

Discovery of Novel Antimicrobial-Active Compounds and Their Analogues by In Silico Small Chemical Screening Targeting Staphylococcus aureus MurB


Abstract

1. Introduction
2. Results
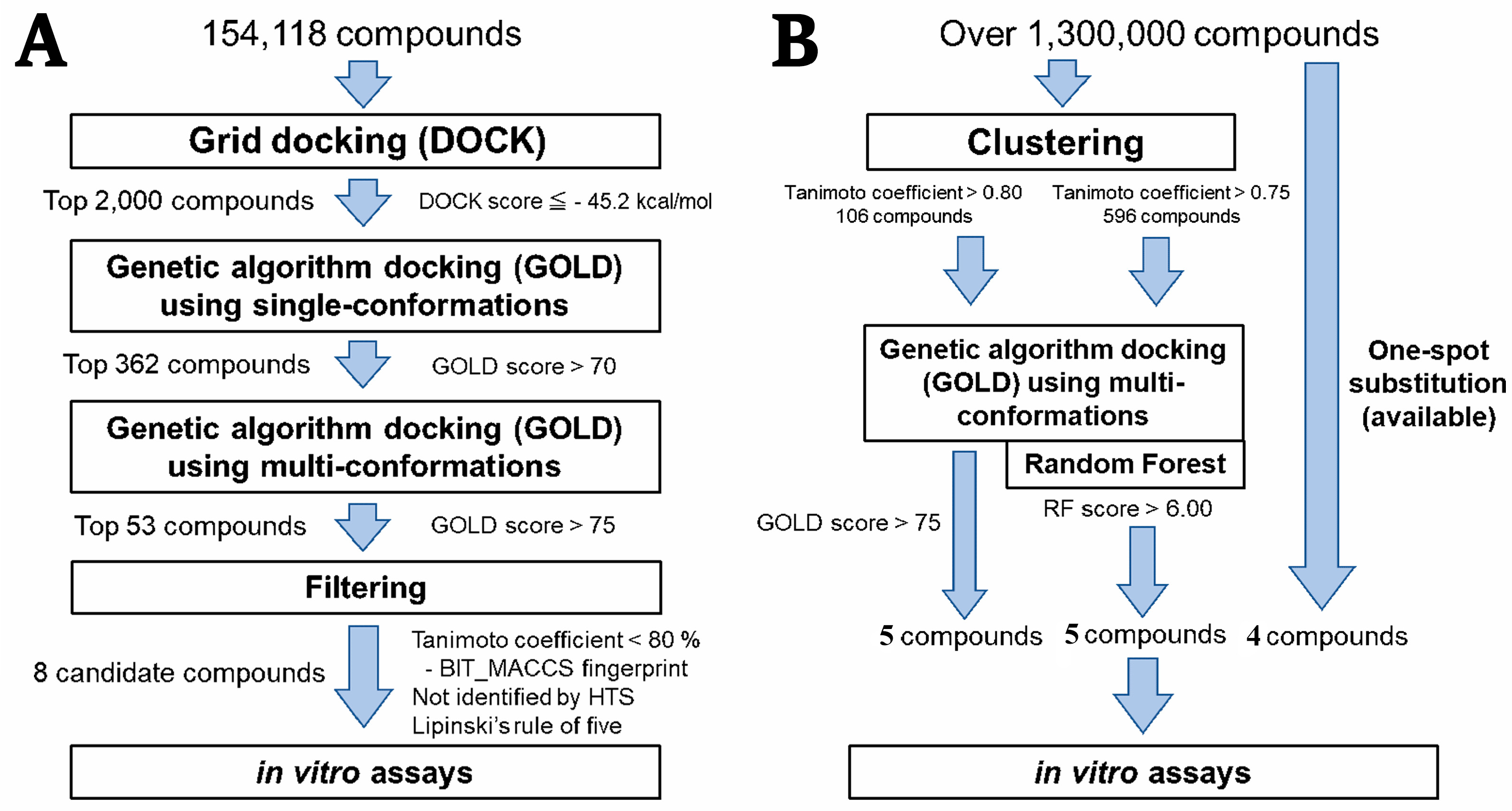
2.1. Hierarchical In Silico SBDS
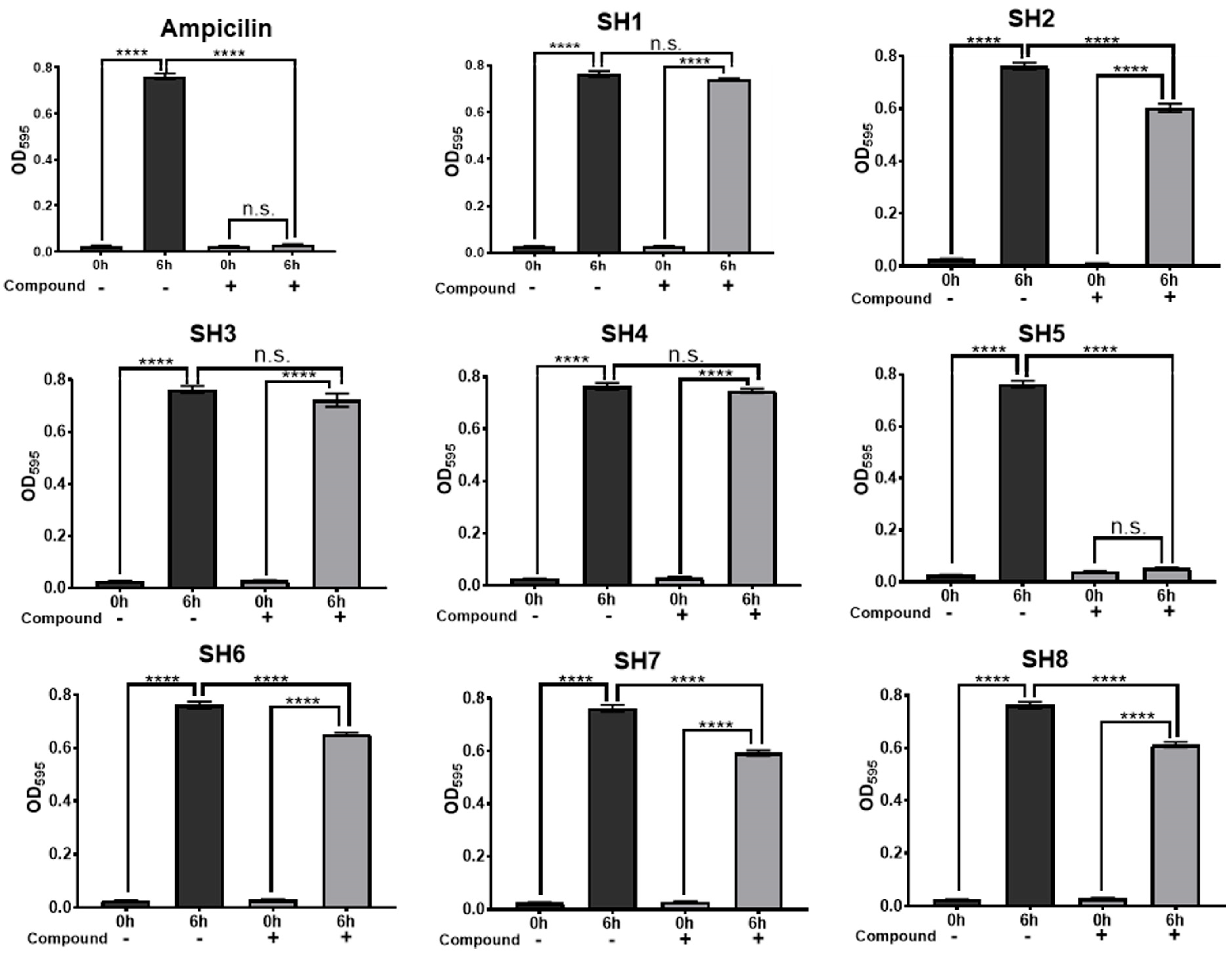
2.2. Growth Inhibition Activity of Compounds for Staphylococcus Bacteria
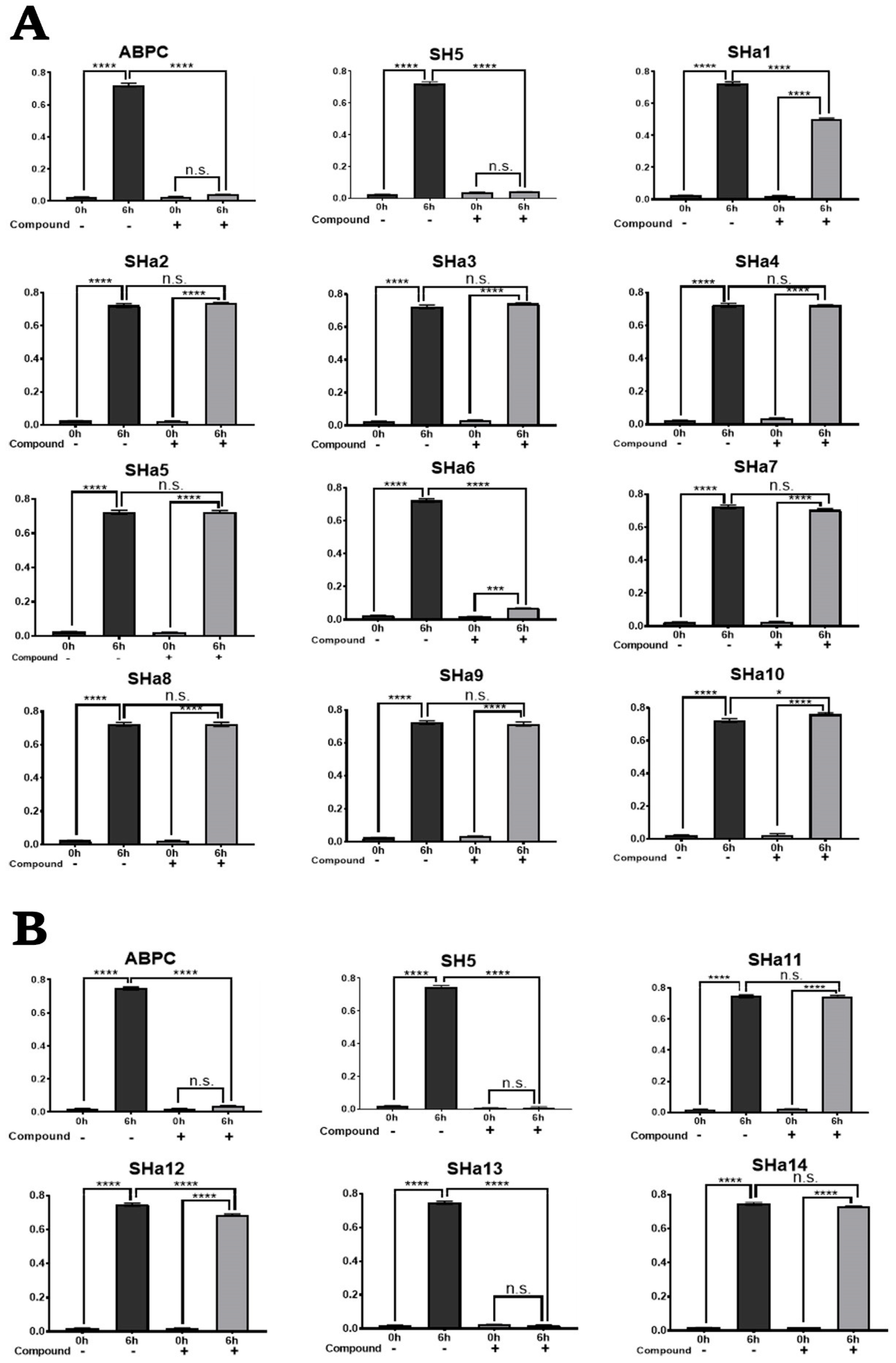
2.3. Screening for Analogues of Active Compounds
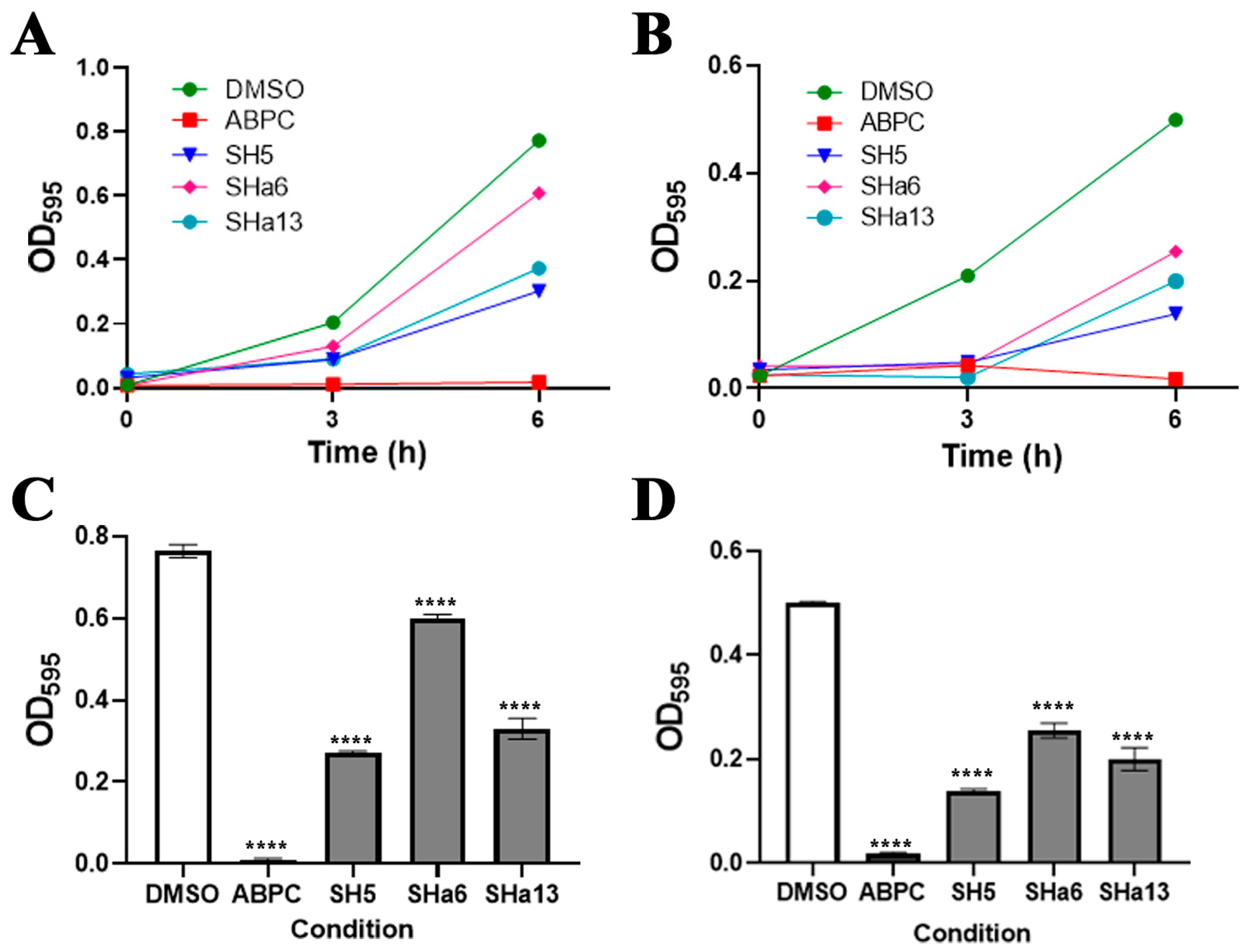
2.4. In Vitro Growth Inhibition Assay of SH5 Analogues Against S. epidermidis
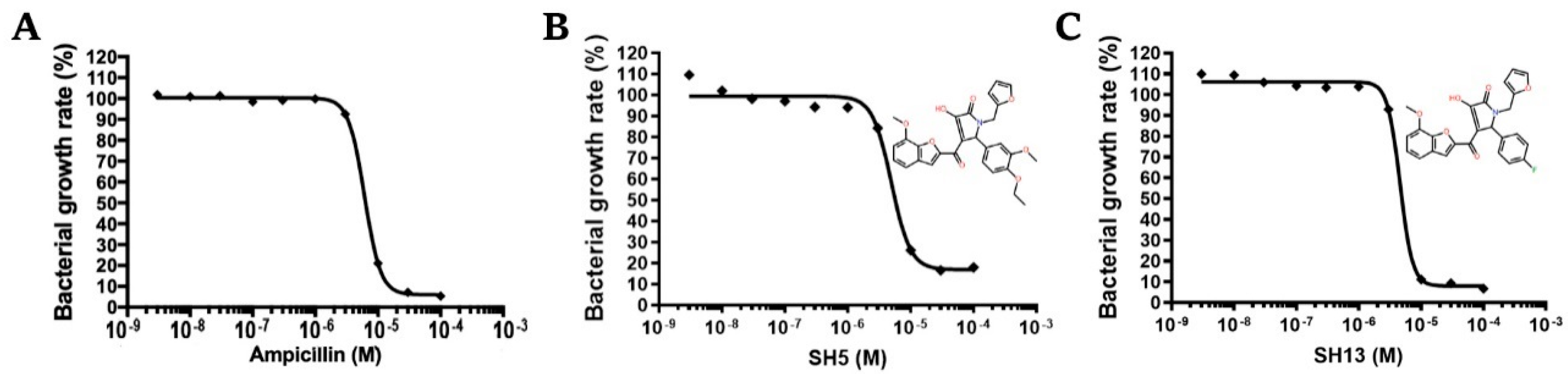
2.5. Dose-Dependent Growth Inhibition Assay Against S. epidermidis
2.6. In Vitro Assay for Escherichia coli
2.7. In Vitro Toxicity Assay for Human Cells
2.8. Prediction of Binding Mode of Active Compounds to SaMurB
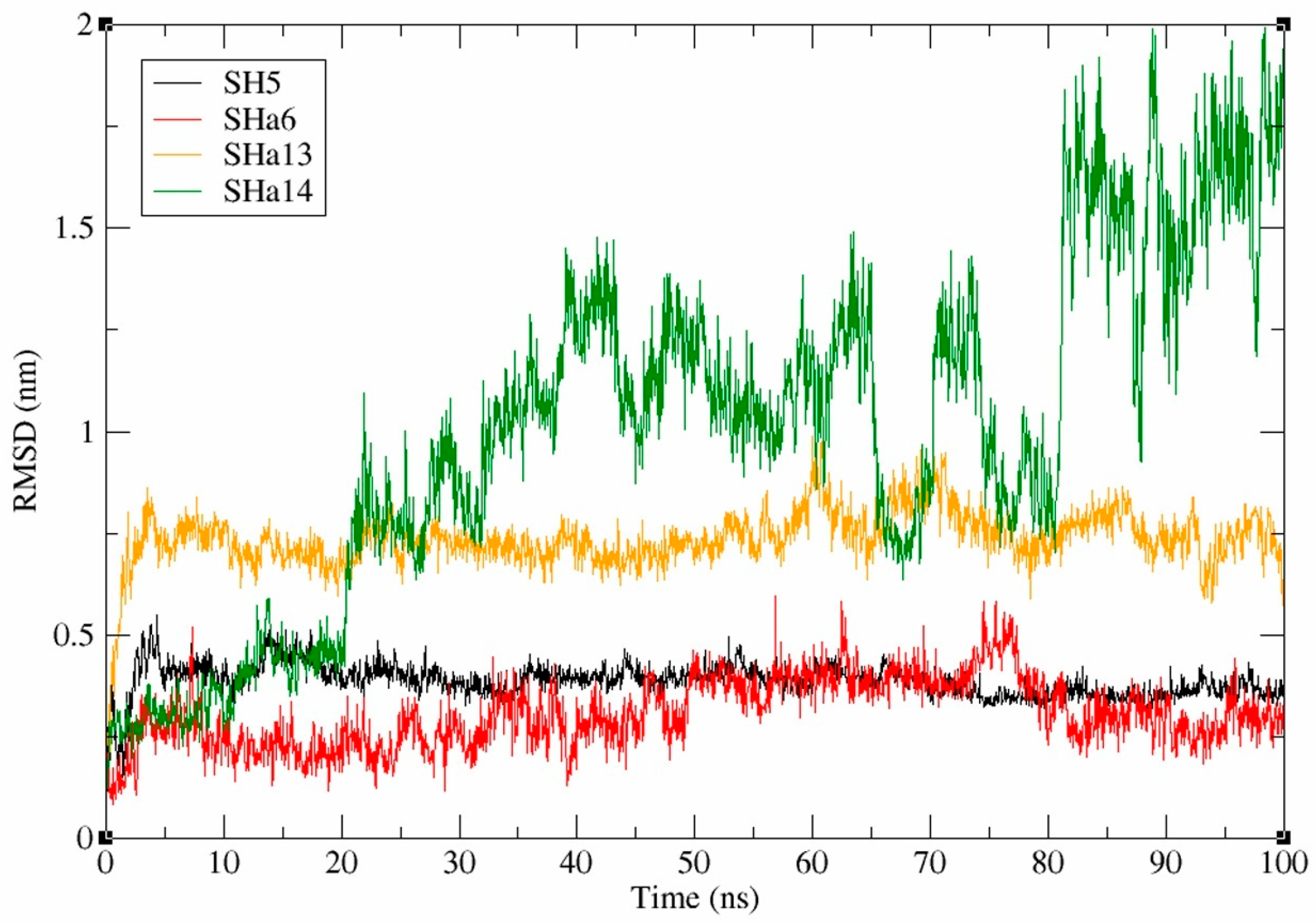
2.9. Analysis of Molecular Dynamics Simulation Data for SaMurB-Compound Complex
2.10. Pharmacochemical Evaluation and Toxicity Prediction of Antimicrobial-Active Compounds
3. Discussion
4. Materials and Methods
4.1. Compound Structure Data Library
4.2. Pretreatment of Target Proteins
4.3. Molecular Surface Extraction and Search for Binding Sites
4.4. Three-Step In Silico SBDS
4.5. Search for Analogous Compounds
4.6. Evaluation of Docking Simulation Accuracy
4.7. Molecular Dynamics Simulation
4.8. Bacterial Species and Compounds
4.9. Bacterial Growth Inhibition Assay
4.10. Toxicity Assay on Human Cells
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lowy, F.D. Staphylococcus aureus Infections. N. Engl. J. Med. 1998, 339, 520–532. [Google Scholar] [CrossRef]
- Chalmers, S.J.; Wylam, M.E. Methicillin-Resistant Staphylococcus aureus Infection and Treatment Options. Methods Mol. Biol. 2020, 2069, 229–251. [Google Scholar] [CrossRef] [PubMed]
- Kluytmans, J.; Van Belkum, A.; Verbrugh, H. Nasal Carriage of Staphylococcus aureus: Epidemiology, Underlying Mechanisms, and Associated Risks. Clin. Microbiol. Rev. 1997, 10, 505–520. [Google Scholar] [CrossRef] [PubMed]
- David, M.Z.; Daum, R.S. Treatment of Staphylococcus aureus Infections. Curr. Top. Microbiol. Immunol. 2017, 409, 325–383. [Google Scholar] [CrossRef] [PubMed]
- Howden, B.P.; Giulieri, S.G.; Wong Fok Lung, T.; Baines, S.L.; Sharkey, L.K.; Lee, J.Y.H.; Hachani, A.; Monk, I.R.; Stinear, T.P. Staphylococcus aureus Host Interactions and Adaptation. Nat. Rev. Microbiol. 2023, 21, 380–395. [Google Scholar] [CrossRef]
- Jiang, J.H.; Cameron, D.R.; Nethercott, C.; Aires-De-Sousa, M.; Peleg, A.Y. Virulence Attributes of Successful Methicillin-Resistant Staphylococcus aureus Lineages. Clin. Microbiol. Rev. 2023, 36, e00148-22. [Google Scholar] [CrossRef]
- Li, Y.; You, S.; Li, S.; Li, S.; Jia, A.; Xiang, X.; Shen, S.; Cai, Y. Exploration the Therapeutic Effects of Sodium Houttuyfonate Combined with Penicillin G on Methicillin Resistant Staphylococcus aureus Infected Wounds. Front. Pharmacol. 2025, 16, 1530217. [Google Scholar] [CrossRef]
- Khosravi, A.D.; Jenabi, A.; Montazeri, E.A. Distribution of Genes Encoding Resistance to Aminoglycoside Modifying Enzymes in Methicillin-Resistant Staphylococcus aureus (MRSA) Strains. Kaohsiung J. Med. Sci. 2017, 33, 587–593. [Google Scholar] [CrossRef]
- Hassoun, A.; Linden, P.K.; Friedman, B. Incidence, Prevalence, and Management of MRSA Bacteremia across Patient Populations-a Review of Recent Developments in MRSA Management and Treatment. Crit. Care 2017, 21, 211. [Google Scholar] [CrossRef]
- McGuinness, W.A.; Malachowa, N.; DeLeo, F.R. Vancomycin Resistance in Staphylococcus aureus. Yale J. Biol. Med. 2017, 90, 269. [Google Scholar] [CrossRef]
- Mediavilla, J.R.; Chen, L.; Mathema, B.; Kreiswirth, B.N. Global Epidemiology of Community-Associated Methicillin Resistant Staphylococcus aureus (CA-MRSA). Curr. Top. Microbiol. 2012, 15, 588–595. [Google Scholar] [CrossRef]
- Parvez, M.A.K.; Ferdous, R.N.; Rahman, M.S.; Islam, S. Healthcare-Associated (HA) and Community-Associated (CA) Methicillin Resistant Staphylococcus aureus (MRSA) in Bangladesh–Source, Diagnosis and Treatment. J. Genet. Eng. Biotechnol. 2018, 16, 473. [Google Scholar] [CrossRef]
- Facts About Antibiotic Resistance. Available online: https://www.idsociety.org/public-health/antimicrobial-resistance/archive-antimicrobial-resistance/facts-about-antibiotic-resistance/ (accessed on 21 January 2025).
- Qureshi, S.I.; Chaudhari, H.K. Design, Synthesis, in-Silico Studies and Biological Screening of Quinazolinone Analogues as Potential Antibacterial Agents against MRSA. Bioorganic Med. Chem. 2019, 27, 2676–2688. [Google Scholar] [CrossRef]
- Thappeta, K.R.V.; Zhao, L.N.; Nge, C.E.; Crasta, S.; Leong, C.Y.; Ng, V.; Kanagasundaram, Y.; Fan, H.; Ng, S.B. In-Silico Identified New Natural Sortase A Inhibitors Disrupt S. Aureus Biofilm Formation. Int. J. Mol. Sci. 2020, 21, 8601. [Google Scholar] [CrossRef]
- Turner, R.D.; Vollmer, W.; Foster, S.J. Different Walls for Rods and Balls: The Diversity of Peptidoglycan. Mol. Microbiol. 2014, 91, 862–874. [Google Scholar] [CrossRef]
- Pasquina-Lemonche, L.; Burns, J.; Turner, R.D.; Kumar, S.; Tank, R.; Mullin, N.; Wilson, J.S.; Chakrabarti, B.; Bullough, P.A.; Foster, S.J.; et al. The Architecture of the Gram-Positive Bacterial Cell Wall. Nature 2020, 582, 294–297. [Google Scholar] [CrossRef]
- Du, W.; Brown, J.R.; Sylvester, D.R.; Huang, J.; Chalker, A.F.; So, C.Y.; Holmes, D.J.; Payne, D.J.; Wallis, N.G. Two Active Forms of UDP-N-Acetylglucosamine Enolpyruvyl Transferase in Gram-Positive Bacteria. J. Bacteriol. 2000, 182, 4146–4152. [Google Scholar]
- Brown, E.D.; Vivas, E.I.; Walsh, C.T.; Kolter, R. MurA (MurZ), the Enzyme That Catalyzes the First Committed Step in Peptidoglycan Biosynthesis, Is Essential in Escherichia Coli. J. Bacteriol. 1995, 177, 4194–4197. [Google Scholar] [CrossRef]
- Benson, T.E.; Harris, M.S.; Choi, G.H.; Cialdella, J.I.; Herberg, J.T.; Martin, J.P.; Baldwin, E.T. A Structural Variation for MurB: X-Ray Crystal Structure of Staphylococcus aureus UDP-N-Acetylenolpyruvylglucosamine Reductase (MurB). Biochemistry 2001, 40, 2340–2350. [Google Scholar]
- Pucci, M.J.; Discotto, L.F.; Dougherty, T.J. Cloning and Identification of the Escherichia Coli MurB DNA Sequence, Which Encodes UDP-N-Acetylenolpyruvoylglucosamine Reductase. J. Bacteriol. 1992, 174, 1690–1693. [Google Scholar] [CrossRef]
- Andres, C.J.; Bronson, J.J.; D’Andrea, S.V.; Deshpande, M.S.; Falk, P.J.; Grant-Young, K.A.; Harte, W.E.; Ho, H.T.; Misco, P.F.; Robertson, J.G.; et al. 4-Thiazolidinones: Novel Inhibitors of the Bacterial Enzyme MurB. Bioorganic Med. Chem. Lett. 2000, 10, 715–717. [Google Scholar] [CrossRef] [PubMed]
- Bronson, J.J.; DenBleyker, K.L.; Falk, P.J.; Mate, R.A.; Ho, H.T.; Pucci, M.J.; Snyder, L.B. Discovery of the First Antibacterial Small Molecule Inhibitors of MurB. Bioorganic Med. Chem. Lett. 2003, 13, 873–875. [Google Scholar] [CrossRef] [PubMed]
- Medical*Online-E. All the Japanese Articles Searchable in English, Downloadable in PDF Format, and Readable in Multiple Languages. Available online: https://mol.medicalonline.jp/en/ (accessed on 23 January 2025).
- Taira, J.; Ito, T.; Nakatani, H.; Umei, T.; Baba, H.; Kawashima, S.; Maruoka, T.; Komatsu, H.; Sakamoto, H.; Aoki, S. In Silico Structure-Based Drug Screening of Novel Antimycobacterial Pharmacophores by DOCK-GOLD Tandem Screening. Int. J. Mycobacteriol. 2017, 6, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Taira, J.; Morita, K.; Kawashima, S.; Umei, T.; Baba, H.; Maruoka, T.; Komatsu, H.; Sakamoto, H.; Sacchettini, J.C.; Aoki, S. Identification of a Novel Class of Small Compounds with Anti-Tuberculosis Activity by in Silico Structure-Based Drug Screening. J. Antibiot. 2017, 70, 1057–1064. [Google Scholar] [CrossRef]
- Kobayashi, M.; Kinjo, T.; Koseki, Y.; Bourne, C.R.; Barrow, W.W.; Aoki, S. Identification of Novel Potential Antibiotics against Staphylococcus Using Structure-Based Drug Screening Targeting Dihydrofolate Reductase. J. Chem. Inf. Model. 2014, 54, 1242–1253. [Google Scholar] [CrossRef]
- Koseki, Y.; Kinjo, T.; Kobayashi, M.; Aoki, S. Identification of Novel Antimycobacterial Chemical Agents through the in Silico Multi-Conformational Structure-Based Drug Screening of a Large-Scale Chemical Library. Eur. J. Med. Chem. 2013, 60, 333–339. [Google Scholar] [CrossRef]
- Kinjo, T.; Koseki, Y.; Kobayashi, M.; Yamada, A.; Morita, K.; Yamaguchi, K.; Tsurusawa, R.; Gulten, G.; Komatsu, H.; Sakamoto, H.; et al. Identification of Compounds with Potential Antibacterial Activity against Mycobacterium through Structure-Based Drug Screening. J. Chem. Inf. Model. 2013, 53, 1200–1212. [Google Scholar] [CrossRef]
- Izumizono, Y.; Arevalo, S.; Koseki, Y.; Kuroki, M.; Aoki, S. Identification of Novel Potential Antibiotics for Tuberculosis by in Silico Structure-Based Drug Screening. Eur. J. Med. Chem. 2011, 46, 1849–1856. [Google Scholar] [CrossRef]
- Lang, P.T.; Brozell, S.R.; Mukherjee, S.; Pettersen, E.F.; Meng, E.C.; Thomas, V.; Rizzo, R.C.; Case, D.A.; James, T.L.; Kuntz, I.D. DOCK 6: Combining Techniques to Model RNA–Small Molecule Complexes. RNA 2009, 15, 1219–1230. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and Validation of a Genetic Algorithm for Flexible Docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization and Multithreading. J. Comput. Chem. 2010, 31, 455. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Carmona, S.; Alvarez-Garcia, D.; Foloppe, N.; Garmendia-Doval, A.B.; Juhos, S.; Schmidtke, P.; Barril, X.; Hubbard, R.E.; Morley, S.D. RDock: A Fast, Versatile and Open Source Program for Docking Ligands to Proteins and Nucleic Acids. PLOS Comput. Biol. 2014, 10, e1003571. [Google Scholar] [CrossRef]
- Miteva, M.A.; Lee, W.H.; Montes, M.O.; Villoutreix, B.O. Fast Structure-Based Virtual Ligand Screening Combining FRED, DOCK, and Surflex. J. Med. Chem. 2005, 48, 6012–6022. [Google Scholar] [CrossRef] [PubMed]
- Koes, D.R.; Baumgartner, M.P.; Camacho, C.J. Lessons Learned in Empirical Scoring with Smina from the CSAR 2011 Benchmarking Exercise. J. Chem. Inf. Model. 2013, 53, 1893–1904. [Google Scholar] [CrossRef]
- McGregor, M.J.; Pallai, P.V. Clustering of Large Databases of Compounds: Using the MDL “Keys” as Structural Descriptors. J. Chem. Inf. Comput. Sci. 1997, 37, 443–448. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- PubChem. Available online: https://pubchem.ncbi.nlm.nih.gov/ (accessed on 21 January 2025).
- BLAST: Basic Local Alignment Search Tool. Available online: https://blast.ncbi.nlm.nih.gov/Blast.cgi (accessed on 21 January 2025).
- Wójcikowski, M.; Ballester, P.J.; Siedlecki, P. Performance of Machine-Learning Scoring Functions in Structure-Based Virtual Screening. Sci. Rep. 2017, 7, 46710. [Google Scholar] [CrossRef]
- Vilar, S.; Cozza, G.; Moro, S. Medicinal Chemistry and the Molecular Operating Environment (MOE): Application of QSAR and Molecular Docking to Drug Discovery. Curr. Top. Med. Chem. 2008, 8, 1555–1572. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, Flexible, and Free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Miller, B.R.; McGee, T.D.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.Py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef] [PubMed]
- Valdés-Tresanco, M.S.; Valdés-Tresanco, M.E.; Valiente, P.A.; Moreno, E. Gmx_MMPBSA: A New Tool to Perform End-State Free Energy Calculations with GROMACS. J. Chem. Theory Comput. 2021, 17, 6281–6291. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Banerjee, P.; Kemmler, E.; Dunkel, M.; Preissner, R. ProTox 3.0: A Webserver for the Prediction of Toxicity of Chemicals. Nucleic Acids Res. 2024, 52, W513–W520. [Google Scholar] [CrossRef]
- Nishida, S.; Kurokawa, K.; Matsuo, M.; Sakamoto, K.; Ueno, K.; Kita, K.; Sekimizu, K. Identification and Characterization of Amino Acid Residues Essential for the Active Site of UDP-N-Acetylenolpyruvylglucosamine Reductase (MurB) from Staphylococcus aureus. J. Biol. Chem. 2006, 281, 1714–1724. [Google Scholar] [CrossRef]
- Benson, T.E.; Walsh, C.T.; Hogle, J.M. X-Ray Crystal Structures of the S229A Mutant and Wild-Type MurB in the Presence of the Substrate Enolpyruvyl-UDP-N-Acetylglucosamine at 1.8-A Resolution. Biochemistry 1997, 36, 806–811. [Google Scholar] [CrossRef]
- Kim, M.K.; Cho, M.K.; Song, H.E.; Kim, D.; Park, B.H.; Lee, J.H.; Kang, G.B.; Kim, S.H.; Im, Y.J.; Lee, D.S.; et al. Crystal Structure of UDP-N-Acetylenolpyruvylglucosamine Reductase (MurB) from Thermus Caldophilus. Proteins Struct. Funct. Bioinform. 2007, 66, 751–754. [Google Scholar] [CrossRef]
- Chen, M.W.; Lohkamp, B.; Schnell, R.; Lescar, J.; Schneider, G. Substrate Channel Flexibility in Pseudomonas Aeruginosa MurB Accommodates Two Distinct Substrates. PLoS ONE 2013, 8, e66936. [Google Scholar] [CrossRef]
- Eniyan, K.; Dharavath, S.; Vijayan, R.; Bajpai, U.; Gourinath, S. Crystal Structure of UDP-N-Acetylglucosamine-Enolpyruvate Reductase (MurB) from Mycobacterium Tuberculosis. Biochim. Biophys. Acta Proteins. Proteom. 2018, 1866, 397–406. [Google Scholar] [CrossRef]
- Yang, Y.; Severin, A.; Chopra, R.; Krishnamurthy, G.; Singh, G.; Hu, W.; Keeney, D.; Svenson, K.; Petersen, P.J.; Labthavikul, P.; et al. 3,5-Dioxopyrazolidines, Novel Inhibitors of UDP-N-Acetylenolpyruvylglucosamine Reductase (MurB) with Activity against Gram-Positive Bacteria. Antioxid. Agents Chemother. 2006, 50, 556. [Google Scholar] [CrossRef]
- Hashemian, S.M.R.; Farhadi, T.; Ganjparvar, M. Linezolid: A Review of Its Properties, Function, and Use in Critical Care. Drug Des. Dev. Ther. 2018, 12, 1759–1767. [Google Scholar] [CrossRef] [PubMed]
- Ethical Drug: Linezolid (Linezolid Injection 600mg “Sawai”). Available online: https://www.kegg.jp/medicus-bin/japic_med?japic_code=00067122 (accessed on 7 March 2025).
- RPBS. Available online: https://bioserv.rpbs.univ-paris-diderot.fr/index.html (accessed on 21 January 2025).
- Yu, H.; Adedoyin, A. ADME–Tox in Drug Discovery: Integration of Experimental and Computational Technologies. Drug Discov. Today 2003, 8, 852–861. [Google Scholar] [CrossRef]
- MOE|MOLSIS Inc. Available online: https://www.molsis.co.jp/lifescience/moe/ (accessed on 21 January 2025).
- Dms. Available online: https://www.cgl.ucsf.edu/chimera/docs/UsersGuide/midas/dms1.html (accessed on 21 January 2025).
- Kuntz, I.D.; Blaney, J.M.; Oatley, S.J.; Langridge, R.; Ferrin, T.E. A Geometric Approach to Macromolecule-Ligand Interactions. J. Mol. Biol. 1982, 161, 269–288. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Lucke, A.J.; Fairlie, D.P. Comparing Sixteen Scoring Functions for Predicting Biological Activities of Ligands for Protein Targets. J. Mol. Graph. Model. 2015, 57, 76–88. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindah, E. GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A Web-Based Graphical User Interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Brooks, B.R.; Brooks, C.L.; Mackerell, A.D.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The Biomolecular Simulation Program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y.; et al. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; De Groot, B.L.; Grubmüller, H.; MacKerell, A.D. CHARMM36m: An Improved Force Field for Folded and Intrinsically Disordered Proteins. Nat. Methods 2016, 14, 71–73. [Google Scholar] [CrossRef]
- Microbe Division (JCM) (RIKEN BRC). Available online: https://jcm.brc.riken.jp/en/ (accessed on 23 January 2025).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (R or S)- | Compound Name | Predicted Interaction Residue 1 | IC50 (µM) 2 |
|---|---|---|---|
| (R)- | SH5 | Arg188, Arg225, Lys228, Gln229 | 5.67 ± 0.51 |
| (S)- | Ala154, Gln156, Arg188, Arg242 | ||
| (R)- | SHa6 | Gln241, Arg242, Ala248, His271, Ala272 | <100 |
| (S)- | Ala154, Gln158, Arg188, Phe240, Arg242, Gly273 | ||
| (R)- | SHa13 | Arg188, Arg242 | 1.64 ± 0.01 |
| (S)- | Gly156, Gln158, Arg188 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Okubo, S.; Hirose, S.; Aoki, S. Discovery of Novel Antimicrobial-Active Compounds and Their Analogues by In Silico Small Chemical Screening Targeting Staphylococcus aureus MurB. Molecules 2025, 30, 1477. https://doi.org/10.3390/molecules30071477
Okubo S, Hirose S, Aoki S. Discovery of Novel Antimicrobial-Active Compounds and Their Analogues by In Silico Small Chemical Screening Targeting Staphylococcus aureus MurB. Molecules. 2025; 30(7):1477. https://doi.org/10.3390/molecules30071477
Chicago/Turabian StyleOkubo, Saya, Shoki Hirose, and Shunsuke Aoki. 2025. "Discovery of Novel Antimicrobial-Active Compounds and Their Analogues by In Silico Small Chemical Screening Targeting Staphylococcus aureus MurB" Molecules 30, no. 7: 1477. https://doi.org/10.3390/molecules30071477
APA StyleOkubo, S., Hirose, S., & Aoki, S. (2025). Discovery of Novel Antimicrobial-Active Compounds and Their Analogues by In Silico Small Chemical Screening Targeting Staphylococcus aureus MurB. Molecules, 30(7), 1477. https://doi.org/10.3390/molecules30071477