


Antimicrobial Peptide Databases as the Guiding Resource in New Antimicrobial Agent Identification via Computational Methods



Abstract
1. Introduction
1.1. Antimicrobial Peptides (AMPs)—Natural Defenders with Therapeutic Applications
1.2. Mechanisms of Action and Immunomodulatory Properties of AMPs
1.3. Classification and Sources of Antimicrobial Peptides
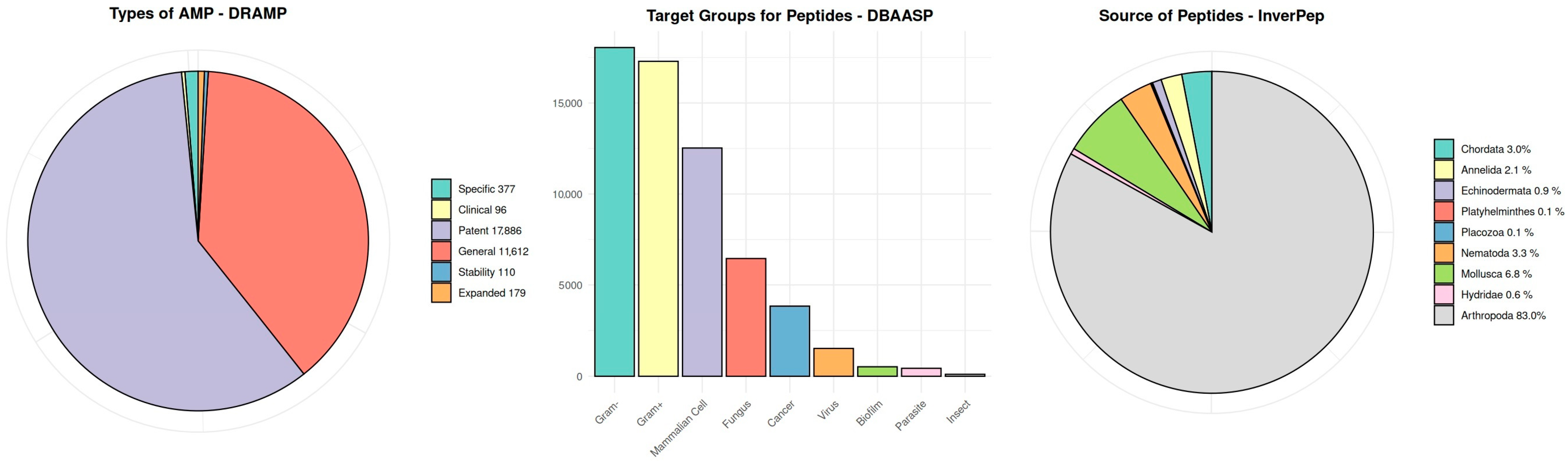
1.4. Antimicrobial Peptide Databases
1.5. Antimicrobial Peptide Databases—Availability
1.6. Antimicrobial Peptide Databases—Reference Database
1.7. Antimicrobial Peptide Databases—Review
- APD is one of the first databases on natural antimicrobial peptides, containing information on the sequences and activities of peptides derived from various organisms. Peptides are classified according to their biological properties, such as antibacterial, antiviral, or anticancer activities [18];
- BaAMPs focuses on peptides tested against biofilms, providing verified experimental data [19];
- CAMP is a database that collects data on AMP sequences, their origin, and biological activity, including synthetic AMPs [20];
- CyBase offers data on cyclic proteins, supporting research on their structures and functions [21];
- dadp collects data on antimicrobial peptides, focusing on precursor sequences and their bioactive fragments [22];
- DBAASP collects data on antimicrobial peptides, providing information on their structures, conditions of action, and molecular targets. It also includes predictive tools supporting peptide design [23];
- DRAMP is a database containing peptides with defined sequences, categorized into general and patent sets, with data on toxicity and hemolytic activity [24];
- InverPep focuses on peptides derived from invertebrates, offering data on sequences, structures, and physicochemical properties [25];
- ParaPep specializes in antiprotozoal peptides, offering information on their structures and mechanisms of action [26];
- SATPdb contains data on therapeutic peptides, enabling sequence and structure similarity searches [27].
1.8. Diamond–High-Throughput Protein Alignment
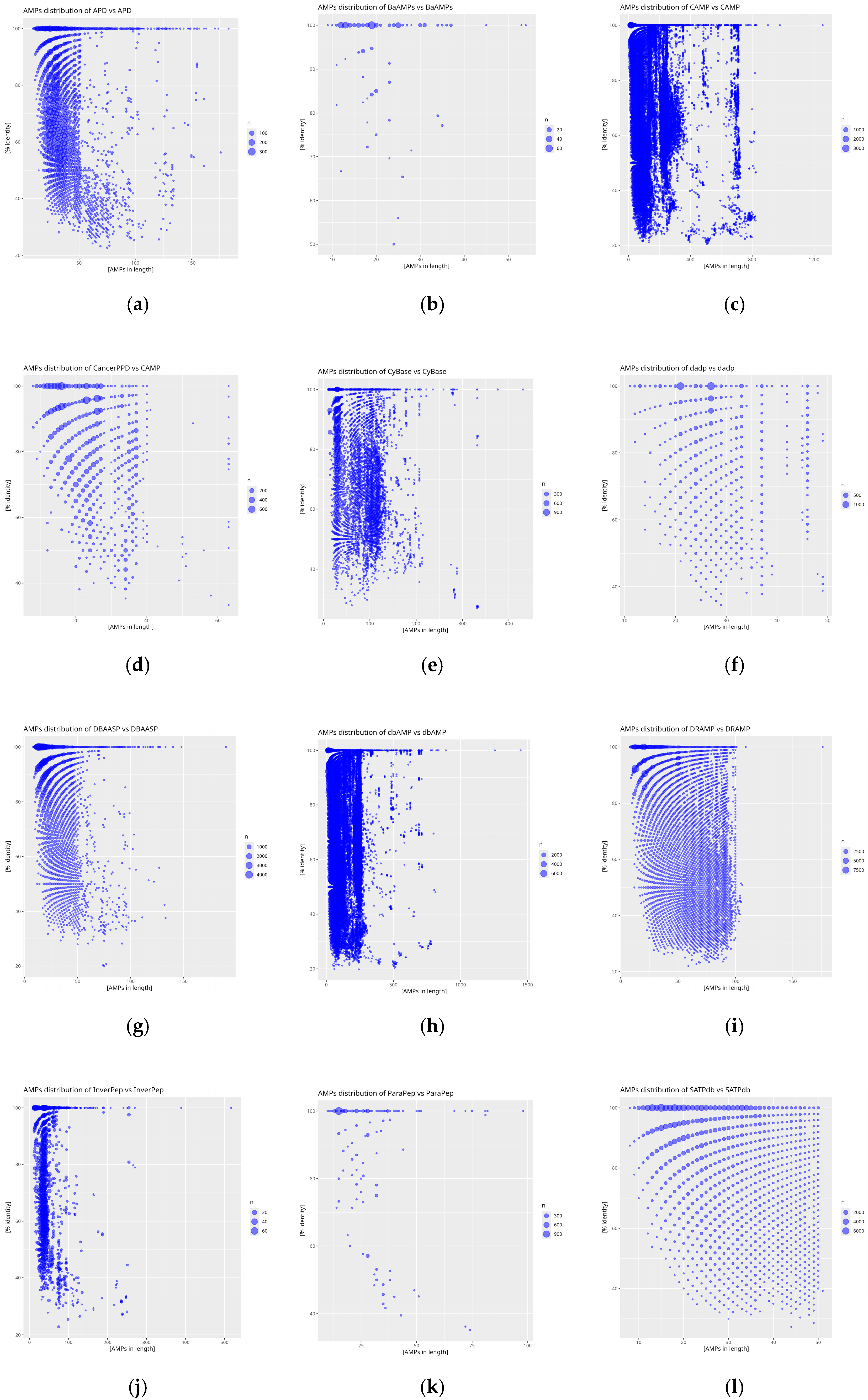
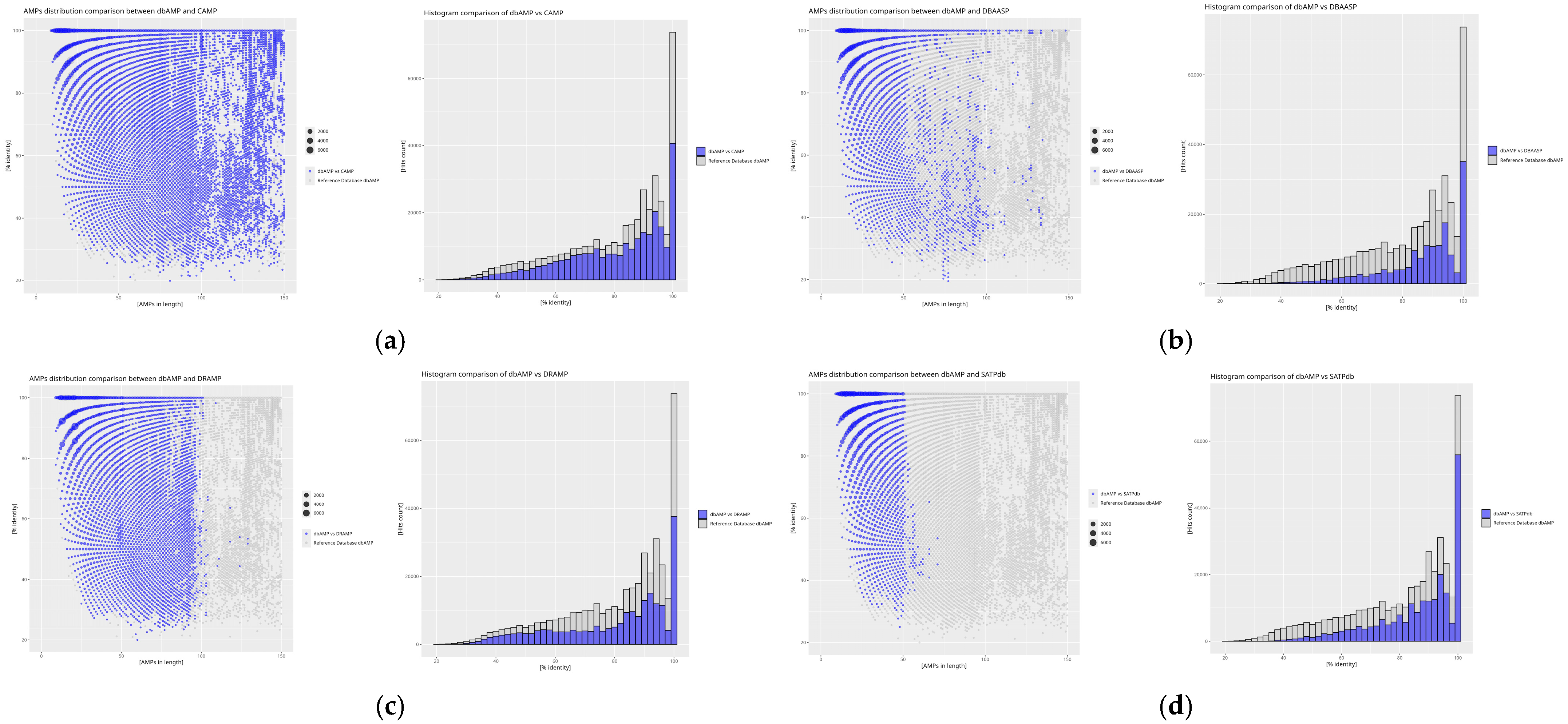
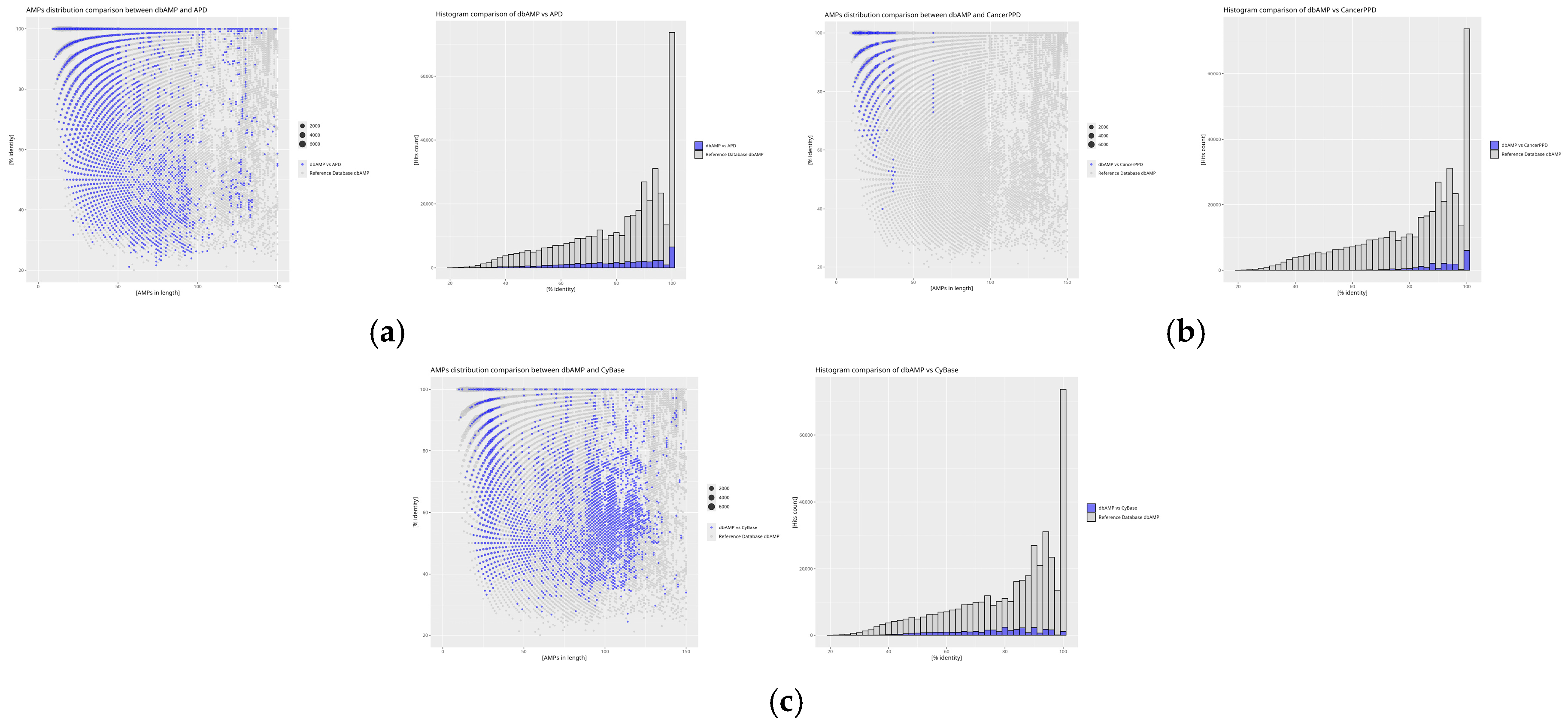
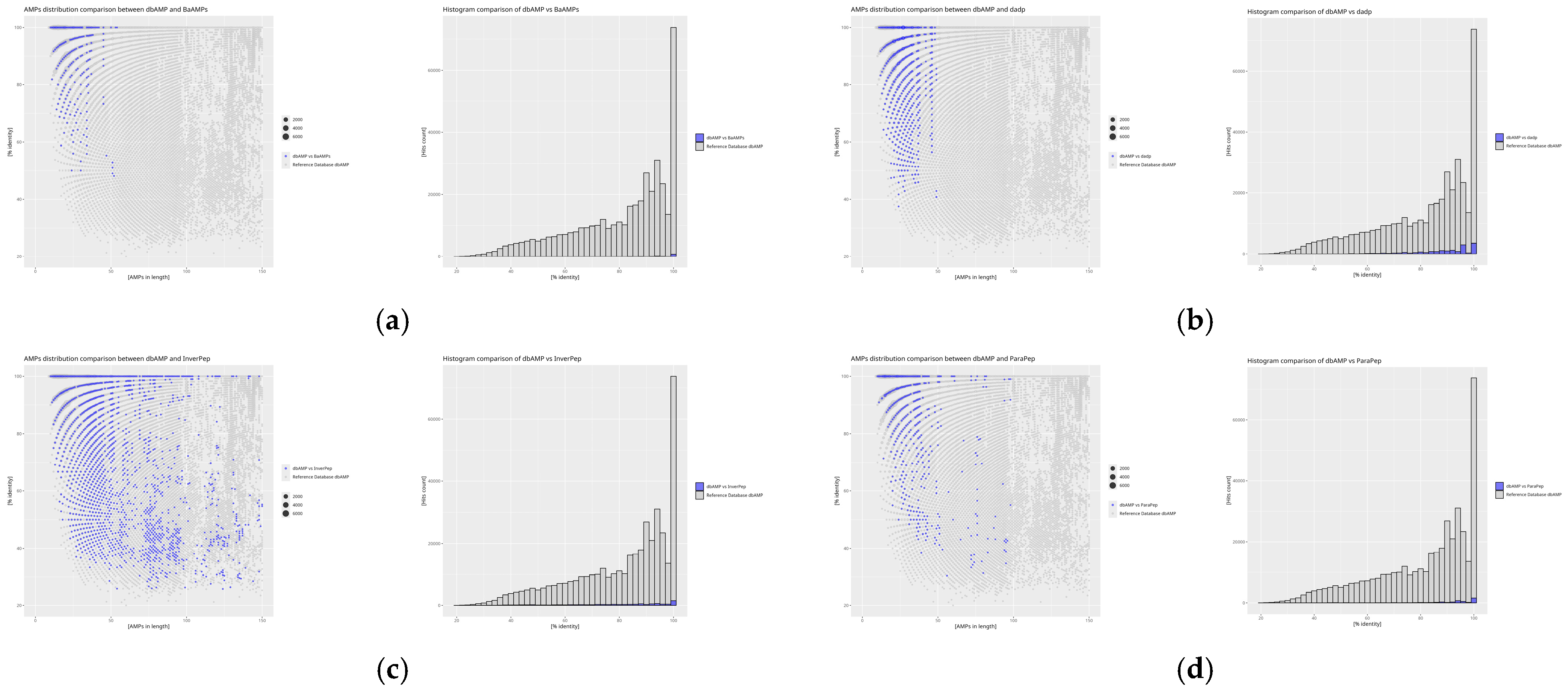
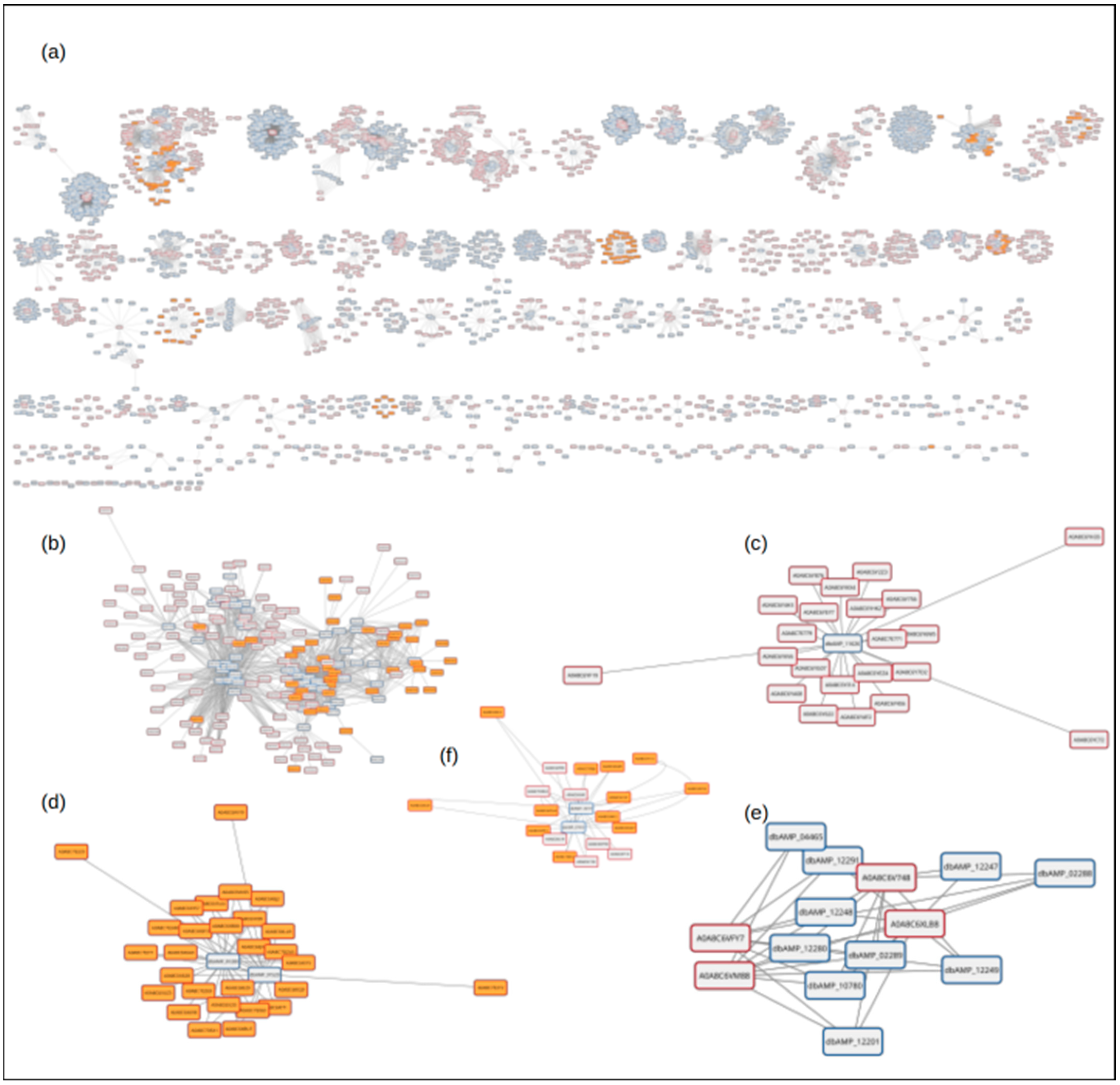
2. Results
3. Discussion
4. Materials and Methods
4.1. Diamond— A Useful Tool in Database Analysis
4.2. Data Preprocessing and Sequence Compatibility for Diamond Tool Analysis
4.3. Efficient Sequence Comparison with Diamond: Converting Fasta to dmnd and Interpreting Results in tsv
4.4. Calculation of DAIRId and IDAIRId Indices
4.5. Graphical Analysis in Rstudio
4.6. Naja naja Proteome as an AMP Potential Source Analysis
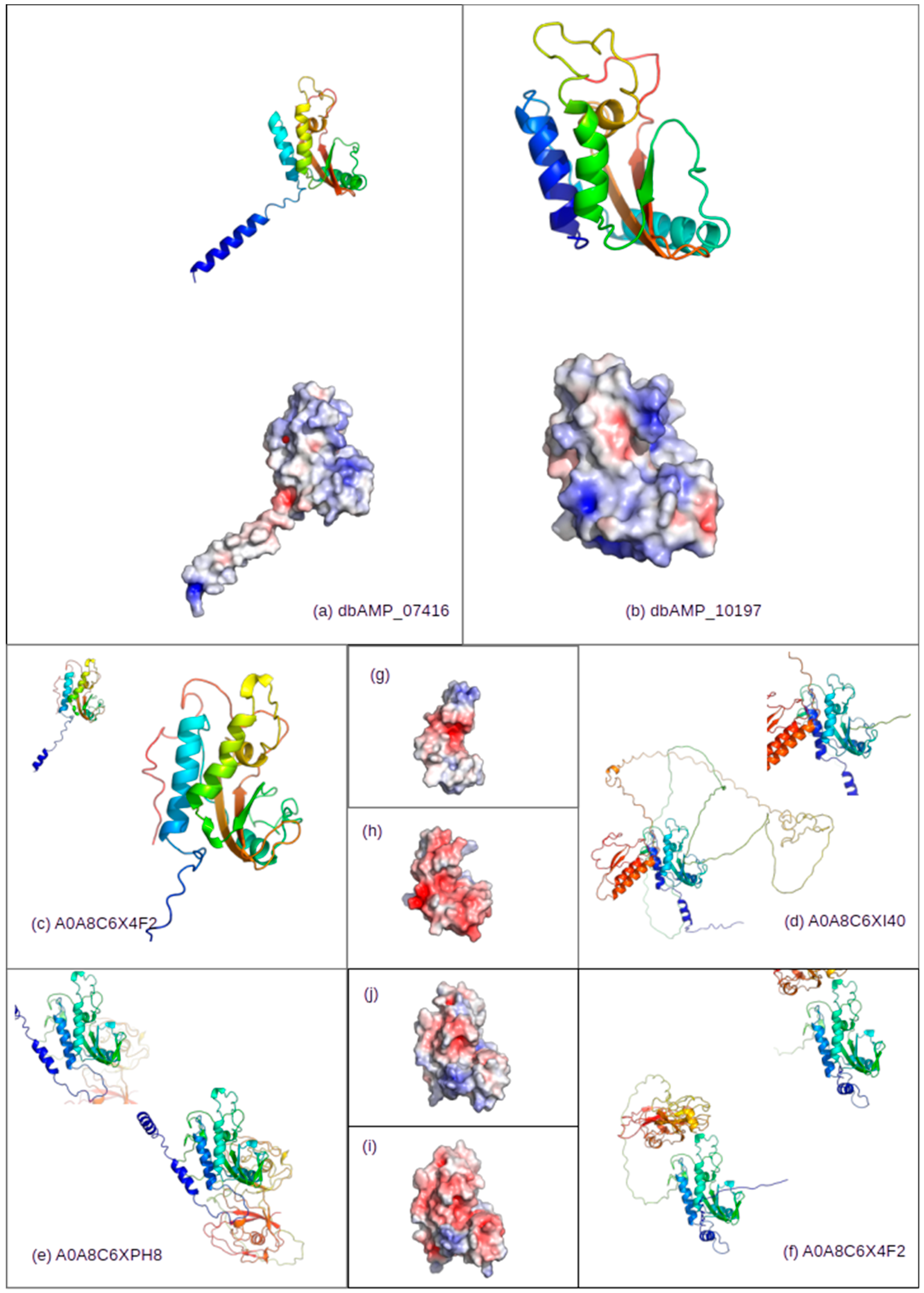
4.7. Naja naja Proteome Hits—Structure Analysis and Visualization
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| DAIRId | Database Absolute-Identity Repeatability Index diamond |
| IDAIRId | Inter-Database Absolute-Identity Repeatability Index diamond |
References
- Seyfi, R.; Kahaki, F.A.; Ebrahimi, T.; Montazersaheb, S.; Eyvazi, S.; Babaeipour, V.; Tarhriz, V. Antimicrobial Peptides (AMPs): Roles, Functions and Mechanism of Action. Int. J. Pept. Res. Ther. 2020, 26, 1451–1463. [Google Scholar] [CrossRef]
- Jenssen, H.; Hamill, P.; Hancock, R.E.W. Peptide Antimicrobial Agents. Clin. Microbiol. Rev. 2006, 19, 491–511. [Google Scholar] [CrossRef]
- Chung, C.-R.; Jhong, J.-H.; Wang, Z.; Chen, S.; Wan, Y.; Horng, J.-T.; Lee, T.-Y. Characterization and Identification of Natural Antimicrobial Peptides on Different Organisms. Int. J. Mol. Sci. 2020, 21, 986. [Google Scholar] [CrossRef]
- Sancho-Vaello, E.; François, P.; Bonetti, E.-J.; Lilie, H.; Finger, S.; Gil-Ortiz, F.; Gil-Carton, D.; Zeth, K. Structural Remodeling and Oligomerization of Human Cathelicidin on Membranes Suggest Fibril-like Structures as Active Species. Sci. Rep. 2017, 7, 15371. [Google Scholar] [CrossRef]
- De Smet, K.; Contreras, R. Human Antimicrobial Peptides: Defensins, Cathelicidins and Histatins. Biotechnol. Lett. 2005, 27, 1337–1347. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Song, Y. Mechanism of Antimicrobial Peptides: Antimicrobial, Anti-Inflammatory and Antibiofilm Activities. Int. J. Mol. Sci. 2021, 22, 11401. [Google Scholar] [CrossRef] [PubMed]
- Grage, S.L.; Afonin, S.; Kara, S.; Kara, G.; Kara, A.S. Frontiers|Membrane Thinning and Thickening Induced by Membrane-Active Amphipathic Peptides. Front. Cell Dev. Biol. 2016, 4, 65. Available online: https://www.frontiersin.org/journals/cell-and-developmental-biology/articles/10.3389/fcell.2016.00065/full (accessed on 22 February 2025).
- Le, C.-F.; Fang, C.-M.; Sekaran, S.D. Intracellular Targeting Mechanisms by Antimicrobial Peptides. Antimicrob. Agents Chemother. 2017, 61, 10–1128. [Google Scholar] [CrossRef]
- Huan, Y.; Kong, Q.; Mou, H.; Yi, H. Antimicrobial Peptides: Classification, Design, Application and Research Progress in Multiple Fields. Front. Microbiol. 2020, 11, 582779. [Google Scholar] [CrossRef]
- Zhang, F.; Cui, X.; Fu, Y.; Zhang, J.; Zhou, Y.; Sun, Y.; Wang, X.; Li, Y.; Liu, Q.; Chen, T. Antimicrobial Activity and Mechanism of the Human Milk-Sourced Peptide Casein201. Biochem. Biophys. Res. Commun. 2017, 485, 698–704. [Google Scholar] [CrossRef]
- Rollins-Smith, L.A. The Role of Amphibian Antimicrobial Peptides in Protection of Amphibians from Pathogens Linked to Global Amphibian Declines. Biochim. Biophys. Acta BBA Biomembr. 2009, 1788, 1593–1599. [Google Scholar] [CrossRef]
- Bulet, P.; Stocklin, R. Insect Antimicrobial Peptides: Structures, Properties and Gene Regulation. Protein Pept. Lett. 2005, 12, 3–11. [Google Scholar] [CrossRef]
- Bin Hafeez, A.; Jiang, X.; Bergen, P.J.; Zhu, Y. Antimicrobial Peptides: An Update on Classifications and Databases. Int. J. Mol. Sci. 2021, 22, 11691. [Google Scholar] [CrossRef]
- Ramazi, S.; Mohammadi, N.; Allahverdi, A.; Khalili, E.; Abdolmaleki, P. A Review on Antimicrobial Peptides Databases and the Computational Tools. Database 2022, 2022, baac011. [Google Scholar] [CrossRef] [PubMed]
- Jhong, J.-H.; Chi, Y.-H.; Li, W.-C.; Lin, T.-H.; Huang, K.-Y.; Lee, T.-Y. dbAMP: An Integrated Resource for Exploring Antimicrobial Peptides with Functional Activities and Physicochemical Properties on Transcriptome and Proteome Data. Nucleic Acids Res. 2019, 47, D285–D297. [Google Scholar] [CrossRef]
- Jhong, J.-H.; Yao, L.; Pang, Y.; Li, Z.; Chung, C.-R.; Wang, R.; Li, S.; Li, W.; Luo, M.; Ma, R.; et al. dbAMP 2.0: Updated Resource for Antimicrobial Peptides with an Enhanced Scanning Method for Genomic and Proteomic Data. Nucleic Acids Res. 2022, 50, D460–D470. [Google Scholar] [CrossRef] [PubMed]
- dbAMP 3.0: Updated Resource of Antimicrobial Activity and Structural Annotation of Peptides in the Post-Pandemic Era|Nucleic Acids Research|Oxford Academic. Available online: https://academic.oup.com/nar/article/53/D1/D364/7900191 (accessed on 22 January 2025).
- APD3: The Antimicrobial Peptide Database as a Tool for Research and Education|Nucleic Acids Research|Oxford Academic. Available online: https://academic.oup.com/nar/article/44/D1/D1087/2503090 (accessed on 4 January 2025).
- Di Luca, M.; Maccari, G.; Maisetta, G.; Batoni, G. BaAMPs: The Database of Biofilm-Active Antimicrobial Peptides. Biofouling 2015, 31, 193–199. [Google Scholar] [CrossRef]
- Gawde, U.; Chakraborty, S.; Waghu, F.H.; Barai, R.S.; Khanderkar, A.; Indraguru, R.; Shirsat, T.; Idicula-Thomas, S. CAMPR4: A Database of Natural and Synthetic Antimicrobial Peptides. Nucleic Acids Res. 2023, 51, D377–D383. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.K.L.; Kaas, Q.; Chiche, L.; Craik, D.J. CyBase: A Database of Cyclic Protein Sequences and Structures, with Applications in Protein Discovery and Engineering. Nucleic Acids Res. 2008, 36, D206–D210. [Google Scholar] [CrossRef]
- Novković, M.; Simunić, J.; Bojović, V.; Tossi, A.; Juretić, D. DADP: The Database of Anuran Defense Peptides. Bioinformatics 2012, 28, 1406–1407. [Google Scholar] [CrossRef]
- Pirtskhalava, M.; Amstrong, A.A.; Grigolava, M.; Chubinidze, M.; Alimbarashvili, E.; Vishnepolsky, B.; Gabrielian, A.; Rosenthal, A.; Hurt, D.E.; Tartakovsky, M. DBAASP v3: Database of Antimicrobial/Cytotoxic Activity and Structure of Peptides as a Resource for Development of New Therapeutics. Nucleic Acids Res. 2021, 49, D288–D297. [Google Scholar] [CrossRef]
- Shi, G.; Kang, X.; Dong, F.; Liu, Y.; Zhu, N.; Hu, Y.; Xu, H.; Lao, X.; Zheng, H. DRAMP 3.0: An Enhanced Comprehensive Data Repository of Antimicrobial Peptides. Nucleic Acids Res. 2022, 50, D488–D496. [Google Scholar] [CrossRef] [PubMed]
- Gómez, E.A.; Giraldo, P.; Orduz, S. InverPep: A Database of Invertebrate Antimicrobial Peptides. J. Glob. Antimicrob. Resist. 2017, 8, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Mehta, D.; Anand, P.; Kumar, V.; Joshi, A.; Mathur, D.; Singh, S.; Tuknait, A.; Chaudhary, K.; Gautam, S.K.; Gautam, A.; et al. ParaPep: A Web Resource for Experimentally Validated Antiparasitic Peptide Sequences and Their Structures. Database 2014, 2014, bau051. [Google Scholar] [CrossRef]
- Singh, S.; Chaudhary, K.; Dhanda, S.K.; Bhalla, S.; Usmani, S.S.; Gautam, A.; Tuknait, A.; Agrawal, P.; Mathur, D.; Raghava, G.P.S. SATPdb: A Database of Structurally Annotated Therapeutic Peptides. Nucleic Acids Res. 2016, 44, D1119–D1126. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and Sensitive Protein Alignment Using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Buchfink, B.; Reuter, K.; Drost, H.-G. Sensitive Protein Alignments at Tree-of-Life Scale Using DIAMOND. Nat. Methods 2021, 18, 366–368. [Google Scholar] [CrossRef]
- Suryamohan, K.; Krishnankutty, S.P.; Guillory, J.; Jevit, M.; Schröder, M.S.; Wu, M.; Kuriakose, B.; Mathew, O.K.; Perumal, R.C.; Koludarov, I.; et al. The Indian Cobra Reference Genome and Transcriptome Enables Comprehensive Identification of Venom Toxins. Nat. Genet. 2020, 52, 106–117. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology Modelling of Protein Structures and Complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- The UniProt Consortium UniProt: The Universal Protein Knowledgebase in 2023. Nucleic Acids Res. 2023, 51, D523–D531. [CrossRef]
- The PyMOL Molecular Graphics System, Version 3.0; Schrödinger, LLC.: New York, NY, USA, 2024.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Database Type | Description | Examples |
|---|---|---|
| General databases | They contain various types of AMPs, regardless of peptide family | APD, CAMP, dbAMP |
| Specific databases | They focus on specific classes of AMPs, such as defensins, cyclotides, or anticancer peptides | CancerPPD, ParaPep |
| Experimental and predictive databases | They offer both natural and predicted AMPs | CyBase, SATPdb, DBAASP, DRAMP |
| Database | Status | Number of AMPs (Data from Article 03.2022) | Number of AMPs (Data Current 11.2024) | URL * |
|---|---|---|---|---|
| APD | Active | 1230 | 5099 | https://aps.unmc.edu |
| BaAMPs | Active ** | 237 | 237 | https://baamps.it/ |
| CAMP | Active | 8160 | 24,243 | https://camp.bicnirrh.res.in |
| CancerPPD | Active | 3490 | 3491 | http://crdd.osdd.net/raghava/cancerppd/index.php |
| CyBase | Active | 1270 | 1818 | https://www.cybase.org.au/index.php |
| dadp | Active | 2571 | 2571 | http://split4.pmfst.hr/dadp/ |
| DBAASP | Active | 15,700 | 22,622 | https://dbaasp.org/home |
| dbAMP | Active | 26,440 | 35,518 | https://awi.cuhk.edu.cn/dbAMP/index.php |
| DRAMP | Active | 22,250 | 30,260 | http://dramp.cpu-bioinfor.org/ |
| InverPep | Active | 774 | 774 | https://ciencias.medellin.unal.edu.co/gruposdeinvestigacion/prospeccionydisenobiomoleculas/InverPep/public/home_en |
| ParaPep | Active | 863 | 863 | https://webs.iiitd.edu.in/raghava/parapep/home.php |
| SATPdb | Active | 2525 | 19,192 | https://webs.iiitd.edu.in/raghava/satpdb/index.html |
| ADAM | Inactive | - | ||
| BACTIBASE | Inactive | - | ||
| Defensins Knowledgebase | Inactive | - | ||
| LAMP | Inactive | - | ||
| Database | Theoretical Number of Sequences | Obtained Number of Sequences | Number of Total diamond Comparisons with the Reference Database dbAMP |
|---|---|---|---|
| APD | 5099 | 3167 | 44,881 |
| BaAMPs | 237 | 221 | 1504 |
| CAMP | 24,243 | 20,750 | 269,680 |
| CancerPPD | 3491 | 2849 | 20,161 |
| CyBase | 1818 | 1757 | 33,185 |
| dadp | 2571 | 933 | 18,015 |
| DBAASP | 22,622 | 22,004 | 22,622 |
| dbAMP | 35,518 | 34,811 | 419,123 |
| DRAMP | 30,260 | 28,302 | 211,244 |
| InverPep | 774 | 773 | 9976 |
| ParaPep | 863 | 513 | 5053 |
| SATPdb | 28,373 | 25,885 | 223,633 |
| Database | DAIRId [%] | IDAIRId Relative to the Reference Database (dbAMP) [%] |
|---|---|---|
| APD | 11.70 | 1.75 |
| BaAMPs | 82.66 | 0.29 |
| CAMP | 15.00 | 27.80 |
| CancerPPD | 70.23 | 1.47 |
| CyBase | 8.99 | 0.28 |
| dadp | 28.65 | 0.97 |
| DBAASP | 26.12 | 27.92 |
| dbAMP | 17.21 | - |
| DRAMP | 24.76 | 17.12 |
| InverPep | 13.20 | 0.55 |
| ParaPep | 73.05 | 0.30 |
| SATPdb | 34.62 | 20.05 |
| UniProt Description | dbAMP _07416 | dbAMP _10197 | Cleavage Site Available | ||
|---|---|---|---|---|---|
| % Identity | Seq. Match Start/Stop (Total Length) | % Identity | Seq. Match Start/Stop (Total Length) | ||
| A0A8C6VDU4_NAJNA R3H domain-containing like | 32.5 | 67/219 (253) | 32.5 | 67/219 (253) | |
| A0A8C6VGD7_NAJNA R3H domain-containing like | 32.5 | 63/215 (249) | 32.5 | 63/215 (249) | |
| A0A8C6VPF7_NAJNA SCP domain-containing protein | 31.9 | 38/188 (270) | 31.9 | 38/188 (270) | |
| A0A8C6VPR5_NAJNA SCP domain-containing protein | 30.1 | 38/162 (206) | 30.1 | 38/162 (206) | |
| A0A8C6 × 1V1_NAJNA R3H domain-containing like | 32.5 | 65/217 (251) | 32.5 | 65/217 (251) | |
| A0A8C6X4F2_NAJNA SCP domain-containing protein OS = Naja naja | 36.8 | 90/173 (302) | 36.8 | 90/173 (302) | yes |
| A0A8C6X4Z0_NAJNA SCP domain-containing protein | 29.8 | 40/181 (181) | 29.8 | 40/181 (181) | |
| A0A8C6XI40_NAJNA SCP domain-containing protein | 39.2 | 58/177 (580) | 39.2 | 58/177 (580) | yes |
| A0A8C6XLY9_NAJNA Peptidase inhibitor 15 | 32.9 | 63/223 (258) | 33.1 | 64/223 (258) | |
| A0A8C6XPH8_NAJNA Cysteine rich secretory protein LCCL domain containing 1 | 35.0 | 58/216 (503) | 35.0 | 58/216 (503) | yes |
| A0A8C6XXU9_NAJNA ShKT domain-containing protein | 26.6 | 26/179 (239) | 26.6 | 30/179 (239) | |
| A0A8C6XXV5_NAJNA ShKT domain-containing protein | 27.2 | 26/179 (239) | 27.3 | 30/179 (239) | |
| A0A8C6XZL9_NAJNA ShKT domain-containing protein | 26.6 | 26/179 (239) | 27.3 | 34/179 (239) | |
| A0A8C6Y1Y2_NAJNA ShKT domain-containing protein | 27.8 | 26/179 (239) | 28.7 | 34/179 (239) | |
| A0A8C6Y1Z2_NAJNA SCP domain-containing protein | 32.4 | 41/178 (219) | 32.4 | 41/178 (219) | |
| A0A8C6YF13_NAJNA Cysteine rich secretory protein LCCL domain containing | 30.9 | 55/215 (495) | 31.1 | 56/215 (495) | yes |
| A0A8C7DRJ6_NAJNA SCP domain-containing | 31.0 | 36/177 (217) | 31.0 | 36/177 (217) | |
| A0A8C7DRK4_NAJNA GLI pathogenesis related 2 OS = Naja naja | 30.7 | 11/145 (154) | 30.7 | 11/145 (154) | |
| A0A8C7E3S7_NAJNA ShKT domain-containing protein | 32.4 | 41/178 (238) | 32.4 | 41/178 (238) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marczak, B.; Bocian, A.; Łyskowski, A. Antimicrobial Peptide Databases as the Guiding Resource in New Antimicrobial Agent Identification via Computational Methods. Molecules 2025, 30, 1318. https://doi.org/10.3390/molecules30061318
Marczak B, Bocian A, Łyskowski A. Antimicrobial Peptide Databases as the Guiding Resource in New Antimicrobial Agent Identification via Computational Methods. Molecules. 2025; 30(6):1318. https://doi.org/10.3390/molecules30061318
Chicago/Turabian StyleMarczak, Bogdan, Aleksandra Bocian, and Andrzej Łyskowski. 2025. "Antimicrobial Peptide Databases as the Guiding Resource in New Antimicrobial Agent Identification via Computational Methods" Molecules 30, no. 6: 1318. https://doi.org/10.3390/molecules30061318
APA StyleMarczak, B., Bocian, A., & Łyskowski, A. (2025). Antimicrobial Peptide Databases as the Guiding Resource in New Antimicrobial Agent Identification via Computational Methods. Molecules, 30(6), 1318. https://doi.org/10.3390/molecules30061318