchor, M.; Wang, D.; Zhi, L.; Zhang, H. Enabling heterogeneous catalysis to achieve carbon neutrality: Directional catalytic conversion of CO2 into carboxylic acids. Carbon Energy 2024, 6, e362. [Google Scholar]
- Zhao, Q.; Lei, K.; Xia, B.Y.; Crespo-Otero, R.; Di Tommaso, D. Molecular engineering binuclear copper catalysts for selective CO2 reduction to C2 products. J. Energy Chem. 2024, 93, 166–173. [Google Scholar]
- Chang, X.; Wang, T.; Yang, P.; Zhang, G.; Gong, J. The development of cocatalysts for photoelectrochemical CO2 reduction. Adv. Mater. 2019, 31, 1804710. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Zhang, Y. Recent Advances in Electrocatalytic Conversion of CO2-to-Ethylene: From Reaction Mechanisms to Tuning Strategies. Appl. Catal. B-Environ. Energy 2024, 363, 124822. [Google Scholar] [CrossRef]
- Xu, W.; Shang, H.; Guan, J.; Yang, X.; Jin, X.; Tao, L.; Shao, Z. Research Progress of Dual-Site Tandem Catalysts in the Preparation of Multi Carbon Products by Electro Reduction of CO2. Adv. Funct. Mater. 2024, 35, 2412812. [Google Scholar] [CrossRef]
- Liu, C.; Guo, R.-T.; Zhu, H.-w.; Cui, H.-f.; Liu, M.-y.; Pan, W.-G. Cu2O-based catalysts applied for CO2 electrocatalytic reduction: A review. J. Mater. Chem. A. 2024, 12, 31769–31796. [Google Scholar] [CrossRef]
- Yang, Y.; Xie, F.; Chen, J.; Qiu, S.; Qiang, N.; Lu, M.; Peng, Z.; Yang, J.; Liu, G. Electrocatalytic Reduction of CO2 to CO by Molecular Cobalt-Polypyridine Diamine Complexes. Molecules 2024, 29, 1694. [Google Scholar] [CrossRef]
- Zhang, S.; Fan, Q.; Xia, R.; Meyer, T.J. CO2 reduction: From homogeneous to heterogeneous electrocatalysis. Acc. Chem. Res. 2020, 53, 255–264. [Google Scholar] [CrossRef]
- Skavenborg, M.L.; Møller, M.S.; Miller, C.J.; Hjelm, J.; Waite, T.D.; McKenzie, C.J. Electrocatalysis of the Oxygen Reduction Reaction by Copper Complexes with Tetradentate Tripodal Ligands. Inorg. Chem. 2023, 62, 18219–18227. [Google Scholar] [CrossRef]
- Branch, K.L.; Johnson, E.R.; Nichols, E.M. Porphyrin Aggregation under Homogeneous Conditions Inhibits Electrocatalysis: A Case Study on CO2 Reduction. ACS Cent. Sci. 2024, 10, 1251–1261. [Google Scholar] [CrossRef]
- Narouz, M.R.; De La Torre, P.; An, L.; Chang, C.J. Multifunctional Charge and Hydrogen-Bond Effects of Second-Sphere Imidazolium Pendants Promote Capture and Electrochemical Reduction of CO2 in Water Catalyzed by Iron Porphyrins. Angew. Chem. Int. Ed. 2022, 61, e202207666. [Google Scholar] [CrossRef]
- Franco, F.; Pinto, M.F.; Royo, B.; Lloret-Fillol, J. A Highly Active N-Heterocyclic Carbene Manganese (I) Complex for Selective Electrocatalytic CO2 Reduction to CO. Angew. Chem. 2018, 130, 4693–4696. [Google Scholar] [CrossRef]
- Wawrzyniak, A.; Koper, M.T. Electrocatalytic CO2 Reduction to Methanol on Pt(111) Modified with a Pd Monolayer. ACS Catal. 2025, 15, 1514–1521. [Google Scholar] [PubMed]
- Yang, Y.; Ji, Y.; Li, G.Y.; Li, Y.Y.; Jia, B.H.; Yuan, J.Q.; Ma, T.Y.; Liu, S.Z. IrOx@In2O3 Heterojunction from Individually Crystallized Oxides for Weak-Light-Promoted Electrocatalytic Water Oxidation. Angew. Chem. Int. Ed. 2021, 60, 26790–26797. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Shan, H.Y.; Li, J.; Wang, Y.; Xu, H.; Wang, C.Y.; Guo, J.; Du, Y.K. Ultralow Ru doping induced interface engineering in MOF derived ruthenium-cobalt oxide hollow nanobox for efficient water oxidation electrocatalysis. Chem. Eng. J. 2021, 420, 129805. [Google Scholar]
- Zhang, L.; Wei, Z.C.; Thanneeru, S.; Meng, M.; Kruzyk, M.; Ung, G.; Liu, B.; He, J. A Polymer Solution to Prevent Nanoclustering and Improve the Selectivity of Metal Nanoparticles for Electrocatalytic CO2 Reduction. Angew. Chem. Int. Ed. 2019, 58, 15834–15840. [Google Scholar] [CrossRef]
- Saha, S.; Sahil, S.T.; Mazumder, M.M.R.; Stephens, A.M.; Cronin, B.; Duin, E.C.; Jurss, J.W.; Farnum, B.H. Synthesis, characterization, and electrocatalytic activity of bis (pyridylimino) isoindoline Cu (II) and Ni (II) complexes. Dalton Trans. 2021, 50, 926–935. [Google Scholar]
- Fisher, K.J.; Materna, K.L.; Mercado, B.Q.; Crabtree, R.H.; Brudvig, G.W. Electrocatalytic water oxidation by a copper (II) complex of an oxidation-resistant ligand. ACS Catal. 2017, 7, 3384–3387. [Google Scholar]
- Rudshteyn, B.; Fisher, K.J.; Lant, H.M.; Yang, K.R.; Mercado, B.Q.; Brudvig, G.W.; Crabtree, R.H.; Batista, V.S. Water-nucleophilic attack mechanism for the CuII (pyalk) 2 water-oxidation catalyst. ACS Catal. 2018, 8, 7952–7960. [Google Scholar]
- Makhado, T.; Das, B.; Kriek, R.; Vosloo, H.; Swarts, A. Chemical and electrochemical water oxidation mediated by bis (pyrazol-1-ylmethyl) pyridine-ligated Cu (i) complexes. Sustain. Energy Fuels 2021, 5, 2771–2780. [Google Scholar]
- Yang, Z.; Deng, B.; Hu, L.; Du, K.; Yin, H.; Wang, D. Selective CO2 Electroreduction with enhanced oxygen evolution efficiency in affordable borate-mediated molten electrolyte. ACS Energy Lett. 2023, 8, 1762–1771. [Google Scholar]
- Serna, Z.; Barandika, M.G.; Cortés, R.; Urtiaga, M.K.; Arriortua, M.I. Crystal structure and esr spectra of two M (II)-dpk-NCS coordination compounds (M= Mn, Cu and dpk= di-2-pyridylketone). Polyhedron 1998, 18, 249–255. [Google Scholar]
- Latham, K.; White, K.F.; Szpakolski, K.B.; Rix, C.J.; White, J.M. Synthesis, crystal structure and luminescent behaviour of coordination complexes of copper with bi-and tridentate amines and phosphonic acids. Inorg. Chim. Acta 2009, 362, 1872–1886. [Google Scholar]
- Szpakolski, K.B.; Latham, K.; Rix, C.J.; White, J.M. Di (2-pyridyl) Ketone Complexes of CuI-and CuII-Containing Iodide and Thiocyanate Ligands: An Unusual Case of a Mixed-Aldol Condensation. Eur. J. Inorg. Chem. 2010, 2010, 5660–5667. [Google Scholar]
- Domínguez, S.; Torres, J.; González-Platas, J.; Hummert, M.; Schumann∥, H.; Kremer, C. Thermodynamic stability and crystal structure of lanthanide complexes with di-2-pyridyl ketone. J. Coord. Chem. 2009, 62, 108–119. [Google Scholar]
- Feller, M.; Robson, R. Complexes of Di-2-pyridyl ketone. I. Complexes of first-row transition elements in normal oxidation states. Aust. J. Chem. 1968, 21, 2919–2927. [Google Scholar]
- Kulkarni, P.; Padhye, S.; Sinn, E.; Anson, C.E.; Powell, A.K. Comparative studies on copper (I) complexes: Synthesis, X-ray crystallography and electrochemical properties of [CuI (dafone) nX] complexes (dafone= 4, 5-diaza-fluoren-9-one, X= Br, I, SCN). Inorg. Chim. Acta 2002, 332, 167–175. [Google Scholar] [CrossRef]
- Morpurgo, G.O.; Dessy, G.; Fares, V. Crystal structures and spectroscopic properties of the polymeric adducts formed from Cu (CN) and Cu (NCS) with 2, 9-dimethyl-1, 10-phenanthroline. J. Chem. Soc. Dalton Trans. 1984, 5, 785–791. [Google Scholar]
- Bakir, M.; McKenzie, J.A. Electrochemical reactions of CO2 with fac-Re (dpk)(CO)3Cl (dpk= di-2-pyridyl ketone). J. Electroanal. Chem. 1997, 425, 61–66. [Google Scholar]
- Bakir, M.; Hassan, I.; Green, O. Manganese carbonyl compounds of N, N-bidentate di-2-pyridylketone (dpk) and N, O, N-tridentate hydroxybis (2-pyridyl) methanolato (dpkO, OH). The structure of fac- [Mn (CO)3 (dpkO, OH)]. J. Mol. Struct. 2003, 657, 75–83. [Google Scholar]
- Zhao, Y.; Lin, J.; Liu, Y.; Ma, B.; Ding, Y.; Chen, M. Efficient light-driven water oxidation catalyzed by a mononuclear cobalt (iii) complex. Chem. Commun. 2015, 51, 17309–17312. [Google Scholar]
- Lin, J.; Liang, X.; Cao, X.; Wei, N.; Ding, Y. An octanuclear Cu (ii) cluster with a bio-inspired Cu4O4cubic fragment for efficient photocatalytic water oxidation. Chem. Commun. 2018, 54, 12515–12518. [Google Scholar]
- Stamatatos, T.C.; Efthymiou, C.G.; Stoumpos, C.C.; Perlepes, S.P. Adventures in the Coordination Chemistry of Di-2-pyridyl Ketone and Related Ligands: From High-Spin Molecules and Single-Molecule Magnets to Coordination Polymers, and from Structural Aesthetics to an Exciting New Reactivity Chemistry of Coordinated Ligands. Eur. J. Inorg. Chem. 2009, 2009, 3361–3391. [Google Scholar]
- Serna, Z.E.; Cortés, R.; Urtiaga, M.K.; Barandika, M.G.; Lezama, L.; Arriortua, M.I.; Rojo, T. Investigation of the CuII/NCS−/dpk Reaction System in CH3OH [dpk= Di (2-pyridyl) Ketone]: Isolation, Structural Analysis and Magnetic Properties of a Dimer and a 1D Polymer with the Same Empirical Formula [Cu (NCS)2 (dpk· CH3OH)]. Eur. J. Inorg. Chem. 2001, 2001, 865–872. [Google Scholar] [CrossRef]
- Bakir, M.; McKenzie, J.A. Rhenium (I) di-and tri-carbonyl compounds of polypyridyl-like ligands: Electrochemical reactions of fac- [Re (CO)3 (dpk) Cl](dpk= di-2-pyridyl ketone) with electrophiles and Group I and II metal ions. J. Chem. Soc. Dalton Trans. 1997, 3571–3578. [Google Scholar] [CrossRef]
- Zhang, T.; Wang, C.; Liu, S.; Wang, J.-L.; Lin, W. A biomimetic copper water oxidation catalyst with low overpotential. J. Am. Chem. Soc. 2014, 136, 273–281. [Google Scholar] [CrossRef]
- Li, P.; Liu, J.B.; Han, S.; Deng, W.; Yao, Z.J. Half-sandwich Ir (III) and Rh (III) complexes as catalysts for water oxidation with double-site. Appl. Organomet. Chem. 2019, 33, 5040. [Google Scholar] [CrossRef]
- Gerlach, D.L.; Bhagan, S.; Cruce, A.A.; Burks, D.B.; Nieto, I.; Truong, H.T.; Kelley, S.P.; Herbst-Gervasoni, C.J.; Jernigan, K.L.; Bowman, M.K. Studies of the pathways open to copper water oxidation catalysts containing proximal hydroxy groups during basic electrocatalysis. Inorg. Chem. 2014, 53, 12689–12698. [Google Scholar] [CrossRef]
- Lee, H.; Wu, X.; Sun, L. Copper-based homogeneous and heterogeneous catalysts for electrochemical water oxidation. Nanoscale 2020, 12, 4187–4218. [Google Scholar] [CrossRef]
- Binaeizadeh, M.R.; Amiri, A.; Shayesteh, A.; Fadaei-Tirani, F. Carboxamide Fe (III) complex as the electrocatalyst of water oxidation reaction: WNA and I2M O–O bond formation pathways. Int. J. Hydrog. Energy 2024, 51, 709–721. [Google Scholar] [CrossRef]
- Shee, U.; Sinha, D.; Mondal, S.; Rajak, K.K. Electrochemical water oxidation reaction by dinuclear Re (v) oxo complexes with a 1, 4-benzoquinone core via the redox induced electron transfer (RIET) process. Dalton Trans. 2024, 53, 8254–8263. [Google Scholar] [CrossRef]
- Kamlesh; Aggarwal, P.; Mudgal, M.; Srivastava, A.K.; Raizada, P.; Singh, P.; Paul, A.; Singh, A. Electrochemistry of Nickelocene-Ferrocene Organometallic Complexes for Electrodeposition of Nickel–Iron–Based Nanostructured Film under Ambient Conditions for Oxygen Evolution Reaction. ACS Appl. Nano Mater. 2024, 7, 24455–24468. [Google Scholar] [CrossRef]
- Chen, X.; Sun, W.; Meng, X.; Gao, Y. Assembly of a Highly Efficient Molecular Device with (CNCbl)-MWCNT/CP as Electrode for CO2 Reduction Coupled to Water Oxidation. ChemElectroChem 2021, 8, 3567–3571. [Google Scholar]
- Guo, C.; Gao, S.; Li, J.; Zhou, M.; Abdukayum, A.; Kong, Q.; Zhou, Y.; Hu, G. P-tuned FeN2 binuclear sites for boosted CO2 electro-reduction. J. Energy Chem. 2025, 101, 816–824. [Google Scholar]
- Yin, X.M.; Zhang, S.F.; Wang, J.M.; Li, J.J.; Chen, F.F.; Yao, S.; Fan, Y.H.; Wang, M. Bioinspired cobalt molecular electrocatalyst for water oxidation coupled with carbon dioxide reduction. Appl. Organomet. Chem. 2021, 35, 6371. [Google Scholar]
- Hooe, S.L.; Dressel, J.M.; Dickie, D.A.; Machan, C.W. Highly Efficient Electrocatalytic Reduction of CO2 to CO by a Molecular Chromium Complex. ACS Catal. 2020, 10, 1146–1151. [Google Scholar]
- Ahsan, H.M.; Breedlove, B.K.; Piangrawee, S.; Mian, M.R.; Fetoh, A.; Cosquer, G.; Yamashita, M. Enhancement of electrocatalytic abilities for reducing carbon dioxide: Functionalization with a redox-active ligand-coordinated metal complex. Dalton Trans. 2018, 47, 11313–11316. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, S.; Liu, T.; Huang, W.; Zhang, C.; Wang, M. Electrocatalytic CO2 Reduction Coupled with Water Oxidation by bi- and Tetranuclear Copper Complexes Based on di-2-pyridyl Ketone Ligand. Molecules 2025, 30, 1544. https://doi.org/10.3390/molecules30071544
Yang S, Liu T, Huang W, Zhang C, Wang M. Electrocatalytic CO2 Reduction Coupled with Water Oxidation by bi- and Tetranuclear Copper Complexes Based on di-2-pyridyl Ketone Ligand. Molecules. 2025; 30(7):1544. https://doi.org/10.3390/molecules30071544
Chicago/Turabian StyleYang, Siyuan, Tian Liu, Wenbo Huang, Chengwen Zhang, and Mei Wang. 2025. "Electrocatalytic CO2 Reduction Coupled with Water Oxidation by bi- and Tetranuclear Copper Complexes Based on di-2-pyridyl Ketone Ligand" Molecules 30, no. 7: 1544. https://doi.org/10.3390/molecules30071544
APA StyleYang, S., Liu, T., Huang, W., Zhang, C., & Wang, M. (2025). Electrocatalytic CO2 Reduction Coupled with Water Oxidation by bi- and Tetranuclear Copper Complexes Based on di-2-pyridyl Ketone Ligand. Molecules, 30(7), 1544. https://doi.org/10.3390/molecules30071544