
Spherical Amides with C3 Symmetry: Improved Synthetic Approach and Structural/Optical Analysis

 ,
,
Abstract

1. Introduction
2. Results and Discussion
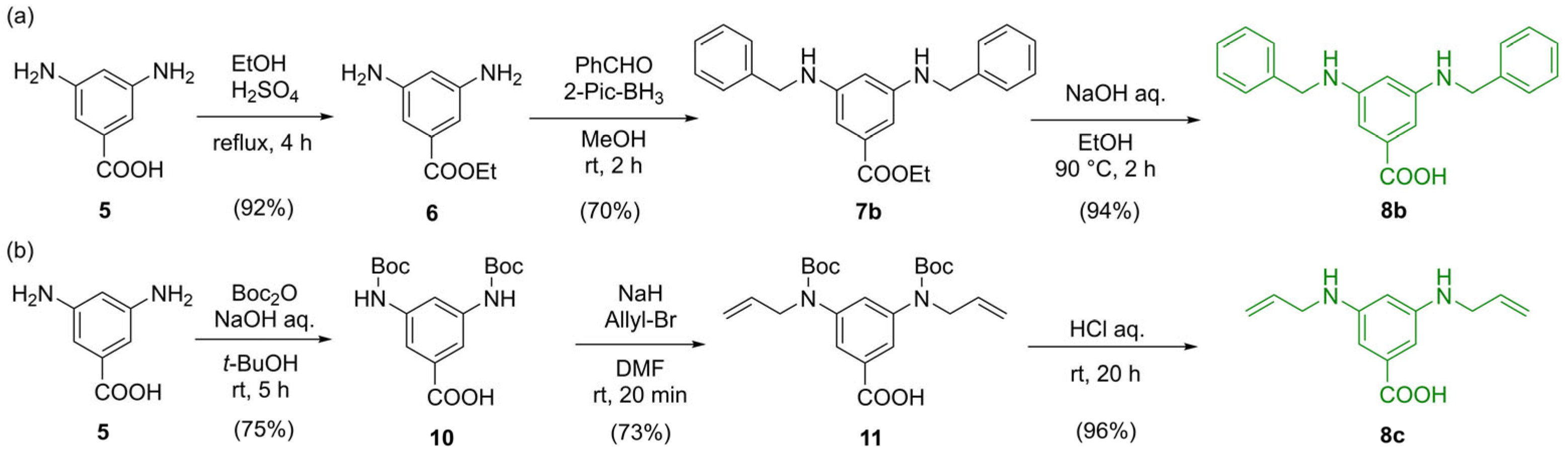
2.1. Synthesis of Spherical Amides
2.2. NMR Spectroscopic Analysis
2.3. Optical Resolution and ECD Spectral Analysis
2.4. X-Ray Crystallographic Analysis
3. Materials and Methods
3.1. General
3.2. Synthesis
- Ethyl 3,5-bis(benzylamino)benzoate (7b)
- 3,5-Bis(benzylamino)benzoic acid (8b)
- 3,5-Bis[(tert-butoxycarbonyl)amino]benzoic acid (10)
- 3,5-Bis[allyl(tert-butoxycarbonyl)amino]benzoic acid (11)
- 3,5-Bis(allylamino)benzoic acid (8c)
- General procedure (cyclization)
- N-Ethyl macrocycle (1a)
- N-Benzyl macrocycle (1b)
- N-Allyl macrocycle (1c)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gutsche, C.D.; Muthukrishnan, R. Calixarenes. 1. Analysis of the Product Mixtures Produced by the Base-Catalyzed Condensation of Formaldehyde with Para-Substituted Phenols. J. Org. Chem. 1978, 43, 4905–4906. [Google Scholar] [CrossRef]
- Chen, Y.; Tian, J.H.; Tian, H.W.; Ma, R.; Wang, Z.H.; Pan, Y.C.; Hu, X.Y.; Guo, D.S. Calixarene-Based Supramolecular Sensor Array for Pesticide Discrimination. Sensors 2024, 24, 3743. [Google Scholar] [CrossRef] [PubMed]
- Alshahateet, S.F.; Altarawneh, R.M.; Al-Trawneh, S.A.; Al-Saraireh, Y.M.; Al-Tawarh, W.M.; Abuawad, K.R.; Abuhalaweh, Y.M.; Zerrouk, M.; Mansour, A.A.; Salghi, R.; et al. Cheminformatics-Based Design and Biomedical Applications of a New Hydroxyphenylcalix[4] Resorcinarene as Anti-Cancer Agent. Sci. Rep. 2024, 14, 30460. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Guo, J.; Lai, Y.; Lin, J.; Liu, J.; Ji, J.; Yin, P.; Wang, W.; Zhao, H.; Chen, G.; et al. Polyoxometalate–Organic Hybrid “Calixarenes” as Supramolecular Hosts. Angew. Chem. Int. Ed. 2024, 63, e202315691. [Google Scholar] [CrossRef]
- Cram, D.J.; Cram Vol, J.M.; Cram, D.J.; Maxwell Cram, J. Bridged Aromatic Compounds; Academic Press: Cambridge, MA, USA, 1959; Volume 20. [Google Scholar]
- Tanaka, K. Catalytic Enantioselective Synthesis of Planar Chiral Cyclophanes. Bull. Chem. Soc. Jpn. 2018, 91, 187–194. [Google Scholar] [CrossRef]
- Ramaiah, D.; Neelakandan, P.P.; Nair, A.K.; Avirah, R.R. Functional Cyclophanes: Promising Hosts for Optical Biomolecular Recognition. Chem. Soc. Rev. 2010, 39, 4158–4168. [Google Scholar] [CrossRef]
- Fang, W.; Zhang, J.; Guo, M.; Zhao, Y.; Sue, A.C.H. Triphenylamine[3]Arenes: Streamlining Synthesis of a Versatile Macrocyclic Platform for Supramolecular Architectures and Functionalities. Angew. Chem. Int. Ed. 2024, 63, e202409120. [Google Scholar] [CrossRef]
- Hartendorp, A.P.T.; De Zwart, F.J.; Bieräugel, H.; De Bruin, B.; Reek, J.N.H.; Van Maarseveen, J.H. Peptide Cyclisation Promoted by Supramolecular Complex Formation. Org. Biomol. Chem. 2022, 20, 575–578. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, Z.; Ma, L.; Wen, H.; Kang, M.; Li, D.; Zhang, W.; Luo, S.; Wang, W.; Zhang, M.; et al. Combining Multiple Photosensitizer Modules into One Supramolecular System for Synergetic Enhanced Photodynamic Therapy. Angew. Chem. Int. Ed. 2024, 63, e202400049. [Google Scholar] [CrossRef]
- Hu, J.; Ni, W.; Han, M.; Zhan, Y.; Li, F.; Huang, H.; Han, J. Machine Learning-Assisted Pattern Recognition and Imaging of Multiplexed Cancer Cells via a Porphyrin-Embedded Dendrimer Array. J. Mater. Chem. B 2024, 13, 207. [Google Scholar] [CrossRef]
- Sun, G.; Zhang, X.; Zheng, Z.; Zhang, Z.-Y.; Dong, M.; Sessler, J.L.; Li, C. Chiral Macrocycles for Enantioselective Recognition. J. Am. Chem. Soc. 2024, 146, 26233–26242. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.F.; Tong, S.; Zhu, J.; Wang, M.X. Inherently Chiral Nor-Heteracalixarenes: Design and Synthesis via Enantioselective Intramolecular Suzuki-Miyaura Reaction. Chem. Sci. 2024, 15, 12517–12522. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, K.; Tabata, Y.; Yamakawa, K.; Mochizuki, T.; Matsui, K.; Hatano, M.; Ishihara, K. Chiral Macrocyclic Catalysts for the Enantioselective Addition of Lithium Acetylides to Ketones. J. Am. Chem. Soc. 2023, 145, 26238–26248. [Google Scholar] [CrossRef]
- Weh, M.; Kroeger, A.A.; Shoyama, K.; Grüne, M.; Karton, A.; Würthner, F. π-π Catalysis Made Asymmetric—Enantiomerization Catalysis Mediated by the Chiral π-System of a Perylene Bisimide Cyclophane. Angew. Chem. Int. Ed. 2023, 62, e202301301. [Google Scholar] [CrossRef]
- Zhang, D.; Wang, L.; Wu, W.; Cao, D.; Tang, H. Macrocyclic Catalysis Mediated by Water: Opportunities and Challenges. Chem. Commun. 2025, 61, 599. [Google Scholar] [CrossRef]
- Kress, C.; Sidler, E.; Downey, P.; Zwick, P.; Fuhr, O.; Fenske, D.; Bernhard, S.; Mayor, M. Circularly Polarized Luminescence of Thiophene-Bridged Macrocyclic Pseudo-Meta [2.2]Paracyclophanes. Chem. Eur. J. 2024, 30, e202303798. [Google Scholar] [CrossRef]
- Sato, S.; Yoshii, A.; Takahashi, S.; Furumi, S.; Takeuchi, M.; Isobe, H. Chiral Intertwined Spirals and Magnetic Transition Dipole Moments Dictated by Cylinder Helicity. Proc. Natl. Acad. Sci. USA 2017, 114, 13097–13101. [Google Scholar] [CrossRef]
- Kroto, H.W. C60: Buckminsterfullerene, The Celestial Sphere That Fell to Earth. Angew. Chem. Int. Ed. 1992, 31, 111–129. [Google Scholar] [CrossRef]
- Abellán-Flos, M.; Tanç, M.; Supuran, C.T.; Vincent, S.P. Exploring Carbonic Anhydrase Inhibition with Multimeric Coumarins Displayed on a Fullerene Scaffold. Org. Biomol. Chem. 2015, 13, 7445–7451. [Google Scholar] [CrossRef]
- Castro, E.; Garcia, A.H.; Zavala, G.; Echegoyen, L. Fullerenes in Biology and Medicine. J. Mater. Chem. B 2017, 5, 6523–6535. [Google Scholar] [CrossRef]
- Fu, C.; Gong, S.; Lin, L.; Bao, Y.; Li, L.; Chen, Q. Characterization and Efficacy of C60 Nano-Photosensitive Drugs in Colorectal Cancer Treatment. Biomed. Pharmacother. 2024, 176, 116828. [Google Scholar] [CrossRef] [PubMed]
- Iijima, S. Helical microtubules of graphitic carbon. Nature 1991, 354, 56–58. [Google Scholar] [CrossRef]
- Liu, Z.; Tabakman, S.; Welsher, K.; Dai, H. Carbon Nanotubes in Biology and Medicine: In Vitro and in Vivo Detection, Imaging and Drug Delivery. Nano Res. 2009, 2, 85–120. [Google Scholar] [CrossRef] [PubMed]
- Imoto, D.; Shudo, H.; Yagi, A.; Itami, K. A Double-walled Noncovalent Carbon Nanotube by Columnar Packing of Nanotube Fragments. Angew. Chem. Int. Ed. 2025, 64, e202413828. [Google Scholar] [CrossRef]
- Andrews, K.G.; Horton, P.N.; Coles, S.J. Programmable Synthesis of Organic Cages with Reduced Symmetry. Chem. Sci. 2024, 15, 6536–6543. [Google Scholar] [CrossRef]
- Lauer, J.C.; Bhat, A.S.; Barwig, C.; Fritz, N.; Kirschbaum, T.; Rominger, F.; Mastalerz, M. [2+3] Amide Cages by Oxidation of [2+3] Imine Cages—Revisiting Molecular Hosts for Highly Efficient Nitrate Binding. Chem. Eur. J. 2022, 28, e202201527. [Google Scholar] [CrossRef]
- Li, Y.; He, J.; Lu, G.; Wang, C.; Fu, M.; Deng, J.; Yang, F.; Jiang, D.; Chen, X.; Yu, Z.; et al. De Novo Construction of Amine-Functionalized Metal-Organic Cages as Heterogenous Catalysts for Microflow Catalysis. Nat. Commun. 2024, 15, 7044. [Google Scholar] [CrossRef]
- Yang, J.; Mao, L.L.; Xiao, H.; Zhang, G.; Zhang, S.; Kang, L.; Lin, Z.; Tung, C.H.; Wu, L.Z.; Cong, H. A Conjugated Phenylene Nanocage with a Guest-Adaptive Deformable Cavity. Angew. Chem. Int. Ed. 2024, 63, e202403062. [Google Scholar] [CrossRef]
- Azumaya, I.; Kagechika, H.; Yamaguchi, K.; Shudo, K. Facile Formation of Aromatic Cyclic N-Methylamides Based on cis Conformational Preference. Tetrahedron Lett. 1996, 37, 5003–5006. [Google Scholar] [CrossRef]
- Yamakado, R.; Mikami, K.; Takagi, K.; Azumaya, I.; Sugimoto, S.; Matsuoka, S.I.; Suzuki, M.; Katagiri, K.; Uchiyama, M.; Muranaka, A. Helicity Induction in Three π-Conjugated Chromophores by Planar Chirality of Calixamide. Chem. Eur. J. 2013, 19, 11853–11857. [Google Scholar] [CrossRef]
- Saito, Y.; Satake, M.; Mori, R.; Okayasu, M.; Masu, H.; Tominaga, M.; Katagiri, K.; Yamaguchi, K.; Kikkawa, S.; Hikawa, H.; et al. Synthesis and Chiroptical Properties of Cylindrical Macrocycles Comprising Two Calix[3]Aramide Moieties. Org. Biomol. Chem. 2020, 18, 230–236. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, A.; Ishii, A.; Ohishi, T.; Kikkawa, S.; Azumaya, I. Synthesis of a Coronene Analogue Containing an Amide Bond by Pd-Mediated Intramolecular C[Sbnd]C Bond Formation of 2-Halogenated 4-(Alkylamino)Benzoic Acid Cyclic Trimer. Tetrahedron Lett. 2021, 62, 152704. [Google Scholar] [CrossRef]
- Itai, A.; Toriumi, Y.; Tomioka, N.; Kagechika, H.; Azumaya, I.; Shudo, K. Stereochemistry of N-Methylbenzanilide and Benzanilide. Tetrahedron Lett. 1989, 30, 6177–6180. [Google Scholar] [CrossRef]
- Azumaya, I.; Yamaguchi, K.; Kagechika, H.; Saito, S.; Itai, A.; Shudo, K. Stereochemistry of Benzanilides and N-Methylbenzanilides. Yakugaku Zasshi 1994, 114, 414–430. [Google Scholar] [CrossRef] [PubMed]
- Imabeppu, F.; Katagiri, K.; Masu, H.; Kato, T.; Tominaga, M.; Therrien, B.; Takayanagi, H.; Kaji, E.; Yamaguchi, K.; Kagechika, H.; et al. Calix[3]amides—Bowl-shaped cyclic trimers toward building block for molecular recognition: Self-complementary dimeric structure in the crystal. Tetrahedron Lett. 2006, 47, 413–416. [Google Scholar] [CrossRef]
- Masu, H.; Katagiri, K.; Kato, T.; Kagechika, H.; Tominaga, M.; Azumaya, I. Chiral Spherical Molecule Constructed from Aromatic Amides: Facile Synthesis and Highly Ordered Network Structure in the Crystal. J. Org. Chem. 2008, 73, 5143–5146. [Google Scholar] [CrossRef]
- Azumaya, I.; Okamoto, T.; Imabeppu, F.; Takayanagi, H. Simple and Convenient Synthesis of Tertiary Benzanilides Using Dichlorotriphenylphosphorane. Tetrahedron 2003, 59, 2325–2331. [Google Scholar] [CrossRef]
- Ikawa, T.; Fujita, Y.; Mizusaki, T.; Betsuin, S.; Takamatsu, H.; Maegawa, T.; Monguchi, Y.; Sajiki, H. Selective N-Alkylation of Amines Using Nitriles under Hydrogenation Conditions: Facile Synthesis of Secondary and Tertiary Amines. Org. Biomol. Chem. 2012, 10, 293–304. [Google Scholar] [CrossRef]
- Koike, D.; Masu, H.; Kikkawa, S.; Chiba, A.; Kamohara, K.; Okuda, A.; Hikawa, H.; Azumaya, I. Chiral Spherical Aromatic Amides: One-Step Synthesis and Their Stereochemical/Chiroptical Properties. Org. Biomol. Chem. 2024, 23, 145. [Google Scholar] [CrossRef]
- Sato, S.; Sakamoto, T.; Miyazawa, E.; Kikugawa, Y. One-Pot Reductive Amination of Aldehydes and Ketones with α-Picoline-Borane in Methanol, in Water, and in Neat Conditions. Tetrahedron 2004, 60, 7899–7906. [Google Scholar] [CrossRef]
- Wu, G.-C.; Tanaka, H.; Sanui, K.; Ogata, N. Reaction Mechanism of Direct Polycondensation with Triphenylphosphine and Hexachloroethane. Polym. J. 1982, 14, 7899–7906. [Google Scholar] [CrossRef]
- Takagi, K.; Miyamoto, D.; Yamaguchi, H.; Azumaya, I. Toward the Synthesis of a Belt-Shaped Cyclic π-Conjugated System Comprising Para-Phenylene Framework and Amide Bridging Unit. Bull. Chem. Soc. Jpn. 2022, 95, 47–51. [Google Scholar] [CrossRef]
- Flack, H.D.; Bernardinelli, G. Absolute structure and absolute configuration. Acta Crystallogr. 1999, A55, 908–915. [Google Scholar] [CrossRef]
- Flack, H.D.; Bernardinelli, G. Reporting and Evaluating Absolute-Structure and Absolute-Configuration Determinations. J. Appl. Crystallogr. 2000, 33, 1143–1148. [Google Scholar] [CrossRef]
- Perrotta, R.R.; Winter, A.H.; Falvey, D.E. Photochemical Heterolysis of 3, 5-Bis(Dimethylamino)Benzyl Alcohols and Esters: Generation of a Benzyl Cation with a Low-Energy Triplet State. Org. Lett. 2011, 13, 212–215. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. 2014, A70, C1437. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. 2015, C71, 3–8. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ∠C(=O)-N-C(H2)-C(sp2) [°] | ||
|---|---|---|
| Molecule A | Molecule B | |
| θ1 | 94.4(5) | 89.8(5) |
| θ2 | −100.4(5) | −128.7(5) |
| θ3 | 104.1(5) | 103.7(5) |
| θ4 | −96.1(5) | −112.6(6) |
| θ5 | −102.2(4) | −73.7(6) |
| θ6 | 124.3(4) | 97.7(5) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koike, D.; Masu, H.; Uno, H.; Kikkawa, S.; Hikawa, H.; Azumaya, I. Spherical Amides with C3 Symmetry: Improved Synthetic Approach and Structural/Optical Analysis. Molecules 2025, 30, 1074. https://doi.org/10.3390/molecules30051074
Koike D, Masu H, Uno H, Kikkawa S, Hikawa H, Azumaya I. Spherical Amides with C3 Symmetry: Improved Synthetic Approach and Structural/Optical Analysis. Molecules. 2025; 30(5):1074. https://doi.org/10.3390/molecules30051074
Chicago/Turabian StyleKoike, Daiki, Hyuma Masu, Haruka Uno, Shoko Kikkawa, Hidemasa Hikawa, and Isao Azumaya. 2025. "Spherical Amides with C3 Symmetry: Improved Synthetic Approach and Structural/Optical Analysis" Molecules 30, no. 5: 1074. https://doi.org/10.3390/molecules30051074
APA StyleKoike, D., Masu, H., Uno, H., Kikkawa, S., Hikawa, H., & Azumaya, I. (2025). Spherical Amides with C3 Symmetry: Improved Synthetic Approach and Structural/Optical Analysis. Molecules, 30(5), 1074. https://doi.org/10.3390/molecules30051074