
Cyclohexane Vibronic States: A Combined VUV Spectroscopy and Theoretical Study

 , , , ,
, , , ,  and
and
Abstract

1. Introduction
2. Structure and Properties of Cyclohexane
3. Discussion
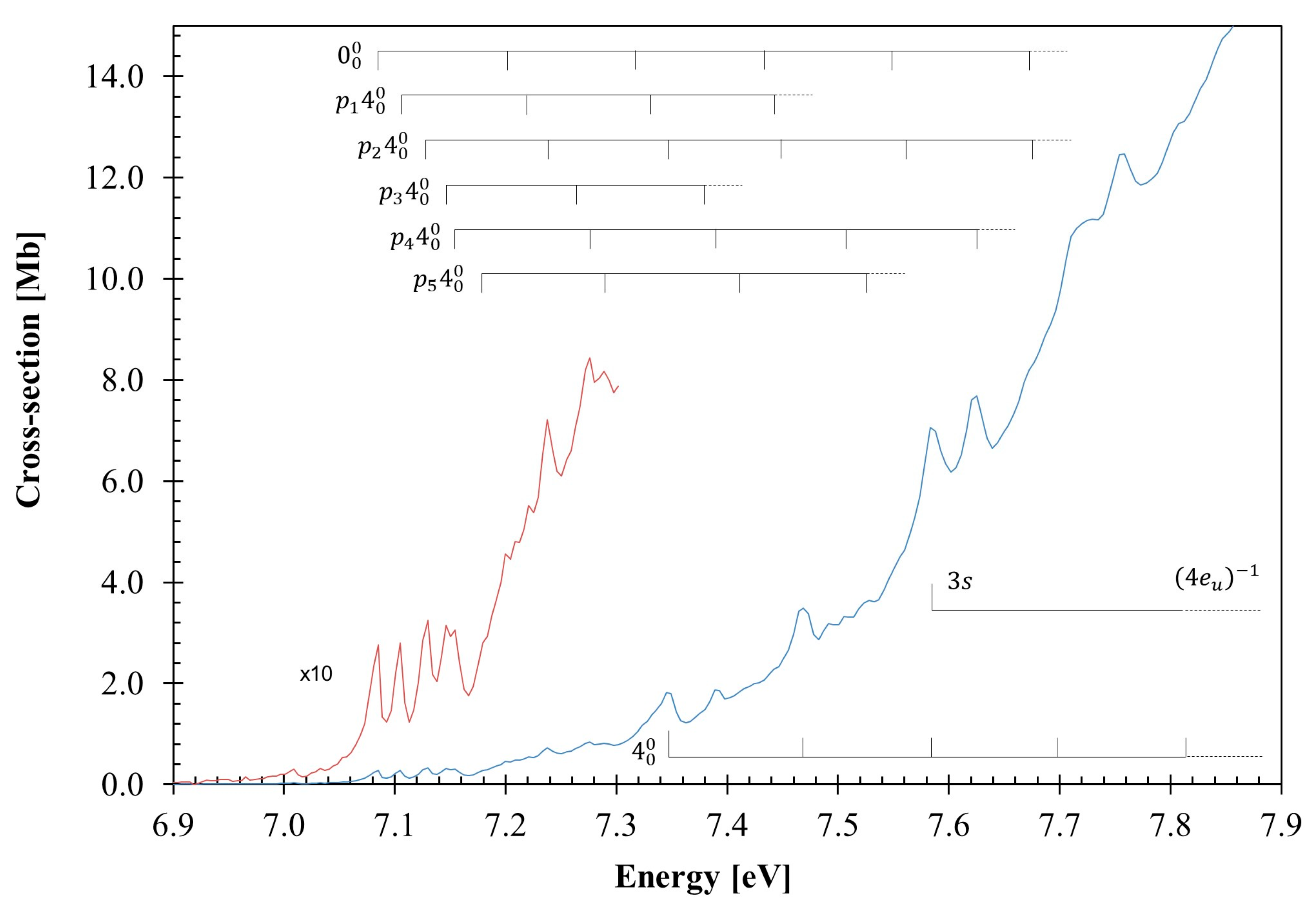
3.1. The 7.0–7.9 eV Photon Energy Range
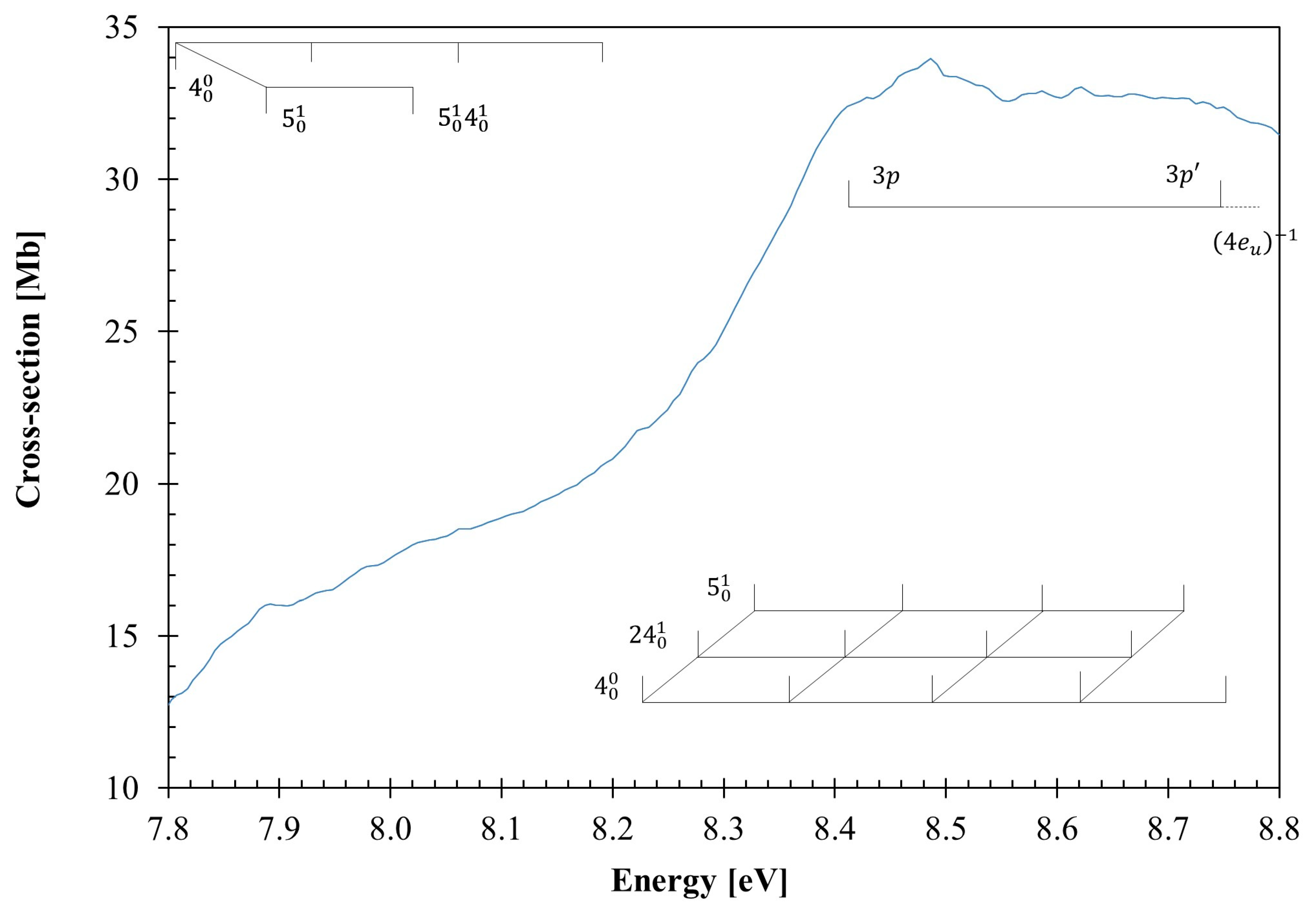
3.2. The 7.8–8.8 eV Photon Energy Range
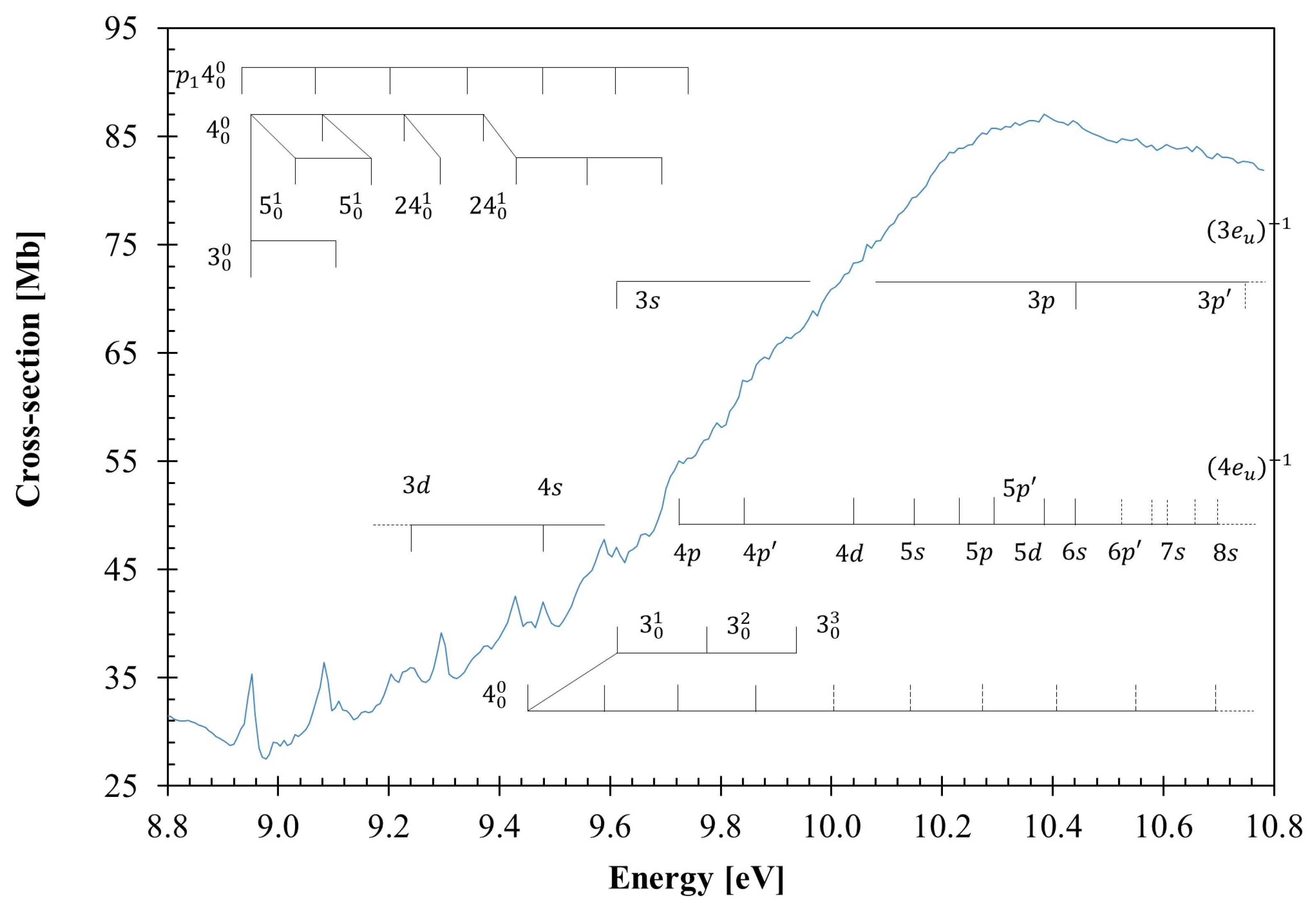
3.3. The 8.8–10.8 eV Photon Energy Range
3.4. Rydberg Transitions
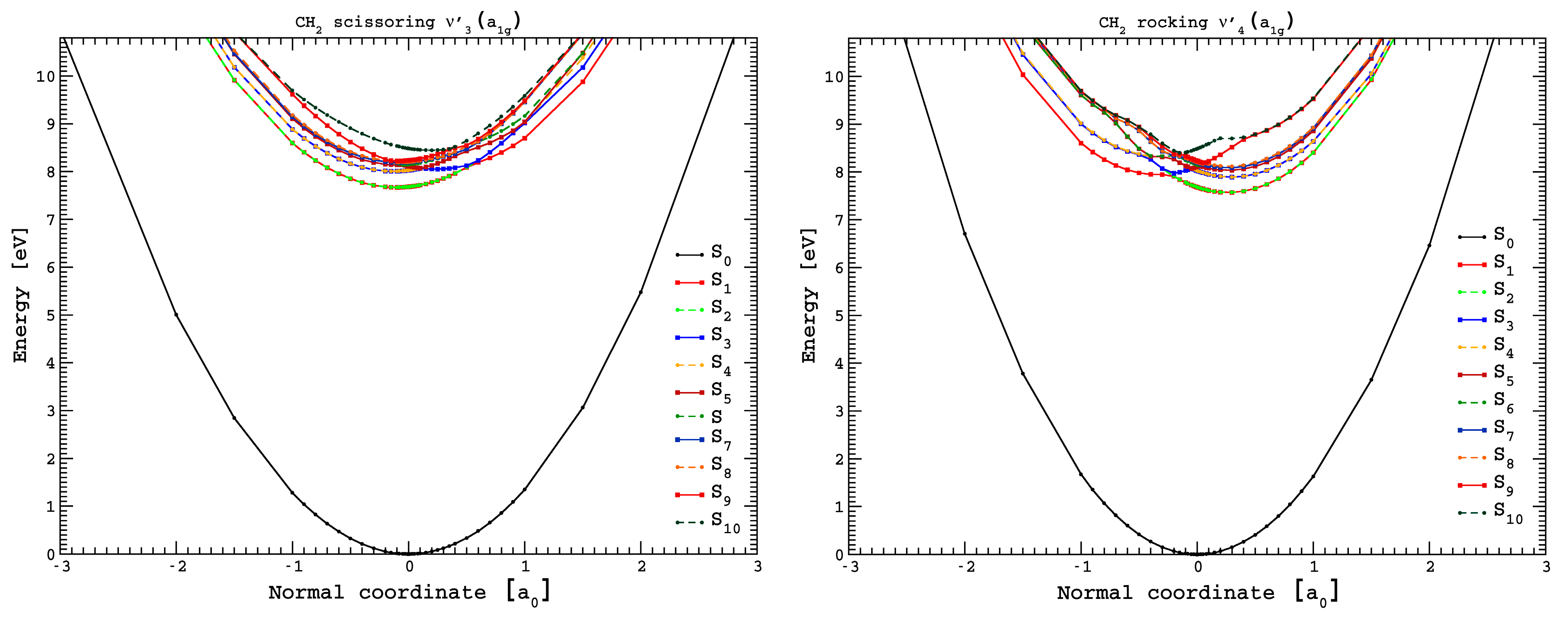
3.5. Potential Energy Curves for CH2 Scissoring and CH2 Rocking Coordinates
3.6. Absolute Photoabsorption Cross-Sections and Atmospheric Photolysis
4. Materials and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Harley, R.A.; Cassi, G.R. Modeling the Atmospheric Concentrations of Individual Volatile Organic Compounds. Atmos. Environ. 1995, 29, 905–922. [Google Scholar]
- Aschmann, S.M.; Chew, A.A.; Arey, J.; Atkinson, R. Products of the Gas-Phase Reaction of OH Radicals with Cyclohexane: Reactions of the Cyclohexoxy Radical. J. Phys. Chem. A 1997, 101, 8042–8048. [Google Scholar]
- McEnally, C.S.; Pfefferle, L.D. Experimental Study of Fuel Decomposition and Hydrocarbon Growth Processes for Cyclohexane and Related Compounds in Nonpremixed Flames. Combust. Flame 2004, 136, 155–167. [Google Scholar] [CrossRef]
- Dahiru, U.H.; Saleem, F.; Zhang, K.; Harvey, A.P. Removal of Cyclohexane as a Toxic Pollutant from Air Using a Non-Thermal Plasma: Influence of Different Parameters. J. Environ. Chem. Eng. 2021, 9, 105023. [Google Scholar] [CrossRef]
- Atkinson, R. Gas-Phase Tropospheric Chemistry of Volatile Organic Compounds: 1. Alkanes and Alkenes. J. Phys. Chem. Ref. Data 1997, 26, 215–290. [Google Scholar] [CrossRef]
- Sage, A.M.; Donahue, N.M. Deconstructing Experimental Rate Constant Measurements: Obtaining Intrinsic Reaction Parameters, Kinetic Isotope Effects, and Tunneling Coefficients from Kinetic Data for OH+methane, Ethane and Cyclohexane. J. Photochem. Photobiol. A Chem. 2005, 176, 238–249. [Google Scholar] [CrossRef]
- Chen, M.W.; Rotavera, B.; Chao, W.; Zádor, J.; Taatjes, C.A. Direct Measurement of OH and HO2 Formation in R + O2 Reactions of Cyclohexane and Tetrahydropyran. Phys. Chem. Chem. Phys. 2018, 20, 10815–10825. [Google Scholar] [CrossRef]
- Voisin, D.; Marchal, A.; Reuillon, M.; Boettner, J.C.; Cathonnet, M. Experimental and Kinetic Modeling Study of Cyclohexane Oxidation in a JSR at High Pressure. Combust. Sci. Technol. 1998, 138, 137–158. [Google Scholar] [CrossRef]
- Rotavera, B.; Savee, J.D.; Antonov, I.O.; Caravan, R.L.; Sheps, L.; Osborn, D.L.; Zádor, J.; Taatjes, C.A. Influence of Oxygenation in Cyclic Hydrocarbons on Chain-Termination Reactions from R + O2: Tetrahydropyran and Cyclohexane. Proc. Combust. Inst. 2017, 36, 597–606. [Google Scholar] [CrossRef]
- Granata, S.; Faravelli, T.; Ranzi, E. A Wide Range Kinetic Modeling Study of the Pyrolysis and Combustion of Naphthenes. Combust. Flame 2003, 132, 533–544. [Google Scholar] [CrossRef]
- Mason, N.J.; Dawes, A.; Mukerji, R.; Drage, E.A.; Vasekova, E.; Webb, S.M.; Limão-Vieira, P. Atmospheric Chemistry with Synchrotron Radiation. J. Phys. B At. Mol. Opt. Phys. 2005, 38, S893–S911. [Google Scholar] [CrossRef]
- Serralheiro, C.; Duflot, D.; Silva, F.F.; Hoffmann, S.V.; Jones, N.C.; Mason, N.J.; Mendes, B.; Limão-Vieira, P. Toluene Valence and Rydberg Excitations as Studied by Ab Initio Calculations and Vacuum Ultraviolet (VUV) Synchrotron Radiation. J. Phys. Chem. A 2015, 119, 9059–9069. [Google Scholar] [PubMed]
- Limão-Vieira, P.; Duflot, D.; Silva, F.F.; Lange, E.; Jones, N.C.; Hoffmann, S.V.; Ǧmiałek, M.A.; Jones, D.B.; Brunger, M.J. Valence and Lowest Rydberg Electronic States of Phenol Investigated by Synchrotron Radiation and Theoretical Methods. J. Chem. Phys. 2016, 145, 034302. [Google Scholar] [CrossRef]
- Śmiałek, M.A.; Hubin-Franskin, M.J.; Delwiche, J.; Duflot, D.; Mason, N.J.; Vrønning-Hoffmann, S.; de Souza, G.G.B.; Ferreira Rodrigues, A.M.; Rodrigues, F.N.; Limão-Vieira, P. Limonene: Electronic State Spectroscopy by High-Resolution Vacuum Ultraviolet Photoabsorption, Electron Scattering, He(I) Photoelectron Spectroscopy and Ab Initio Calculations. Phys. Chem. Chem. Phys. 2012, 14, 2056–2064. [Google Scholar] [CrossRef]
- Martins, G.; Ferreira-Rodrigues, A.M.; Rodrigues, F.N.; de Souza, G.G.B.; Mason, N.J.; Eden, S.; Duflot, D.; Flament, J.-P.; Hoffmann, S.V.; Delwiche, J.; et al. Valence Shell Electronic Spectroscopy of Isoprene Studied by Theoretical Calculations and by Electron Scattering, Photoelectron, and Absolute Photoabsorption Measurements. Phys. Chem. Chem. Phys. 2009, 11, 11219–11231. [Google Scholar] [CrossRef]
- Kubala, D.; Drage, E.A.; Al-Faydhi, A.M.E.; Kočíšek, J.; Papp, P.; Matejčík, V.; Mach, P.; Urban, J.; Limão-Vieira, P.; Hoffmann, S.V.; et al. Electron Impact Ionisation and UV Absorption Study of α- and β-Pinene. Int. J. Mass Spectrom. 2009, 280, 169–173. [Google Scholar] [CrossRef]
- Limão-Vieira, P.; Eden, S.; Kendall, P.A.; Mason, N.J.; Hoffmann, S.V. VUV Photo-Absorption Cross-Section for CCl2F2. Chem. Phys. Lett. 2002, 364, 535–541. [Google Scholar] [CrossRef]
- Arimura, M.; Yoshikawa, Y. Ionization Efficiency and Ionization Energy of Cyclic Compounds by Electron Impact. Mass Spectrom. 1984, 32, 375–380. [Google Scholar]
- Holmes, J.L.; Lossing, F.P. Ionization Energies of Homologous Organic Compounds and Correlation with Molecular Size. Org. Mass Spectrom. 1991, 26, 537–541. [Google Scholar] [CrossRef]
- Jiao, C.Q.; Adams, S.F. Electron Ionization of Selected Cyclohexanes. J. Phys. B At. Mol. Opt. Phys. 2011, 44, 17520. [Google Scholar] [CrossRef]
- Koizumi, H.; Shinsara, K.; Hatano, Y. VUV-Optical Oscillator Strength Distributions of Molecules and Their Implications to Early Events in Radiation Chemistry. Radiat. Phys. Chem 1989, 34, 87–92. [Google Scholar]
- Koizumi, H.; Shinsaka, I.K.; Yostu, T.; Hironaka, K.; Arai, S.; Ug, M.; Morita, M.; Nakazawa, H.; Kimura, A.; Hatano, Y.; et al. Ionization Efficiencies of C3H6, C4H8, C6HI2, C2H6O, and C3H8O Isomers. Radiat. Phys. Chem. 1988, 32, 111–115. [Google Scholar]
- Shimamori, H.; Sunagawa, T. Penning Ionization and Molecular Dissociation in the Deexcitation of Neon Metastable Atoms by Some Polyatomic Molecules. J. Phys. Chem. 1996, 100, 18033–18036. [Google Scholar]
- Doner, A.C.; Christianson, M.G.; Davis, J.C.; Koritzke, A.L.; Larsson, A.; Frandsen, K.; Rotavera, B. Vacuum-Ultraviolet Absorption Cross-Sections of Functionalized Cyclic Hydrocarbons: Six-Membered Rings. J. Quant. Spectrosc. Radiat. Transf. 2019, 236, 106603. [Google Scholar] [CrossRef]
- Pickett, L.W.; Muntz, M.; McPherson, M.M. Vacuum Ultraviolet Absorption Spectra of Cyclic Compounds. I. Cyclohexane, Cyclohexene, Cyclopentane, Cyclopentene and Benzene. J. Am. Chem. Soc. 1951, 73, 4862–4865. [Google Scholar]
- Raymonda, J.W. Rydberg States in Cyclic Alkanes. J. Chem. Phys. 1972, 56, 3912–3920. [Google Scholar] [CrossRef]
- Raymonda, J.W.; Simpson, W.T. Experimental and Theoretical Study of Sigma-Bond Electronic Transitions in Alkanes. J. Chem. Phys. 1967, 47, 430–448. [Google Scholar] [CrossRef]
- Crain, J.; Poon, W.C.-K.; Cairns-Smith, A.; Hatton, P.D. High-Pressure Raman Spectroscopic Study of Cyclohexane C6H12 and C6D12. J. Phys. Chem. 1992, 96, 8168–8173. [Google Scholar]
- Rasmussen, R.S. The Infra-Red Absorption Spectrum and Configuration of Cyclohexane. J. Chem. Phys. 1943, 11, 249–252. [Google Scholar] [CrossRef]
- Takahashi, H.; Shimanouchi, T. Infrared Spectrum and Normal Vibrations of Cyclohexane. J. Mol. Spectrosc. 1964, 13, 43–56. [Google Scholar]
- Shimanouchi, T. Tables of Molecular Vibrational Frequencies; US Government Printing Office: Washington, DC, USA, 1967; Part 1. [Google Scholar]
- Kovac, B.; Klasinc, L. Photoelectron Spectroscopy of Adamantane and Some Adamantanones. Croat. Chem. Acta 1978, 51, 55–74. [Google Scholar]
- Eden, S.; Limão-Vieira, P.; Hoffmann, S.V.; Mason, N.J. VUV Photoabsorption in CF3X (X = Cl, Br, I) Fluoro-Alkanes. Chem. Phys. 2006, 323, 313–333. [Google Scholar] [CrossRef]
- Palmer, M.H.; Ridley, T.; Hoffmann, S.V.; Jones, N.C.; Coreno, M.; De Simone, M.; Grazioli, C.; Biczysko, M.; Baiardi, A.; Limão-Vieira, P. Interpretation of the Vacuum Ultraviolet Photoabsorption Spectrum of Iodobenzene by Ab Initio Computations. J. Chem. Phys. 2015, 142, 134302. [Google Scholar] [CrossRef] [PubMed]
- Bauernschmitt, R.; Ahlrichs, R. Treatment of Electronic Excitations within the Adiabatic Approximation of Time Dependent Density Functional Theory. Chem. Phys. Lett. 1996, 256, 454–464. [Google Scholar]
- Casida, M.E. Time-Dependent Density-Functional Theory for Molecules and Molecular Solids. J. Mol. Struct.-Theochem 2009, 914, 3–18. [Google Scholar]
- Yanai, T.; Tew, D.P.; Handy, N.C. A New Hybrid Exchange-Correlation Functional Using the Coulomb-Attenuating Method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar]
- Barca, G.M.J.; Bertoni, C.; Carrington, L.; Datta, D.; De Silva, N.; Deustua, J.E.; Fedorov, D.G.; Gour, J.R.; Gunina, A.O.; Guidez, E.; et al. Recent Developments in the General Atomic and Molecular Electronic Structure System. J. Chem. Phys. 2020, 152, 154102. [Google Scholar]
- Bode, B.M.; Gordon, M.S. MacMolPlt: A Graphical User Interface for GAMESS. J. Mol. Graphics Mod. 1998, 16, 133–138. [Google Scholar]
- Kashinski, D.O.; Chase, G.M.; Nelson, R.G.; Di Nallo, O.E.; Scales, A.N.; Vanderley, D.L.; Byrd, E.F.C. Harmonic Vibrational Frequencies: Approximate Global Scaling Factors for TPSS, M06, and M11 Functional Families Using Several Common Basis Sets. J. Phys. Chem. A 2017, 121, 2265–2273. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cyclohexane | E (eV) Expt. 2 | Cross- Section (Mb) | ||||
|---|---|---|---|---|---|---|
| State (D3d) | State (C2h) | E (eV) | fL | Dominant Excitations 1 | ||
| 1 1Eu | Bu | 8.026 | 0.000172 | (4a2u) ← (4eg) (90%) | 7.46(9) | 3.49 |
| 1 1A2u | Bu | 8.147 | 0.002014 | ′(5eu) ← (4eg) (44%), ″(5eu) ← (4eg) (44%) | 8.02(0) | 17.99 |
| 2 1Eu | Bu | 8.198 | 0.034207 | (5eu) ← (4eg) (43%), ″(5eu) ← (4eg) (43%) | 8.07(7) | 18.57 |
| 2 1A2u | Bu | 8.487 | 0.221001 | (4a2u) ← (4a1g) (90%) | 8.486 | 33.96 |
| 5 1Eu | Bu | 9.677 | 0.094079 | (5a2u) ← (4eg) (83%) | 9.429 | 42.53 |
| 8 1Eu | Bu | 10.014 | 0.106166 | (5a1g) ← (3eu) (28%), (6a2u) ← (4eg) (33%), (7a2u) ← (4eg) (13%) | 9.92(7) | 66.36 |
| 10 1A2u | Bu | 10.751 | 0.173676 | (7a2u) ← (4a1g) (87%) | 10.393 | 86.81 |
| Assignment | Energy | ΔE (ν3′) | ΔE (ν4′) | ΔE (ν5′) |
|---|---|---|---|---|
| 7.085 | – | – | – | |
| 7.105 | – | – | – | |
| 7.130 | – | – | – | |
| 7.146 | – | – | – | |
| 7.154 | – | – | – | |
| 7.17(9) (s,w) | – | – | 0.094 | |
| 7.20(0) (s) | – | 0.115 | 0.095 | |
| 7.22(1) (w) | – | 0.116 | 0.091 | |
| 7.238 | 0.153 | 0.108 | – | |
| 7.26(3) (s) | – | 0.117 | 0.109 | |
| 7.276 | 0.146 | 0.122 | 0.097 | |
| 7.28(9) (w) | – | 0.110 | – | |
| 7.31(9) (s) | – | 0.119 | 0.098 | |
| 7.32(8) (s) | – | 0.107 | 0.090 | |
| 7.345 | – | 0.107 | – | |
| 7.38(0) (s) | – | 0.117 | 0.104 | |
| 7.389 | 0.151 | 0.113 | – | |
| 7.41(5) (s) | – | 0.126 | 0.096 | |
| 7.42(9) (s,w) | 0.153 | – | 0.101 | |
| 7.43(3) (s) | – | 0.114 | – | |
| 7.44(2) (s) | – | 0.114 | – | |
| 7.44(7) (s) | – | 0.102 | – | |
| 7.51(4) (s,b) | – | 0.125 | 0.099 | |
| 7.52(8) (b,w) | – | 0.113 | 0.099 | |
| 7.54(6) (s) | 0.157 | 0.113 | – | |
| 7.56(0) (s) | – | 0.113 | – | |
| 7.57(0) (s) | 0.141 | – | – | |
| 7.625 | – | 0.111 | 0.097 | |
| 7.67(2) (s) | – | 0.126 | – | |
| 7.67(7) (s) | – | 0.117 | – | |
| 7.69(6) (s,b) | 0.150 | – | – | |
| 7.84(7) (s,w) | 0.151 | – | – | |
| 0.150 | 0.115 | 0.098 | ||
| 7.345 | – | – | – | |
| 7.46(9) | – | 0.124 | – | |
| 7.51(0) (w) | 0.165 | – | – | |
| 7.583 | – | 0.114 | – | |
| 7.67(7) (s) | 0.167 | – | – | |
| 7.69(6) (s,b) | – | 0.113 | – | |
| 7.82(2) (s,b) | – | 0.126 | – | |
| 7.94(3) (s,b) | – | 0.121 | – | |
| 0.153 | 0.120 | – | ||
| Assignment | Energy | ΔE (ν4′) | ΔE (ν5′) | ΔE (ν24′) |
|---|---|---|---|---|
| 7.80(8) (s) | – | – | – | |
| 7.89(2) (s) | – | 0.084 | – | |
| 7.93(3) (s) | 0.125 | – | – | |
| 8.02(0) (s) | 0.128 | – | – | |
| 8.06(7) (s) | 0.134 | – | – | |
| 8.19(5) (s,w) | 0.128 | – | – | |
| 8.22(7) (s) | – | – | – | |
| 8.27(7) (s) | – | – | 0.050 | |
| 8.32(7) (s,w) | – | 0.100 | – | |
| 8.36(0) (s,w) | 0.133 | – | – | |
| 8.41(1) (s,b) | 0.134 | – | 0.051 | |
| 8.46(3) (s,w) | 0.136 | – | – | |
| 8.486 | 0.126 | – | – | |
| 8.53(9) (s,w) | 0.128 | – | 0.053 | |
| 8.58(6) (w) | 0.123 | 0.100 | – | |
| 8.62(2) (b) | 0.136 | – | ||
| 8.67(0) (b,w) | 0.131 | – | 0.048 | |
| 8.71(9) (w) | 0.133 | 0.097 | – | |
| 8.75(0) (b,w) | 0.128 | – | – | |
| 0.130 | 0.095 | 0.051 | ||
| Assignment | Energy | ΔE (ν3′) | ΔE (ν4′) | ΔE (ν5′) | ΔE (ν24′) |
|---|---|---|---|---|---|
| 8.93(3) (s) | – | – | – | – | |
| 8.952 | – | – | – | – | |
| 9.03(7) (w) | – | – | 0.084 | – | |
| 9.06(3) (s) | – | 0.130 | – | – | |
| 9.083 | – | 0.131 | – | – | |
| 9.110 | 0.158 | – | – | – | |
| 9.17(0) (b) | – | 0.133 | 0.087 | – | |
| 9.205 | – | 0.142 | – | – | |
| 9.23(2) (b) | – | 0.149 | – | – | |
| 9.294 | – | 0.124 | – | 0.062 | |
| 9.35(0) (s) | – | 0.145 | – | – | |
| 9.37(1) (w) | – | 0.139 | – | – | |
| 9.429 | – | 0.135 | – | 0.058 | |
| 9.479 | – | 0.129 | – | – | |
| 9.559 | – | 0.130 | – | – | |
| 9.611 | – | 0.132 | – | – | |
| 9.69(4) (s,w) | – | 0.135 | – | – | |
| 9.74(7) (w) | – | 0.129 | – | – | |
| 9.45(7) (w) | – | – | – | – | |
| 9.589 | – | 0.132 | – | – | |
| 9.611 | 0.154 | – | – | – | |
| 9.72(4) (w) | – | 0.135 | – | – | |
| 9.77(8) (s,b) | 0.167 | – | – | – | |
| 9.86(4) (s,w) | – | 0.140 | – | – | |
| 9.92(7) (s,w) | 0.149 | – | – | – | |
| 9.99(9) (s,w) | – | 0.135 | – | – | |
| 10.14(6) (s,w) | – | 0.147 | – | – | |
| 10.28(1) (s,w) | – | 0.135 | – | – | |
| 10.41(0) (s,w) | – | 0.129 | – | – | |
| 10.55(2) (s,w) * | – | 0.142 | – | – | |
| 10.69(8) (s,w) * | – | 0.146 | – | – | |
| 0.157 | 0.136 | 0.086 | 0.060 | ||
| En | δ | Assignment | En | δ | Assignment |
|---|---|---|---|---|---|
| (IE3)v = 10.98 eV | (IE5)v = 13.03 eV | ||||
| (ns ← 4eu) | (ns ← 3eu) | ||||
| 7.583 | 1.00 | 3s | 9.611 | 1.00 | 3s |
| 9.479 | 0.99 | 4s | – | – | – |
| 10.14(6) (s,w) | 0.96 | 5s | – | – | – |
| 10.445 | 0.96 | 6s * | – | – | – |
| 10.60(6) (w) | 0.97 | 7s * | – | – | – |
| 10.69(8) (w) | 1.05 | 8s * | – | – | – |
| (np ← 4eu) | (np ← 3eu) | ||||
| 8.41(1) (s,b) | 0.70 | 3p | 10.445 | 0.70 | 3p * |
| 9.73(2) (s) | 0.70 | 4p | – | – | – |
| 10.23(4) (s,w) | 0.73 | 5p | – | – | – |
| (np′ ← 4eu) | (np′ ← 3eu) | ||||
| 8.75(0) (w) | 0.53 | 3p′ | 10.76(3) (b,w) | 0.55 | 3p′ * |
| 9.84(8) (s) | 0.53 | 4p′ | – | – | – |
| 10.29(8) (s,w) | 0.53 | 5p′ | – | – | – |
| 10.52(5) (w) | 0.53 | 6p′ * | – | – | – |
| 10.66(1) (s,w) | 0.47 | 7p′ * | – | – | – |
| (nd ← 4eu) | (nd ← 3eu) | ||||
| 9.23(9) (b,w) | 0.20 | 3d | – | – | – |
| 10.03(9) (s) | 0.20 | 4d | – | – | – |
| 10.38(4) (w) | 0.22 | 5d | – | – | – |
| 10.57(9) (w) | 0.17 | 6d * | – | – | – |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bandeira, E.; Barbosa, A.S.; Jones, N.C.; Hoffmann, S.V.; Bettega, M.H.F.; Limão-Vieira, P. Cyclohexane Vibronic States: A Combined VUV Spectroscopy and Theoretical Study. Molecules 2025, 30, 1493. https://doi.org/10.3390/molecules30071493
Bandeira E, Barbosa AS, Jones NC, Hoffmann SV, Bettega MHF, Limão-Vieira P. Cyclohexane Vibronic States: A Combined VUV Spectroscopy and Theoretical Study. Molecules. 2025; 30(7):1493. https://doi.org/10.3390/molecules30071493
Chicago/Turabian StyleBandeira, Edvaldo, Alessandra S. Barbosa, Nykola C. Jones, Søren V. Hoffmann, Márcio H. F. Bettega, and Paulo Limão-Vieira. 2025. "Cyclohexane Vibronic States: A Combined VUV Spectroscopy and Theoretical Study" Molecules 30, no. 7: 1493. https://doi.org/10.3390/molecules30071493
APA StyleBandeira, E., Barbosa, A. S., Jones, N. C., Hoffmann, S. V., Bettega, M. H. F., & Limão-Vieira, P. (2025). Cyclohexane Vibronic States: A Combined VUV Spectroscopy and Theoretical Study. Molecules, 30(7), 1493. https://doi.org/10.3390/molecules30071493