
Bioactive Secondary Metabolites of Two Chinese Edible Boletes, Phlebopus portentosus and Butyriboletus roseoflavus

 , and
, and
Abstract
1. Introduction
2. Results
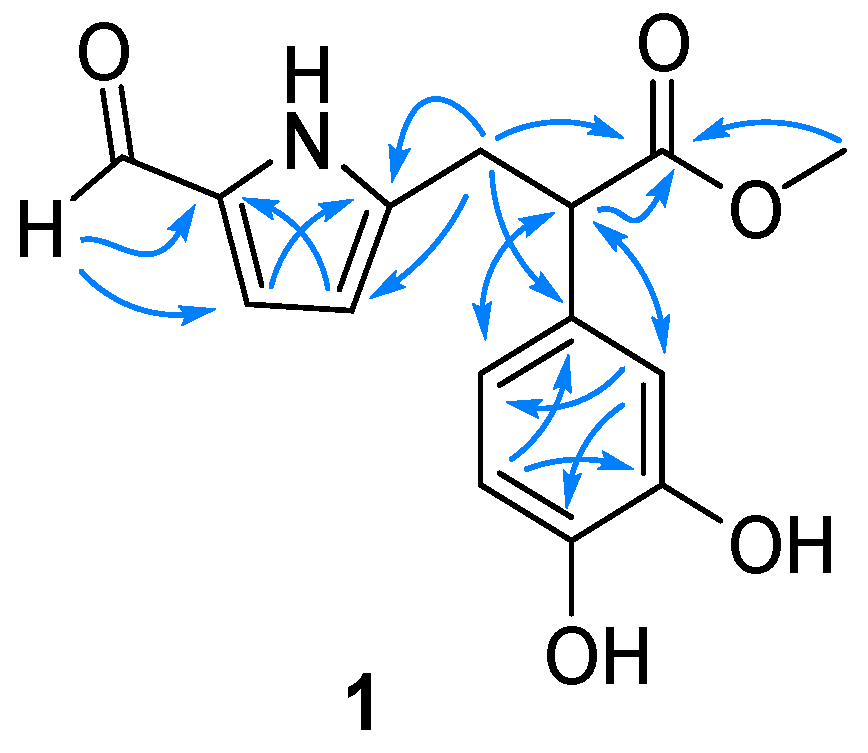
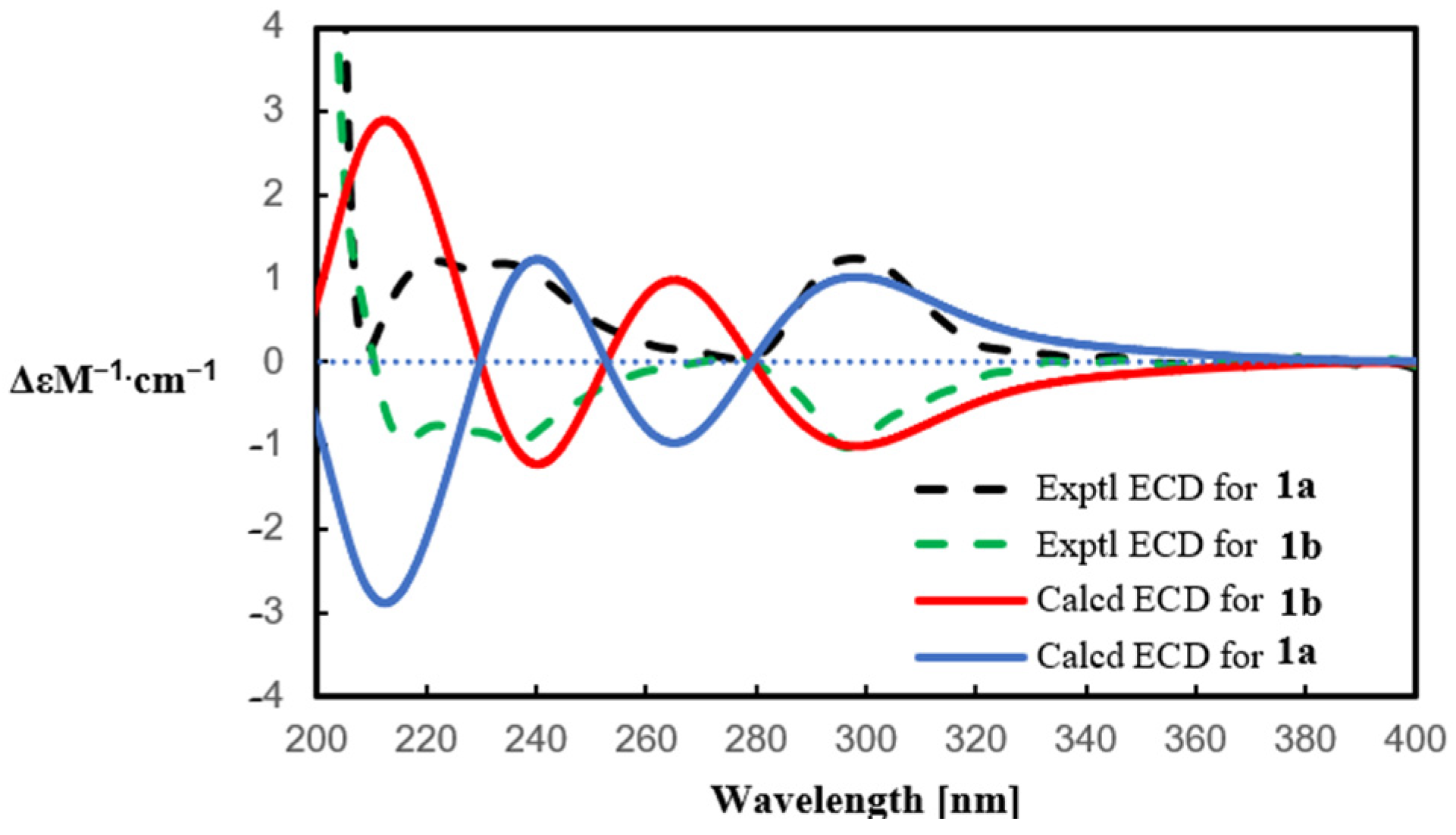
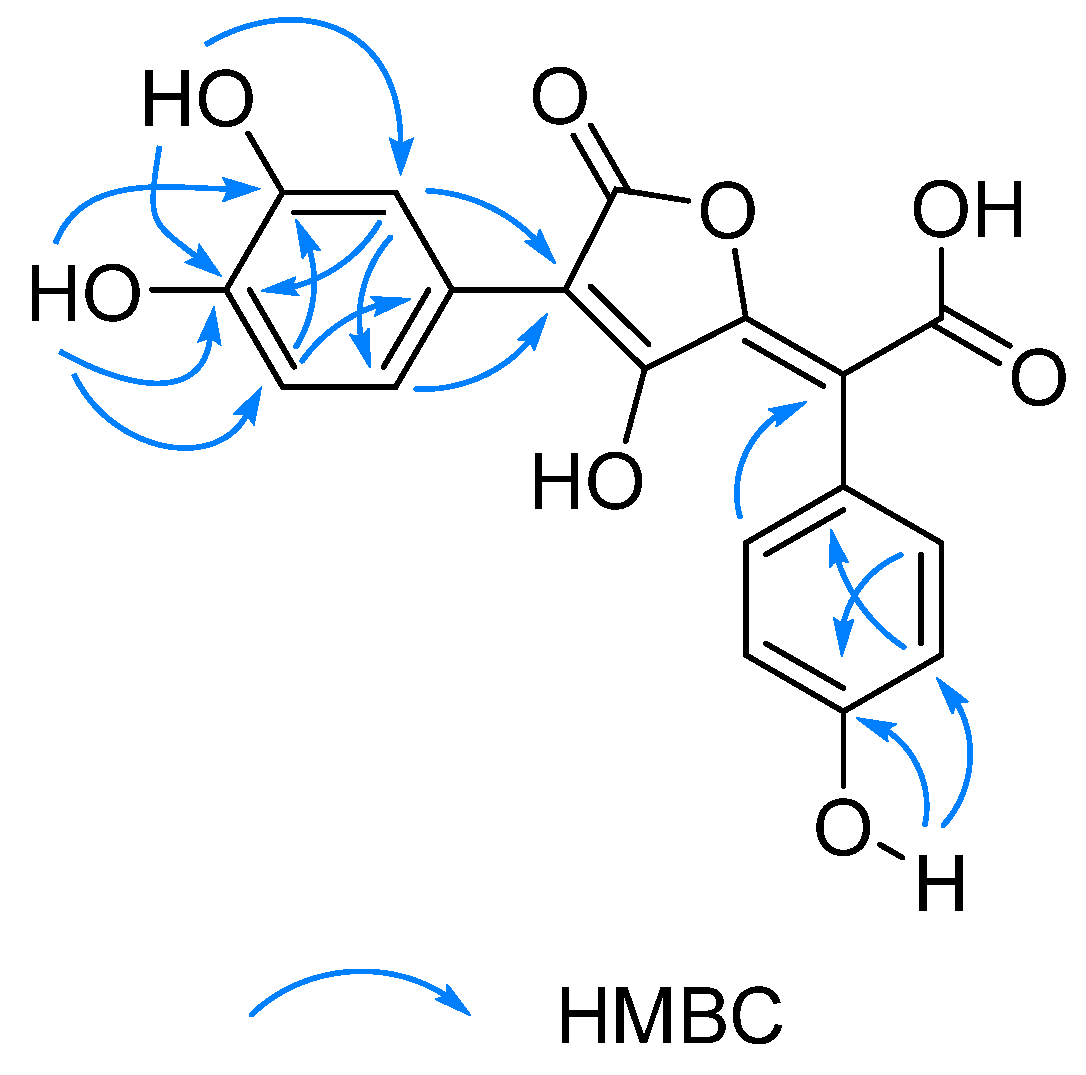
2.1. Structure Elucidation
2.2. Biological Activities
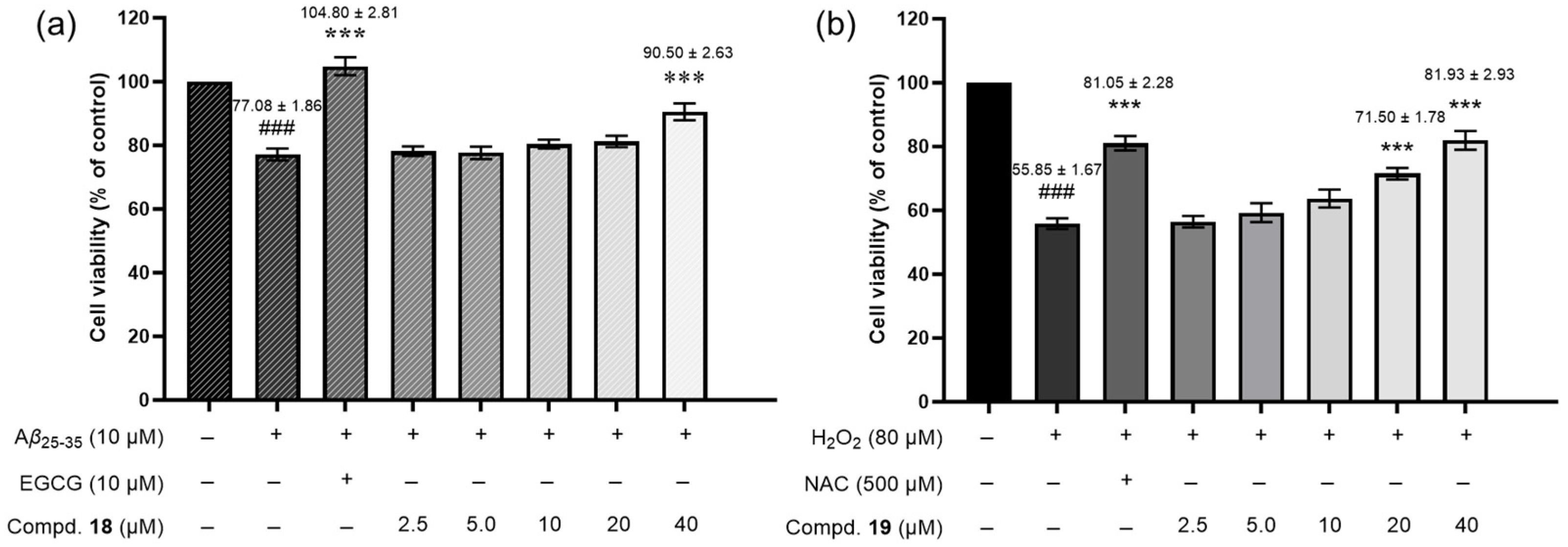
2.2.1. Neuroprotective Activity
2.2.2. Cytotoxicity Activity
2.2.3. Antiviral Activity
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Mushroom Materials
3.3. Extraction and Isolation
3.3.1. Extraction and Isolation of Cultivated P. portentosus
3.3.2. Extraction and Isolation of Wild P. portentosus
3.3.3. Extraction and Isolation of B. roseoflavus
3.3.4. Chiral Separation
3.4. Spectroscopic Data of the Isolated Compounds
3.5. Computational Calculation of ECD Data
3.6. Bioactivity Assays
3.6.1. Neuroprotective Activity Assay
3.6.2. Cytotoxicity Assay
3.6.3. Antiviral Assays
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sun-Waterhouse, D.X.; Chen, X.-Y.; Liu, Z.-H.; Waterhouse, G.I.N.; Kang, W.-Y. Transformation from traditional medicine-food homology to modern food-medicine homology. Food Med. Homol. 2024, 1, 9420014. [Google Scholar] [CrossRef]
- Ai, T.M. Medicinal Flora of China (Volume 13) Glossary of Chinese Medicinal Plants; Peking University Medical Press: Beijing, China, 2021. [Google Scholar]
- National Health Commission of China. Available online: http://www.nhc.gov.cn/sps/s3585/201411/67ac54fb05ed46929adc63f2db31d4bf.shtml (accessed on 4 March 2025).
- Zhou, Y.; Chu, M.; Ahmadi, F.; Agar, O.T.; Barrow, C.J.; Dunshea, F.R.; Suleria, H.A.R. A comprehensive review on phytochemical profiling in mushrooms: Occurrence, biological activities, applications and future prospective. Food Rev. Int. 2023, 40, 924–951. [Google Scholar] [CrossRef]
- Sandargo, B.; Chepkirui, C.; Cheng, T.; Chaverra-Muñoz, L.; Thongbai, B.; Stadler, M.; Hüttel, S. Biological and chemical diversity go hand in hand: Basidomycota as source of new pharmaceuticals and agrochemicals. Biotechnol. Adv. 2019, 37, 107344. [Google Scholar] [CrossRef]
- Li, T.-H.; Song, B. Keys to the Bolete genera occurring in China. Ecol. Sci. 2002, 21, 240–245. [Google Scholar]
- Wu, G.; Miyauchi, S.; Morin, E.; Kuo, A.; Drula, E.; Varga, T.; Kohler, A.; Feng, B.; Cao, Y.; Lipzen, A.; et al. Evolutionary innovations through gain and loss of genes in the ectomycorrhizal Boletales. New Phytol. 2022, 233, 1383–1400. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Li, Y.-C.; Zhu, X.-T.; Zhao, K.; Han, L.-H.; Cui, Y.-Y.; Li, F.; Xu, J.-P.; Yang, Z.-L. One hundred noteworthy boletes from China. Fungal Divers. 2016, 81, 25–188. [Google Scholar] [CrossRef]
- Hellwig, V.; Dasenbrock, D.; Gräf, C.; Kahner, L.; Schumann, S.; Steglich, W. Calopins and cyclocalopins bitter principles from Boletus calopus and related mushrooms. Eur. J. Org. Chem. 2002, 9, 2895–2904. [Google Scholar] [CrossRef]
- Wang, L.; Ma, Q.-Y.; Huang, S.-Z.; Kong, F.-D.; Ding, Q.; Zhao, Y.-X. Research advances in chemical composition and bioactivity of boletes. J. Trop. Biol. 2017, 8, 127–132. [Google Scholar]
- Kumla, J.; Suwannarach, N.; Lumyong, S. Cultivation of edible tropical bolete, Phlebopus spongiosus, in Thailand and yield improvement by high-voltage pulsed stimulation. Agronomy 2022, 12, 115. [Google Scholar] [CrossRef]
- Ji, K.-P.; Cao, Y.; Zhang, C.-X.; He, M.-X.; Liu, J.; Wang, W.-B.; Wang, Y. Cultivation of Phlebopus portentosus in southern China. Mycol. Progress. 2011, 10, 293–300. [Google Scholar] [CrossRef]
- Sun, Z.-C.; Hu, M.-G.; Sun, Z.-H.; Zhu, N.-L.; Yang, J.-S.; Ma, G.-X.; Xu, X.-D. Pyrrole alkaloids from the edible mushroom Phlebopus portentosus with their bioactive activities. Molecules 2018, 23, 1198. [Google Scholar] [CrossRef] [PubMed]
- Qian, Z.-M.; Huang, Q.; Shen, Q.-H.; Xie, M.-X.; Li, W.-J.; Dong, C.-H. Content determination and HPLC fingerprint analysis of uridine and ergosterol in Phlebopus portentosus. J. Fungal Res. 2019, 17, 103–109. [Google Scholar]
- Chen, Z.-L.; Fu, D.-T.; Zhang, L.; Tan, L.-Y.; Fu, X.-J. Chemical constituents from Phlebopus portentosus and their anti-tumor cytotoxic activities. Chin. Tradit. Pat. Med. 2023, 45, 3296–3301. [Google Scholar]
- Chuankid, B.; Schrey, H.; Thongbai, B.; Raspé, O.; Hyde, K.D.; Stadler, M. Secondary metabolites of Phlebopus species from Northern Thailand. Mycol. Progress 2020, 19, 1525–1536. [Google Scholar] [CrossRef]
- Arora, D.; Frank, J.L. Clarifying the butter boletes: A new genus, Butyriboletus, is established to accommodate Boletus sect. Appendiculati, and six new species are described. Mycologia 2014, 106, 464–480. [Google Scholar] [CrossRef]
- Su, L.-H.; Geng, C.-A.; Li, T.-Z.; Huang, X.-Y.; Ma, Y.-B.; Zhang, X.-M.; Wu, G.; Yang, Z.-L.; Chen, J.-J. Spiroseoflosterol, a rearranged ergostane-steroid from the fruiting bodies of Butyriboletus roseoflavus. J. Nat. Prod. 2020, 83, 1706–1710. [Google Scholar] [CrossRef]
- Wood, J.M.; Furkert, D.P.; Brimble, M.A. 2-Formylpyrrole natural products: Origin, structural diversity, bioactivity and synthesis. Nat. Prod. Rep. 2019, 36, 289–306. [Google Scholar] [CrossRef]
- Zhang, L.-Y.; Bai, H.-B.; Shan, W.-G.; Zhan, Z.-J. A new alkaloid from the mycelium of Inonotus obliquus. J. Chem. Res. 2014, 38, 245–246. [Google Scholar] [CrossRef]
- Nelsen, S.F. Bluing components and other pigments of boletes. Fungi 2010, 3, 11–14. [Google Scholar]
- Pradayrol, É.; Séon-Méniel, B.; Oger, S.; Sinniah-Kandiah, B.; Guillot, R.; Franco, R.; Souce, M.; Kasselouri, A.; Maciuk, A.; Evanno, L. Bioinspired synthesis of pulvinic acids including xerocomic acid and fluorescence properties of bis-lactone intermediates. Eur. J. Org. Chem. 2023, 26, e202201152. [Google Scholar] [CrossRef]
- Yang, Y.-P.; Cheng, M.-J.; Teng, C.-M.; Chang, Y.-L.; Tsai, I.-L.; Chen, I.-S. Chemical and anti-platelet constituents from Formosan Zanthoxylum simulans. Phytochemistry 2002, 61, 567–572. [Google Scholar] [CrossRef]
- Yu, J.-G.; Chen, R.-Y.; Yao, Z.-X.; Zhai, Y.-F.; Yang, S.-L.; Ma, J.-L. Studies on constituents of Ganoderam Capense IV. the chemical structure of ganoine, ganodine and ganoderpurine. Acta Pharm. Sin. 1990, 25, 612–616. [Google Scholar]
- Chin, Y.-W.; Lim, S.W.; Kim, S.-H.; Shin, D.-Y.; Suh, Y.-B.; Kim, Y.-C.; Kim, J. Hepatoprotective pyrrole derivatives of Lycium chinense fruits. Bioorg. Med. Chem. Lett. 2003, 13, 79–81. [Google Scholar] [CrossRef]
- Soto-Otero, R.; Méndez-Alvarez, E.; Riguera-Vega, R.; Quiñoá-Cabana, E.; Sánchez-Sellero, I.; López-Rivadulla Lamas, M. Studies on the interaction between 1,2,3,4-tetrahydro-β-carboline and cigarette smoke: A potential mechanism of neuroprotection for Parkinson’s disease. Brain Res. 1998, 802, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Hwang, I.-H.; Kim, S.-Y.; Lee, M.-S. Method for Producing N-Formyltryptoline. Republic of Korea Patent No. KR2024127622 A, 23 August 2024. [Google Scholar]
- Fadaeinasab, M.; Hadi, A.H.A.; Kia, Y.; Basiri, A.; Murugaiyah, V. Cholinesterase enzyme inhibitors from the leaves of Rauvolfia reflexa and their molecular docking study. Molecules 2013, 18, 3779–3788. [Google Scholar] [CrossRef]
- Qi, S.-H.; Zhang, S.; Yang, L.-H.; Qian, P.-Y. Antifouling and antibacterial compounds from the gorgonians Subergorgia suberosa and Scripearia gracillis. Nat. Prod. Res. 2008, 22, 154–166. [Google Scholar] [CrossRef]
- Parameswaran, P.S.; Naik, C.G.; Hegde, V.R. Secondary metabolites from the sponge Tedania anhelans: Isolation and characterization of two novel pyrazole acids and other metabolites. J. Nat. Prod. 1997, 60, 802–803. [Google Scholar] [CrossRef]
- Yang, F.; Chen, R.; Feng, L.; Li, H.-D.; Zhang, H.; Liang, J.-Y. Chemical constituents from the aerial part of Peganum nigellastrum. Chin. J. Nat. Med. 2010, 8, 199–201. [Google Scholar] [CrossRef]
- Chen, M.-H.; Lian, Y.-Y.; Fang, D.-S.; Chen, L.; Jia, J.; Zhang, W.-L.; Jiang, H. Identification and antimicrobial properties of a new alkaloid produced by marine-derived Verrucosispora sp. FIM06-0036. Nat. Prod. Res. 2021, 35, 4211–4217. [Google Scholar] [CrossRef]
- Elliott, M.C.; Williams, E. Synthesis and reactions of partially reduced biisoquinolines. Org. Biomol. Chem. 2003, 1, 3038–3047. [Google Scholar] [CrossRef]
- Chen, S.-D.; Yong, T.-Q.; Xiao, C.; Su, J.-Y.; Zhang, Y.-F.; Jiao, C.-W.; Xie, Y.-Z. Pyrrole alkaloids and ergosterols from Grifola frondosa exert anti-α-glucosidase and anti-proliferative activities. J. Funct. Foods 2018, 43, 196–205. [Google Scholar] [CrossRef]
- Desage-El Murr, M.; Nowaczyk, S.; Le Gall, T.; Mioskowski, C. Synthesis of pulvinic acid and norbadione A analogues by Suzuki–Miyaura cross-coupling of benzylated intermediates. Eur. J. Org. Chem. 2006, 2006, 1489–1498. [Google Scholar] [CrossRef]
- Huang, Y.-T.; Onose, J.; Abe, N.; Yoshikawa, K. In vitro inhibitory effects of pulvinic acid derivatives isolated from Chinese edible mushrooms, Boletus calopus and Suillus bovinus, on cytochrome P450 activity. Biosci. Biotechnol. Biochem. 2009, 73, 855–860. [Google Scholar] [CrossRef] [PubMed]
- Kawagishi, H.; Katsumi, R.; Sazawa, T.; Mizuno, T.; Hagiwara, T.; Nakamura, T. Cytotoxic steroids from the mushroom Agaricus blazei. Phytochemistry 1988, 27, 2777–2779. [Google Scholar] [CrossRef]
- Yaoita, Y.; Amemiya, K.; Ohnuma, H.; Furumura, K.; Masaki, A.; Matsuki, T.; Kikuchi, M. Sterol constituents from five edible mushrooms. Chem. Pharm. Bull. 1998, 46, 944–950. [Google Scholar] [CrossRef]
- Piccialli, V.; Sica, D. Four new trihydroxylated sterols from the sponge Spongionella gracilis. J. Nat. Prod. 1987, 50, 915–920. [Google Scholar] [CrossRef]
- Ishizuka, T.; Yaoita, Y.; Kikuchi, M. Sterol constituents from the fruit bodies of Grifola frondosa (FR.) S. F. Gray. Chem. Pharm. Bull. 1997, 45, 1756–1760. [Google Scholar] [CrossRef]
- Yaoita, Y.; Endo, M.; Tani, Y.; Machida, K.; Amemiya, K.; Furumura, K.; Kikuchi, M. Sterol constituents from seven mushrooms. Chem. Pharm. Bull. 1999, 47, 847–851. [Google Scholar] [CrossRef]
- Yue, J.-M.; Chen, S.-N.; Lin, Z.-W.; Sun, H.-D. Sterols from the fungus Lactarium volemus. Phytochemistry 2001, 56, 801–806. [Google Scholar] [CrossRef]
- Chen, H.-L.; Chiang, H.-C. Constituents of fruit bodies of Tramete orientalis. J. Chin. Chem. Soc. 1995, 42, 97–100. [Google Scholar] [CrossRef]
- Gao, H.; Hong, K.; Chen, G.-D.; Wang, C.-X.; Tang, J.-S.; Yu, Y.; Jiang, M.-M.; Li, M.-M.; Wang, N.-L.; Yao, X.-S. New oxidized sterols from Aspergillus awamori and the endo-boat conformation adopted by the cyclohexene oxide system. Magn. Reson. Chem. 2010, 48, 38–43. [Google Scholar] [CrossRef]
- Liao, Y.-W.; Chen, C.-R.; Hsu, J.-L.; Cheng, H.-L.; Shih, W.-L.; Kuo, Y.-H.; Huang, T.-C.; Chang, C.-I. Sterols from the stems of Momordica charantia. J. Chin. Chem. Soc. 2011, 58, 893–898. [Google Scholar] [CrossRef]
- Shi, Q.-Q.; Huang, Y.-J.; Su, H.-G.; Gao, Y.; Peng, X.-R.; Zhou, L.; Li, X.-N.; Qiu, M.-H. C28 steroids from the fruiting bodies of Ganoderma resinaceum with potential anti-inflammatory activity. Phytochemistry 2019, 168, 112109. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-K.; Kuo, Y.-H.; Chiang, B.-H.; Lo, J.-M.; Sheen, L.-Y. Cytotoxic activities of 9,11-dehydroergosterol peroxide and ergosterol peroxide from the fermentation mycelia of Ganoderma lucidum cultivated in the medium containing leguminous plants on Hep 3B cells. J. Agric. Food Chem. 2009, 57, 5713–5719. [Google Scholar] [CrossRef]
- da Graça Sgarbi, D.B.; da Silva, A.J.R.; Carlos, I.Z.; Silva, C.L.; Angluster, J.; Alviano, C.S. Isolation of ergosterol peroxide and its reversion to ergosterol in the pathogenic fungus Sporothrix schenckii. Mycopathologia 1997, 139, 9–14. [Google Scholar] [CrossRef]
- De Riccardis, F.; Spinella, A.; Izzo, I.; Giordano, A.; Sodano, G. Synthesis of (17R)-17-methylincisterol, a highly degraded marine steroid. Tetrahedron Lett. 1995, 36, 4303–4306. [Google Scholar] [CrossRef]
- Barnham, K.-J.; Masters, C.-L.; Bush, A.-I. Neurodegenerative diseases and oxidative stress. Nat. Rev. Drug Discov. 2004, 3, 205–214. [Google Scholar] [CrossRef]
- Wang, X.-F.; Zhang, J.; Chen, Q.; Zhou, W.; Wu, J. Generation of sulfonylureas under photoredox catalysis and their biological evaluations. Chin. Chem. Lett. 2022, 33, 4860–4864. [Google Scholar] [CrossRef]
- Hung, H.-C.; Tseng, C.-P.; Yang, J.-M.; Ju, Y.-W.; Tseng, S.N.; Chen, Y.-F.; Chao, Y.-S.; Hsieh, S.P.; Shih, S.R.; Hsu, J.T.A. Aurintricarboxylic acid inhibits influenza virus neuraminidase. Antiviral Res. 2009, 81, 123–131. [Google Scholar] [CrossRef]
- Chen, M.-T.; Chen, H.-Y.; Luo, Y.-F.; Zhou, J.; Yang, J.-J.; Jiang, Z.-P.; Huang, L. Highly conjugated ergosterols with anti-influenza virus activity from the marine-derived fungus Eutypella Sp. F0219. Chem. Biodivers. 2024, e202402465. [Google Scholar] [CrossRef]
- Loregian, A.; Mercorelli, B.; Nannetti, G.; Compagnin, C.; Palù, G. Antiviral strategies against influenza virus: Towards new therapeutic approaches. Cell Mol. Life Sci. 2014, 19, 3659–3683. [Google Scholar] [CrossRef] [PubMed]
- Terrier, O.; Slama-Schwok, A. Anti-influenza drug discovery and development: Targeting the virus and its host by all possible means. In Antiviral Drug Discovery and Development; Liu, X., Zhan, P., Menéndez-Arias, L., Poongavanam, V., Eds.; Springer: Singapore, 2021; Volume 1322, pp. 195–218. [Google Scholar]
- Shokry, S.; Hegazy, A.; Abbas, A.-M.; Mostafa, I.; Eissa, I.-H.; Metwaly, A.-M.; Yahya, G.; El-Shazly, A.-M.; Aboshanab, K.-M.; Mostafa, A. Phytoestrogen β-sitosterol exhibits potent in vitro antiviral activity against influenza A viruses. Vaccines 2023, 11, 228. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Fu, Y.; Xiong, J.; Li, M.; Ma, G.-L.; Yang, G.-X.; Wei, B.-G.; Zhao, Y.; Zhang, H.-Y.; Hu, J.-F. Casuarinines A–J, lycodine-type alkaloids from Lycopodiastrum casuarinoides. J. Nat. Prod. 2013, 76, 1475–1484. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | 1 a | 2 b | Inotopyrrole (4) b | |||
|---|---|---|---|---|---|---|
| δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | δC | |
| 1 | 8.52, s | |||||
| 2 | 133.5 | 132.4 | 132.3 | |||
| 3 | 6.87, d (3.9) | 116.4 | 6.92, d (4.0) | 124.2 | 6.94, d (4.2) | 124.6 |
| 4 | 5.99, d (3.9) | 111.3 | 6.19, d (4.0) | 111.3 | 6.18, d (4.2) | 110.1 |
| 5 | 141.8 | 138.9 | 144.7 | |||
| 6 | 3.32, dd (14.6, 7.1) | 32.7 | 4.11, s | 65.6 | 4.30, s | 56.3 |
| 3.00, dd (14.6, 7.1) | ||||||
| 1′ | 130.9 | 138.6 | 138.5 | |||
| 2′ | 6.74, d (2.2) | 115.8 | 7.16, br d (7.7) | 129.0 | 7.11, br d (7.8) | 129.1 |
| 3′ | 146.5 | 7.29, br dd (7.7, 6.9) | 128.5 | 7.27, br t (7.8) | 128.6 | |
| 4′ | 145.9 | 7.23, br t (6.9) | 126.6 | 7.22, br t (7.8) | 126.7 | |
| 5′ | 6.69, d (8.2) | 118.1 | 7.29, br dd (7.7, 6.9) | 128.5 | 7.27, br t (7.8) | 128.6 |
| 6′ | 6.60, dd (8.2, 2.2) | 120.3 | 7.16, br d (7.7) | 129.0 | 7.11, br d (7.8) | 129.1 |
| 7′ | 3.84, dd (9.2, 7.0) | 52.0 | 3.02, t (7.6) | 37.8 | 3.06, t (7.2) | 37.7 |
| 8′ | 175.6 | 4.51, t (7.6) | 47.8 | 4.56, t (7.2) | 47.6 | |
| -CHO | 9.26, s | 179.9 | 9.58, s | 179.4 | 9.59, s | 179.4 |
| -OCH3 | 3.61, s | 52.5 | 3.31, s | 57.9 | ||
| No. | 16 a | 16 b | Xerocomic Acid (18) b | |||
|---|---|---|---|---|---|---|
| δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | δC | |
| 1 | 126.8 | 128.8 | 125.6 | |||
| 2 | 6.99, br d (8.4) | 131.5 | 7.14, br d (8.4) | 132.8 | 7.26, br d (8.4) | 132.6 |
| 3 | 6.71, dd (8.4, 2.0) | 113.8 | 6.78, br d (8.5) | 114.7 | 6.87, br d (8.8) | 115.4 |
| 4 | 156.2 | 157.2 | 158.3 | |||
| 5 | 6.71, dd (8.4, 2.0) | 113.8 | 6.78, br d (8.5) | 114.7 | 6.87, br d (8.8) | 115.4 |
| 6 | 6.99, br d (8.4) | 131.5 | 7.14, br d (8.4) | 132.8 | 7.26, br d (8.4) | 132.6 |
| 7 | 168.9 | 169.8 | 167.1 | |||
| 8 | 95.1 | 97.2 | 104.3 | |||
| 9 | 167.0 | 169.6 | 160.6 | |||
| 10 | 152.0 | 153.4 | 155.2 | |||
| 11 | 117.5 | 119.2 | 117.0 | |||
| 12 | 169.3 | 170.2 | 174.0 | |||
| 1′ | 123.9 | 126.1 | 122.7 | |||
| 2′ | 7.68, br s | 113.3 | 7.89, d (2.0) | 114.2 | 7.76, d (2.0) | 115.4 |
| 3′ | 144.5 | 145.0 | 145.5 | |||
| 4′ | 143.0 | 143.8 | 146.2 | |||
| 5′ | 6.67, br d (8.5) | 115.1 | 6.78, br d (8.5) | 115.5 | 6.87, br d (8.8) | 116.0 |
| 6′ | 7.46, br d (8.5) | 117.0 | 7.71, dd (8.4, 2.0) | 119.2 | 7.61, dd (8.1, 2.0) | 121.0 |
| 3′-OH | 8.78, dd (6.8, 6.4) | |||||
| 4′-OH | 8.62, dd (12.0, 7.1) | |||||
| 4-OH | 9.35, s | |||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Z.; Zhou, W.; He, Y.; Zhao, Z.; Cao, Y.; Luo, S.; Ji, G.; Ji, K.; Chen, J.; Li, J.; et al. Bioactive Secondary Metabolites of Two Chinese Edible Boletes, Phlebopus portentosus and Butyriboletus roseoflavus. Molecules 2025, 30, 1197. https://doi.org/10.3390/molecules30061197
Wang Z, Zhou W, He Y, Zhao Z, Cao Y, Luo S, Ji G, Ji K, Chen J, Li J, et al. Bioactive Secondary Metabolites of Two Chinese Edible Boletes, Phlebopus portentosus and Butyriboletus roseoflavus. Molecules. 2025; 30(6):1197. https://doi.org/10.3390/molecules30061197
Chicago/Turabian StyleWang, Zhixuan, Wei Zhou, Yuhang He, Zeyu Zhao, Yang Cao, Shunzhen Luo, Guangyan Ji, Kaiping Ji, Jing Chen, Jiyang Li, and et al. 2025. "Bioactive Secondary Metabolites of Two Chinese Edible Boletes, Phlebopus portentosus and Butyriboletus roseoflavus" Molecules 30, no. 6: 1197. https://doi.org/10.3390/molecules30061197
APA StyleWang, Z., Zhou, W., He, Y., Zhao, Z., Cao, Y., Luo, S., Ji, G., Ji, K., Chen, J., Li, J., & Xiong, J. (2025). Bioactive Secondary Metabolites of Two Chinese Edible Boletes, Phlebopus portentosus and Butyriboletus roseoflavus. Molecules, 30(6), 1197. https://doi.org/10.3390/molecules30061197