

The Molecular Recognition of Lurasidone by Human Serum Albumin: A Combined Experimental and Computational Approach
 , ,
, , 
Abstract
1. Introduction
2. Results and Discussion
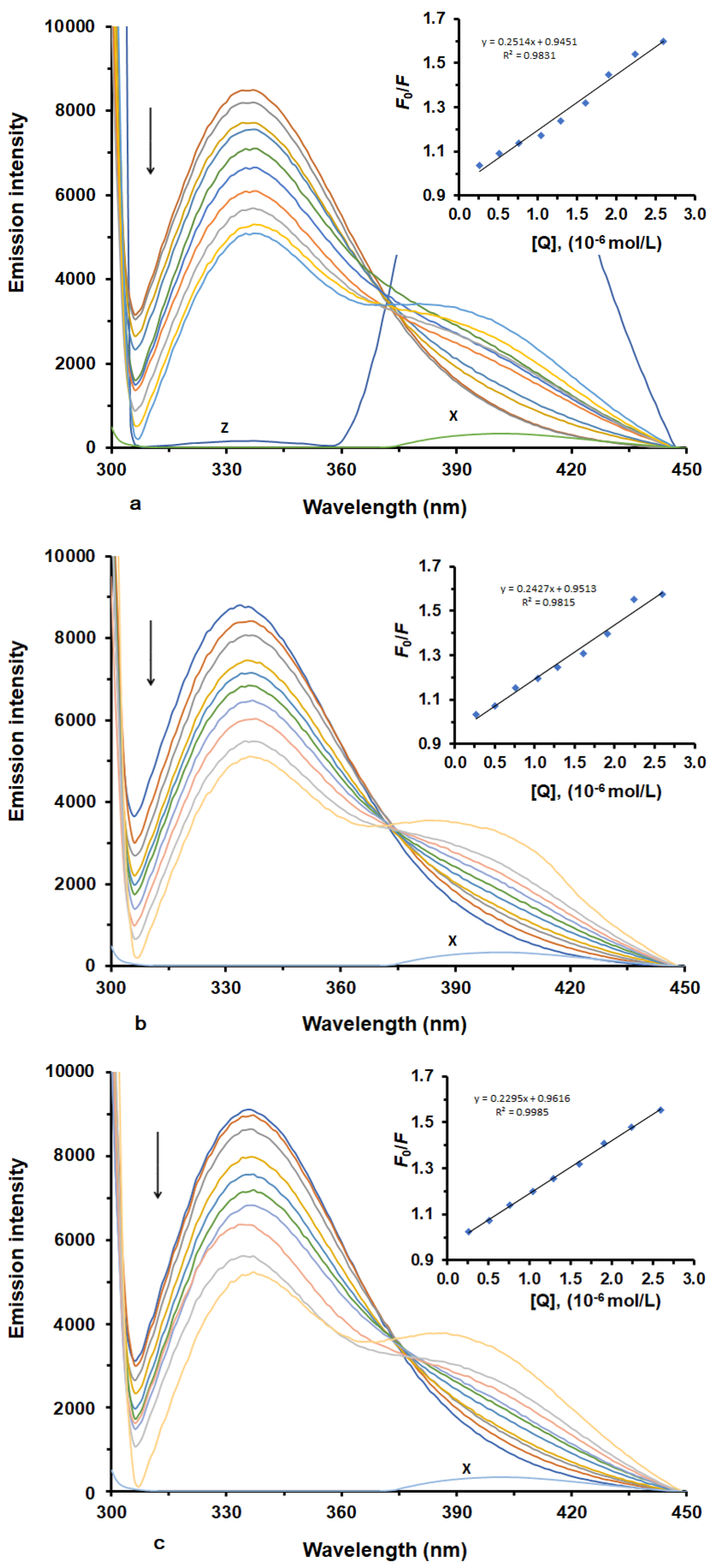
2.1. Characterisation of LUR Binding to HSA
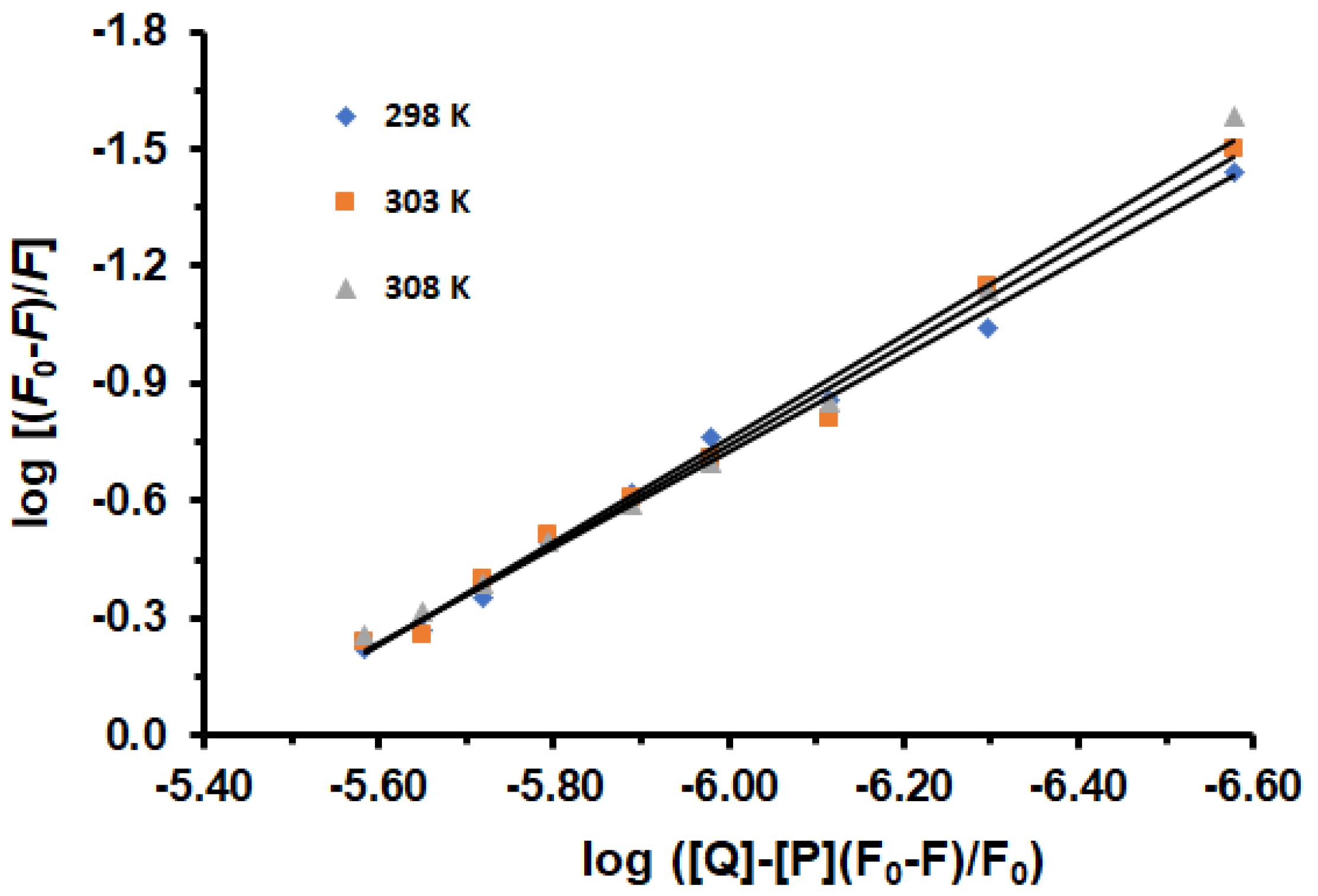
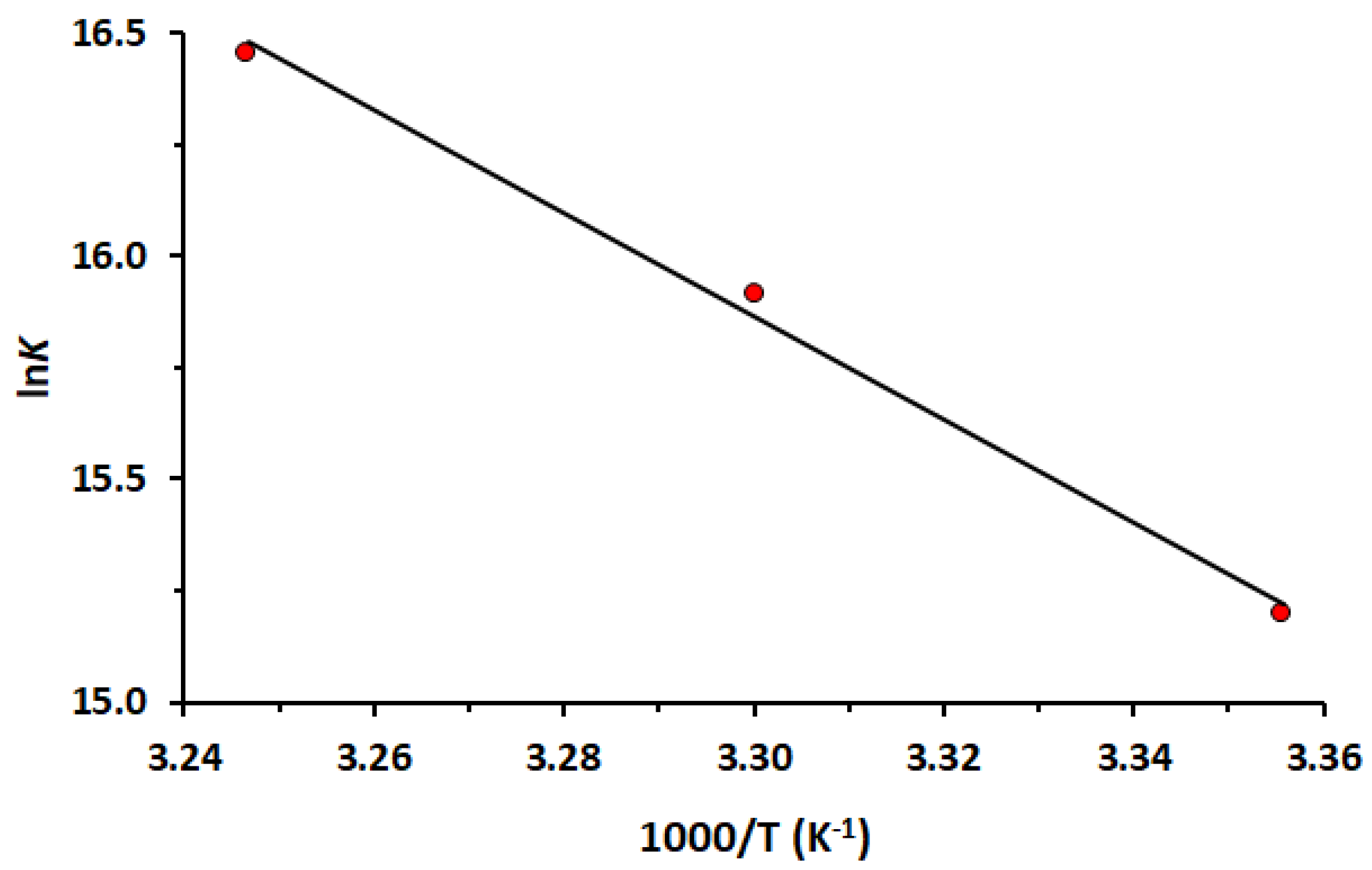
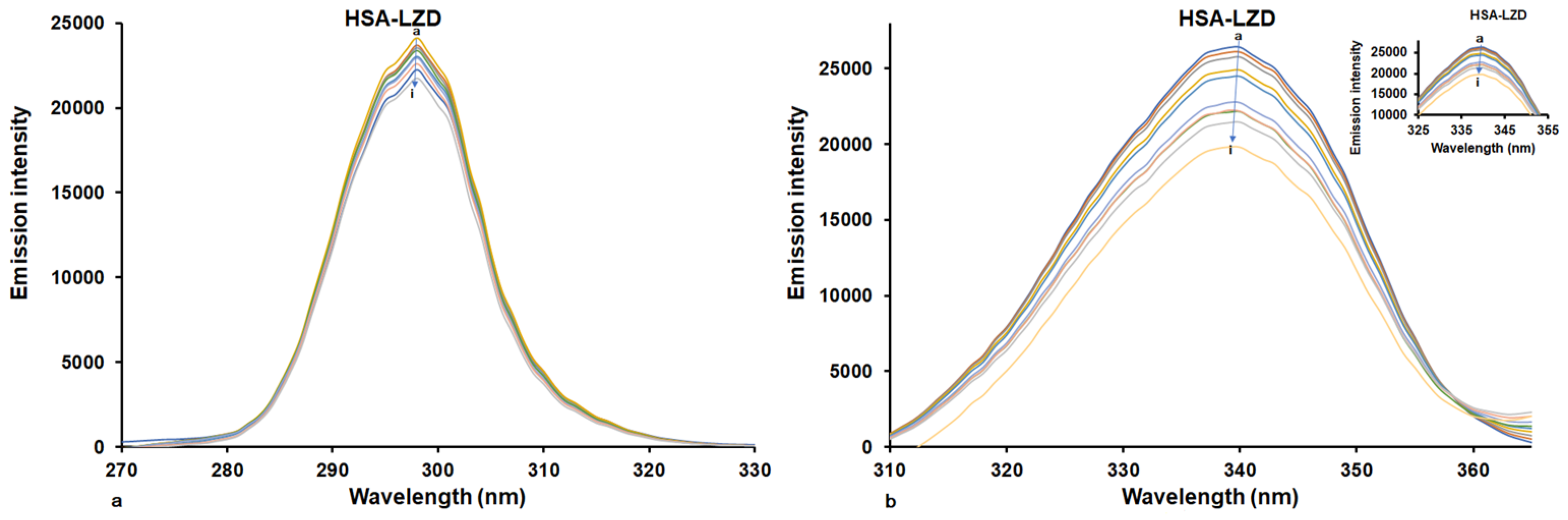
2.2. Determination of Binding Constant and Binding Stoichiometry
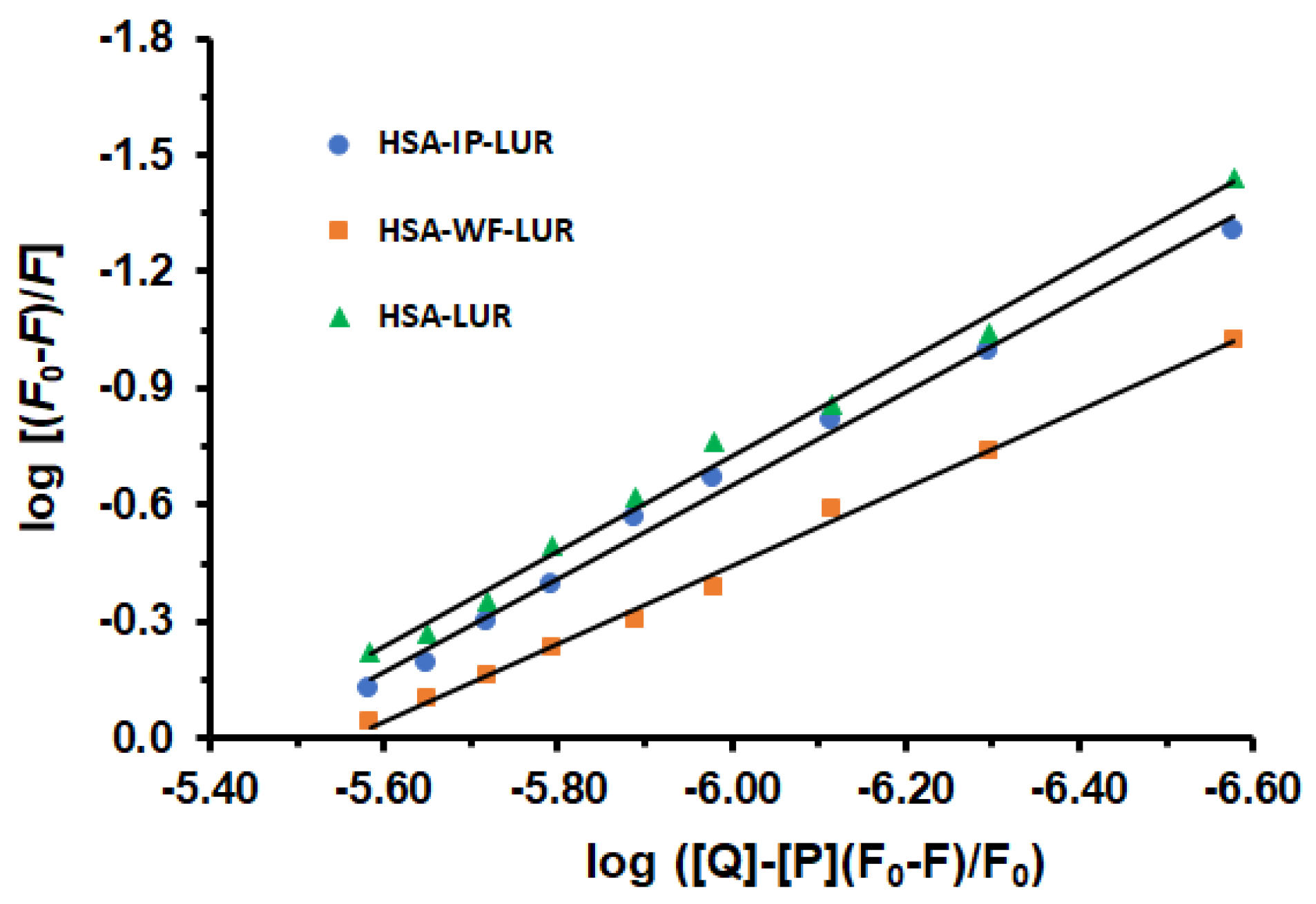
2.3. Site-Selective Binding of LUR to HSA
2.4. Conformational Changes in HSA Induced by the LUR
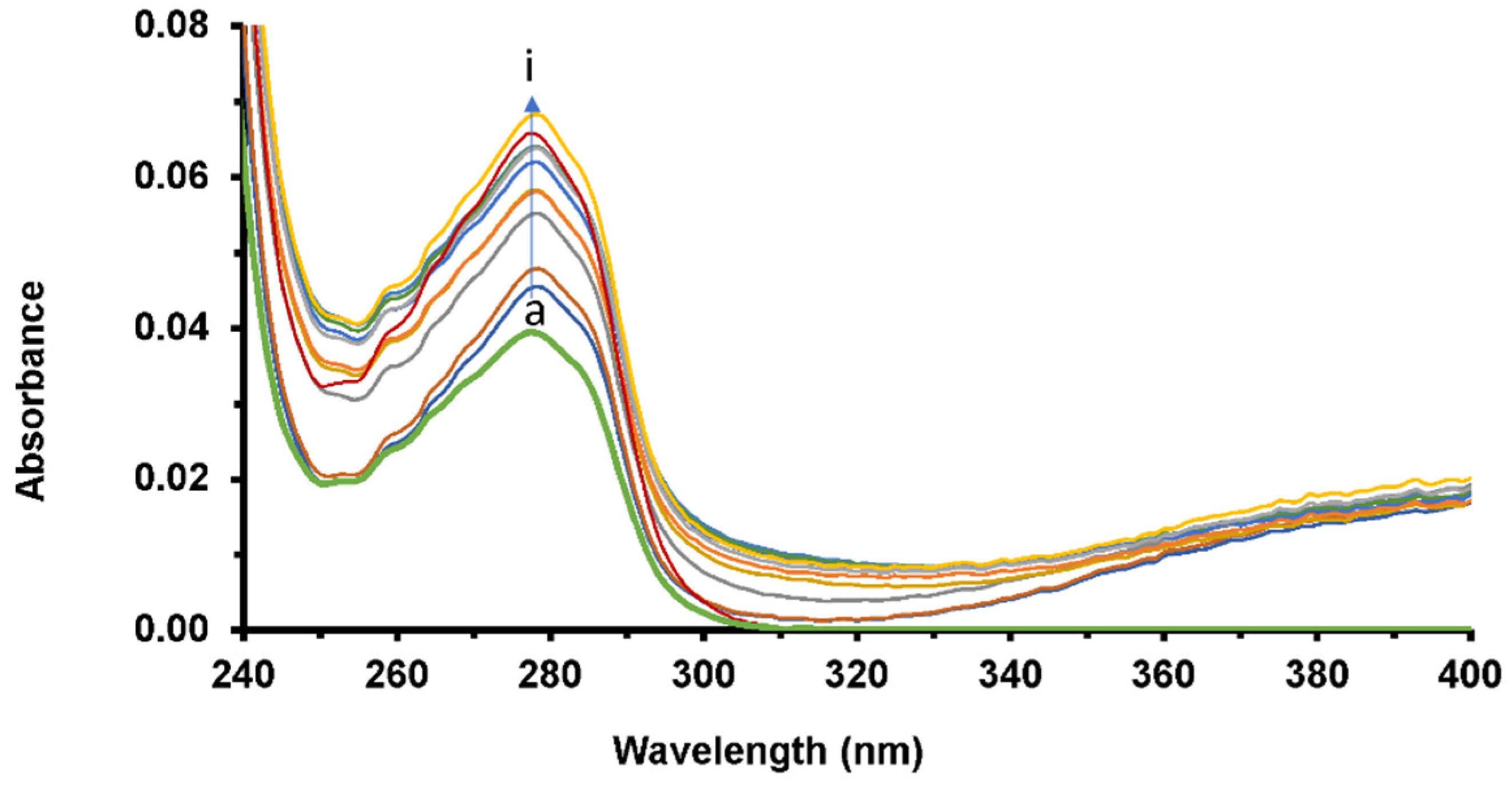
2.4.1. UV-Vis Spectroscopy
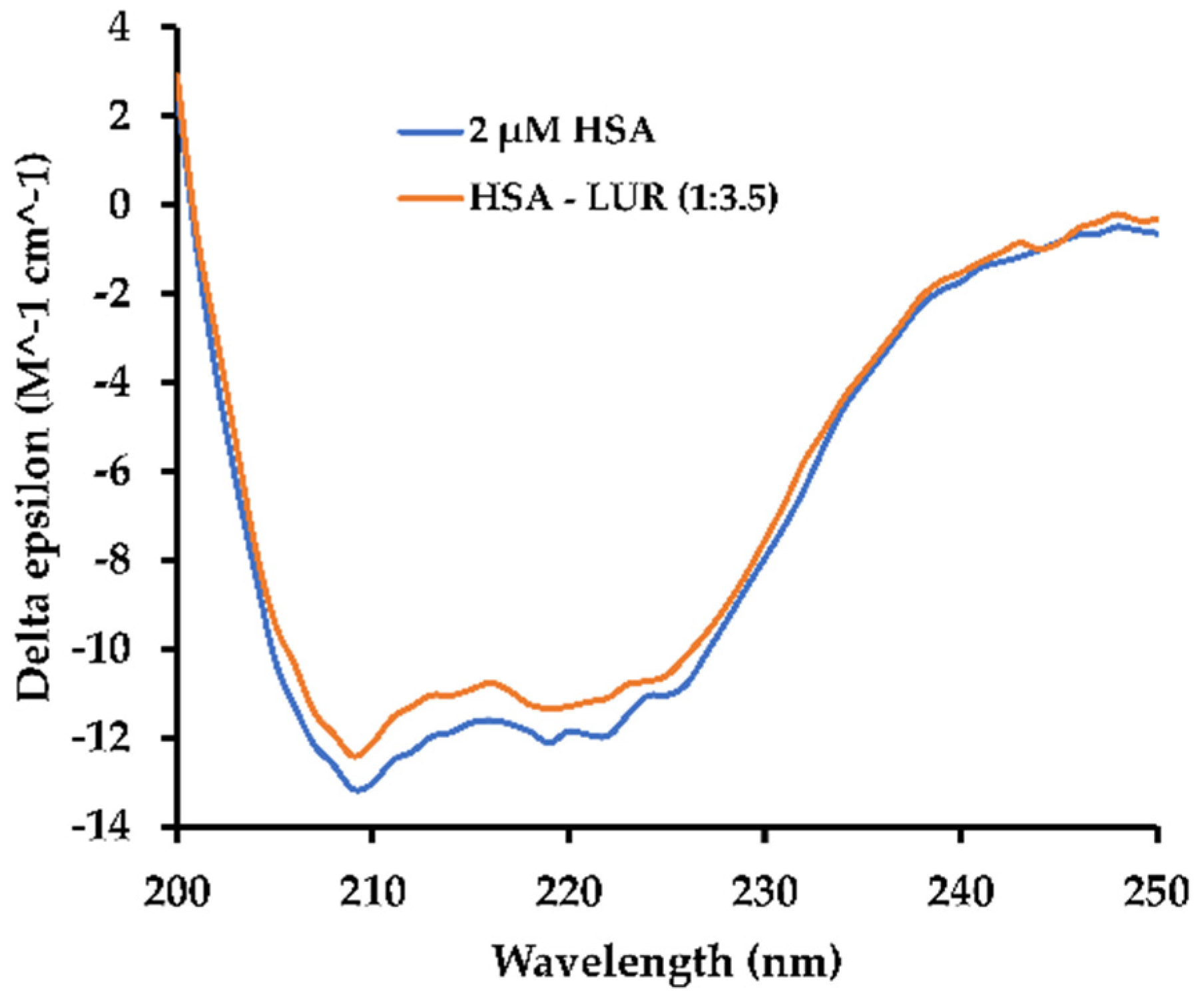
2.4.2. CD Spectral Measurements
2.4.3. Synchronous Fluorescence Spectral Measurements
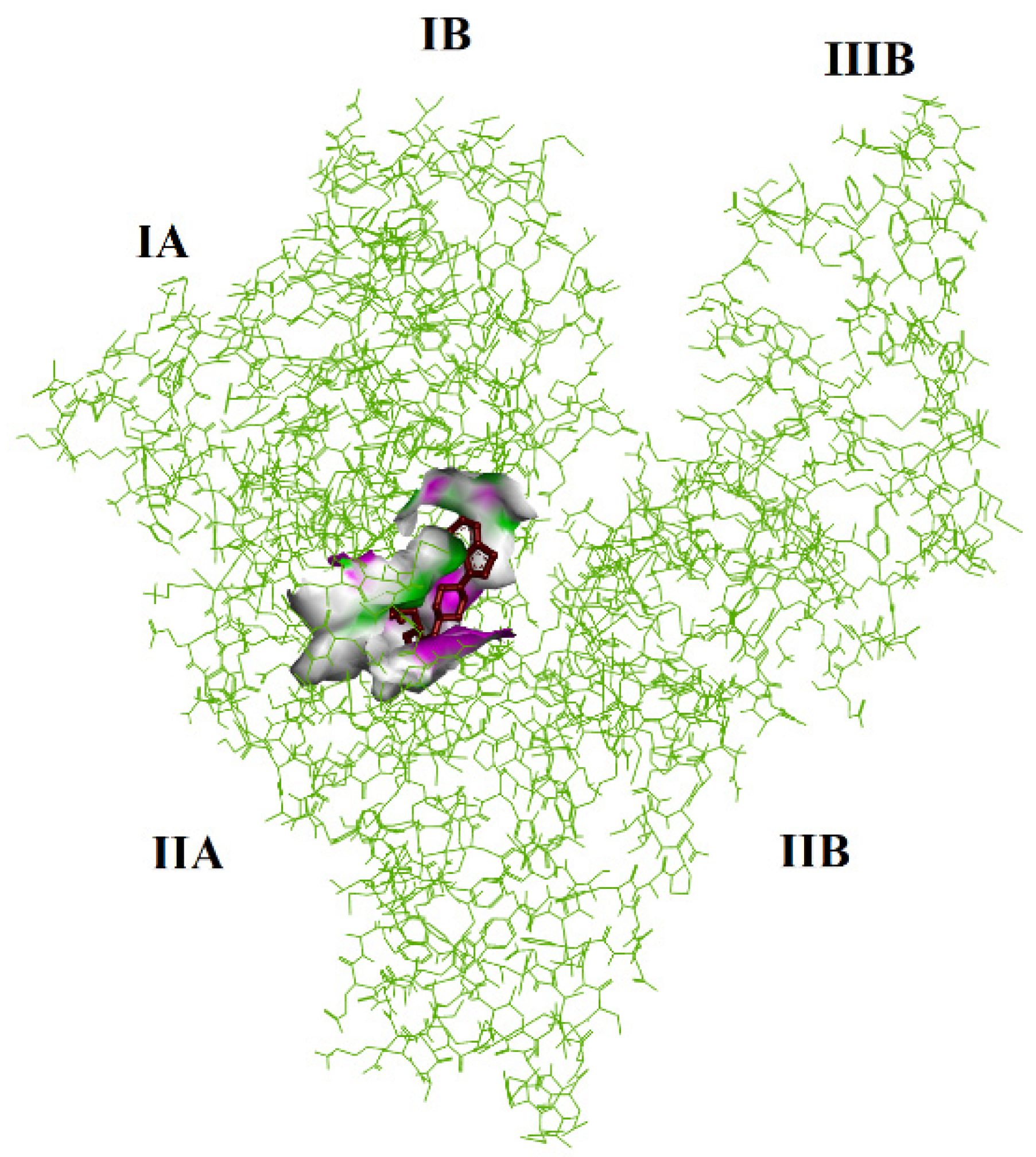
2.5. Molecular Docking
3. Materials and Methods
3.1. The Materials
3.2. Preparation of Protein and Ligand Solutions
3.3. Ligand Binding Studies
3.4. Competitive Ligand Binding Studies
3.5. Analysis of the Binding Data
3.6. Ultra-Violet Spectroscopy
3.7. Circular Dichroism (CD) Spectral Measurement
3.8. Synchronous Fluorescence Spectra
3.9. Molecular Docking
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Du, X.; Li, Y.; Xia, Y.L.; Ai, S.M.; Liang, J.; Sang, P.; Ji, X.L.; Liu, S.Q. Insights into Protein–Ligand Interactions: Mechanisms, Models, and Methods. Int. J. Mol. Sci. 2016, 17, 144. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, S.; Ameen, F.; ur Rehman, S.; Sarwar, T.; Tabish, M. Studying the interaction of drug/ligand with serum albumin. J. Mol. Liq. 2021, 336, 116200. [Google Scholar] [CrossRef]
- Spada, A.; Emami, J.; Tuszynski, J.A.; Lavasanifar, A. The uniqueness of albumin as a carrier in nanodrug delivery. Mol. Pharm. 2021, 18, 1862. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.; Zhu, S.; Hou, Y. Insight on serum albumin: From structure and biological properties to functional biomaterials for bone repair. ACS Biomater. Sci. Eng. 2023, 9, 2235–2250. [Google Scholar] [CrossRef]
- Park, J.; Kim, M.S.; Park, T.; Kim, Y.H.; Shin, D.H. Crystal structure of pharmaceutical-grade human serum albumin. Int. J. Biol. Macromol. 2021, 166, 221–228. [Google Scholar] [CrossRef]
- Zsila, F. Subdomain IB is the third major drug binding region of human serum albumin: Toward the three-sites model. Mol. Pharm. 2013, 10, 1668. [Google Scholar] [CrossRef] [PubMed]
- Radomska, K.; Wolszczak, M. Spontaneous and ionizing radiation-induced aggregation of human serum albumin: Dityrosine as a fluorescent probe. Int. J. Mol. Sci. 2022, 23, 8090. [Google Scholar] [CrossRef]
- Ayimbila, F.; Tantimongcolwat, T.; Ruankham, W.; Pingaew, R.; Prachayasittikul, V.; Worachartcheewan, A.; Prachayasittikul, V.; Prachayasittikul, S.; Phopin, K. Insight into the binding mechanisms of fluorinated 2-aminothiazole sulfonamide and human serum albumin: Spectroscopic and in silico approaches. Int. J. Biol. Macromol. 2024, 277, 134048. [Google Scholar] [CrossRef]
- Heng, W.; Su, M.; Cheng, H.; Shen, P.; Liang, S.; Zhang, L.; Qian, S. Molecular interactions of drugs. Mol. Pharm. 2019, 17, 84. [Google Scholar] [CrossRef]
- Guilera, T.; Pascual, J.P.C.; del Carmen Blasco, M.; Estopiñán, P.C.; González, R.A.P.; Martínez, I.R.; Moyano, C.R.; Pérez, R.P.; Gabarda-Inat, I.; Prados-Ojeda, J.L.; et al. Lurasidone for the treatment of schizophrenia in adult and paediatric populations. Drugs Context. 2023, 12, 2022. [Google Scholar] [CrossRef]
- Ceskova, E. Critical appraisal of lurasidone in the management of schizophrenia. Expert. Opin. Pharmacother. 2022, 23, 1865–1868. [Google Scholar] [CrossRef]
- Stahl, S.M.; Morrissette, D.A. Should high dose or very long-term antipsychotic monotherapy be considered before antipsychotic polypharmacy? In Polypharmacy in Psychiatry Practice: Multiple Medication Use Strategies, 1st ed.; Ritsner, M.S., Ed.; Springer Nature: Cham, Switzerland, 2013; Volume 1, pp. 107–125. [Google Scholar] [CrossRef]
- Caccia, S.; Pasina, L.; Nobili, A. Pharmacokinetics of antipsychotics. Neuropsychiatr. Dis. Treat. 2012, 8, 155–168. [Google Scholar] [CrossRef]
- Danek, P.J.; Wójcikowski, J.; Daniel, W.A. The atypical neuroleptics iloperidone and lurasidone inhibit human cytochrome P450 enzymes in vitro. Evaluation of potential metabolic interactions. Pharmacol. Rep. 2020, 72, 1685–1694. [Google Scholar] [CrossRef]
- Starosta, R.; Santos, F.C.; de Almeida, R.F. Human and bovine serum albumin time-resolved fluorescence: Tryptophan and tyrosine contributions, effect of DMSO and rotational diffusion. J. Mol. Struct. 2020, 1221, 128805. [Google Scholar] [CrossRef]
- Hong, Y.; Feng, C.; Yu, Y.; Liu, J.; Lam, J.W.Y.; Luo, K.Q.; Tang, B.Z. Quantitation, visualization, and monitoring of conformational transitions of human serum albumin by a tetraphenylethene derivative with aggregation-induced emission characteristics. Anal. Chem. 2010, 82, 7035–7043. [Google Scholar] [CrossRef] [PubMed]
- Crouse, H.F.; Petrunak, E.M.; Donovan, A.M.; Merkle, A.C.; Swartz, B.L.; Basu, S. Static and dynamic quenching of tryptophan fluorescence in various proteins by a chromium (III) complex. Spectrosc. Lett. 2011, 44, 369–374. [Google Scholar] [CrossRef]
- Lakowicz, J.R.; Weber, G. Quenching of protein fluorescence by oxygen. Detection of structural fluctuations in proteins on the nanosecond time scale. Biochemistry 1973, 12, 4171–4179. [Google Scholar] [CrossRef]
- Yang, F.; Zhang, Y.; Liang, H. Interactive association of drugs binding to human serum albumin. Int. J. Mol. Sci. 2014, 15, 3580–3595. [Google Scholar] [CrossRef]
- Tian, F.F.; Li, J.H.; Jiang, F.L.; Han, X.L.; Xiang, C.; Ge, Y.S.; Liu, Y. The adsorption of an anticancer hydrazone by protein: An unusual static quenching mechanism. RSC. Adv. 2012, 2, 501–513. [Google Scholar] [CrossRef]
- Feroz, S.R.; Mohamad, S.B.; Bujang, N.; Malek, S.N.A.; Tayyab, S.J. Multispectroscopic and Molecular Modeling Approach to Investigate the Interaction of Flavokawain B with Human Serum Albumin. J. Agric. Food Chem. 2012, 60, 5899–5908. [Google Scholar] [CrossRef]
- Berić, J.D.; Stojanović, S.D.; Mrkalić, E.M.; Matović, Z.D.; Milovanović, D.R.; Sovrlić, M.M.; Jelić, R.M. Interaction of haloperidol with human serum albumin and effect of metal ions on the binding. Monatshefte für Chemie 2018, 149, 2359. [Google Scholar] [CrossRef]
- da Silva Fragoso, V.M.; de Morais Coura, C.P.; Hoppe, L.Y.; Soares, M.A.G.; Silva, D.; Cortez, C.M. Binding of sulpiride to seric albumins. Int. J. Mol. Sci. 2016, 17, 59. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.Y.; Li, X.Y.; Hu, L.Y.; Bai, A.M.; Hu, Y.J. Comparative study of two antipsychotic drugs binding to human serum albumin: By multispectroscopic and molecular docking methods. J. Mol. Liq. 2022, 365, 120084. [Google Scholar] [CrossRef]
- Caraci, F.; Sultana, J.; Drago, F.; Spina, E. Clinically relevant drug interactions with anti-Alzheimer’s drugs. CNS Neurol. Disord. Drug. Targets. 2017, 16, 501–513. [Google Scholar] [CrossRef] [PubMed]
- Sengul, M.C.B.; Karadag, F.; Sengul, C.; Karakulah, K.; Kalkanci, O.; Herken, H. Risk of psychotropic drug interactions in real world settings: A pilot study in patients with schizophrenia and schizoaffective disorder. Klin. Psikofarmakol. B. 2014, 24, 235–247. [Google Scholar] [CrossRef]
- Ross, P.D.; Subramanian, S. Thermodynamics of Protein Association Reactions: Forces Contributing to Stability. Biochemistry 1981, 20, 3096–3102. [Google Scholar] [CrossRef]
- Rezaei-Tavirani, M.; Moghaddamnia, S.H.; Ranjbar, B.; Amani, M.; Marashi, S.A. Conformational study of human serum albumin in pre-denaturation temperatures by differential scanning calorimetry, circular dichroism and UV spectroscopy. BMB Rep. 2006, 39, 530–536. [Google Scholar] [CrossRef]
- Bertucci, C.; Domenici, E. Reversible and covalent binding of drugs to human serum albumin: Methodological approaches and physiological relevance. Curr. Med. Chem. 2002, 9, 1463–1481. [Google Scholar] [CrossRef]
- Ghuman, J.; Zunszain, P.A.; Petitpas, I.; Bhattacharya, A.A.; Otagiri, M. Structural basis of the drug-binding specificity of human serum albumin. J. Mol. Biol. 2005, 353, 38–52. [Google Scholar] [CrossRef]
- Sharma, A.S.; Anandakumar, S.; Ilanchelian, M. A combined spectroscopic and molecular docking study on site selective binding interaction of Toluidine blue O with Human and Bovine serum albumins. J. Lumin. 2014, 151, 206. [Google Scholar] [CrossRef]
- Abdelhameed, A.S.; Alanazi, A.M.; Bakheit, A.H.; Hassan, E.S.; Herqash, R.N.; Almutairi, F.M. Novel BTK inhibitor acalabrutinib (ACP-196) tightly binds to site I of the human serum albumin as observed by spectroscopic and computational studies. Int. J. Biol. Macromol. 2019, 127, 536. [Google Scholar] [CrossRef] [PubMed]
- Jafari-Arvari, H.; Saei-Dehkordi, S.; Farhadian, S. Evaluation of interactions between food colorant, tartrazine, and Apo-transferrin using spectroscopic analysis and docking simulation. J. Mol. Liq. 2021, 339, 116715. [Google Scholar] [CrossRef]
- Nazzaro, A.; Lu, B.; Sawyer, N.; Watkins, A.M.; Arora, P.S. Macrocyclic β-Sheets Stabilized by Hydrogen Bond Surrogates. Angew. Angew. Chem. Int. Ed. 2023, 62, e202303943. [Google Scholar] [CrossRef]
- Zhao, L.; Zhang, J.; Zhang, Y.; Ye, S.; Zhang, G.; Chen, X.; Jiang, J. Accurate machine learning prediction of protein circular dichroism spectra with embedded density descriptors. JACS. Au 2021, 1, 2377–2384. [Google Scholar] [CrossRef]
- Micsonai, A.; Moussong, É.; Wien, F.; Boros, E.; Vadászi, H.; Murvai, N.; Lee, Y.-H.; Molnár, T.; Réfrégiers, M.; Goto, Y.; et al. BeStSel: Webserver for secondary structure and fold prediction for protein CD spectroscopy. Nucleic Acids Res. 2022, 50, W90–W98. [Google Scholar] [CrossRef]
- Amézqueta, S.; Beltrán, J.L.; Bolioli, A.M.; Campos-Vicens, L.; Luque, F.J.; Ràfols, C. Evaluation of the interactions between human serum albumin (HSA) and non-steroidal anti-inflammatory (NSAIDs) drugs by multiwavelength molecular fluorescence, structural and computational analysis. Pharmaceuticals 2021, 14, 214. [Google Scholar] [CrossRef] [PubMed]
- Vesović, M.; Jelić, R.; Nikolić, M.; Nedeljković, N.; Živanović, A.; Bukonjić, A.; Mrkalić, E.; Radić, G.; Ratkovićc, Z.; Kljun, J.; et al. Investigation of the interaction between S-isoalkyl derivatives of the thiosalicylic acid and human serum albumin. J. Biomol. Struct. Dyn. 2024, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy, 3rd ed.; Springer: New York, NY, USA, 2006. [Google Scholar]
- Mrkalić, E.; Jelić, R.; Stojanović, S.; Sovrlić, M. Interaction between olanzapine and human serum albumin and effect of metal ions, caffeine and flavonoids on the binding: A spectroscopic study. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2021, 249, 119295. [Google Scholar] [CrossRef]
- Liu, Y.; Li, Q.Y.; Wang, Y.P.; Liu, Y.M.; Liu, B.; Liu, M.M.; Liu, B.M. Spectroscopic investigation of the anticancer alkaloid piperlongumine binding to human serum albumin from the viewpoint of drug delivery. Luminescence 2018, 33, 305–311. [Google Scholar] [CrossRef]
- Bi, S.; Song, D.; Tian, Y.; Zhou, X.; Liu, Z.; Zhang, H. Molecular spectroscopic study on the interaction of tetracyclines with serum albumins. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2005, 61, 629–636. [Google Scholar] [CrossRef]
- Dassault Systèmes BIOVIA. Discovery Studio; Version 17.2.0; Dassault Systèmes: San Diego, CA, USA, 2016. [Google Scholar]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Below, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective re-ceptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Sanner, M.F. Phyton: A programming language for software integration and development. J. Mol. Graph. Model. 1999, 17, 57–61. [Google Scholar] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | T (K) | KSV × 105 (M−1) | Kq × 1013 (M−1 s−1) | R2 a |
|---|---|---|---|---|
| HSA-LUR | 298 | 2.51 ± 0.04 | 2.51 | 0.986 |
| HSA-LUR | 303 | 2.43 ± 0.04 | 2.43 | 0.982 |
| HSA-LUR | 308 | 2.30 ± 0.02 | 2.30 | 0.998 |
| T (K) | Ka × 106 (M−1) | n | R2 | ΔH0 (kJmol−1) | ΔS0 (JK−1mol−1) | ΔG0 (kJmol−1) |
|---|---|---|---|---|---|---|
| 298 | 3.98 ± 0.06 | 1.22 | 0.993 | −37.71 | ||
| 303 | 8.13 ± 0.07 | 1.28 | 0.992 | 95.97 | 448.58 | −39.95 |
| 308 | 13.99 ± 0.07 | 1.32 | 0.992 | −42.19 |
| System | T a | ΔG b | ΔG c | Kid 10−6 | Site (Subdomain) |
|---|---|---|---|---|---|
| Lurasidone (blind) * | 298 | −9.91 | −41.46 | 5.40 | I/IIA |
| Lurasidone | 298 | −8.17 | −34.18 | 1.02 | I/IIA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Živković, N.; Mrkalić, E.; Jelić, R.; Tomović, J.; Odović, J.; Serafinović, M.Ć.; Sovrlić, M. The Molecular Recognition of Lurasidone by Human Serum Albumin: A Combined Experimental and Computational Approach. Molecules 2025, 30, 1420. https://doi.org/10.3390/molecules30071420
Živković N, Mrkalić E, Jelić R, Tomović J, Odović J, Serafinović MĆ, Sovrlić M. The Molecular Recognition of Lurasidone by Human Serum Albumin: A Combined Experimental and Computational Approach. Molecules. 2025; 30(7):1420. https://doi.org/10.3390/molecules30071420
Chicago/Turabian StyleŽivković, Nevena, Emina Mrkalić, Ratomir Jelić, Jovica Tomović, Jadranka Odović, Marina Ćendić Serafinović, and Miroslav Sovrlić. 2025. "The Molecular Recognition of Lurasidone by Human Serum Albumin: A Combined Experimental and Computational Approach" Molecules 30, no. 7: 1420. https://doi.org/10.3390/molecules30071420
APA StyleŽivković, N., Mrkalić, E., Jelić, R., Tomović, J., Odović, J., Serafinović, M. Ć., & Sovrlić, M. (2025). The Molecular Recognition of Lurasidone by Human Serum Albumin: A Combined Experimental and Computational Approach. Molecules, 30(7), 1420. https://doi.org/10.3390/molecules30071420