
Chemical Looping CH4 Reforming Through Isothermal Two-Step Redox Cycling of SrFeO3 Oxygen Carrier in a Tubular Solar Reactor


Abstract
1. Introduction
2. Results and Discussion
2.1. Influence of Temperature on Syngas Production
2.2. Influence of the Oxidizing Agent in Second Step on Syngas Production
2.3. Influence of CH4 Mole Fraction in First Step on Syngas Production
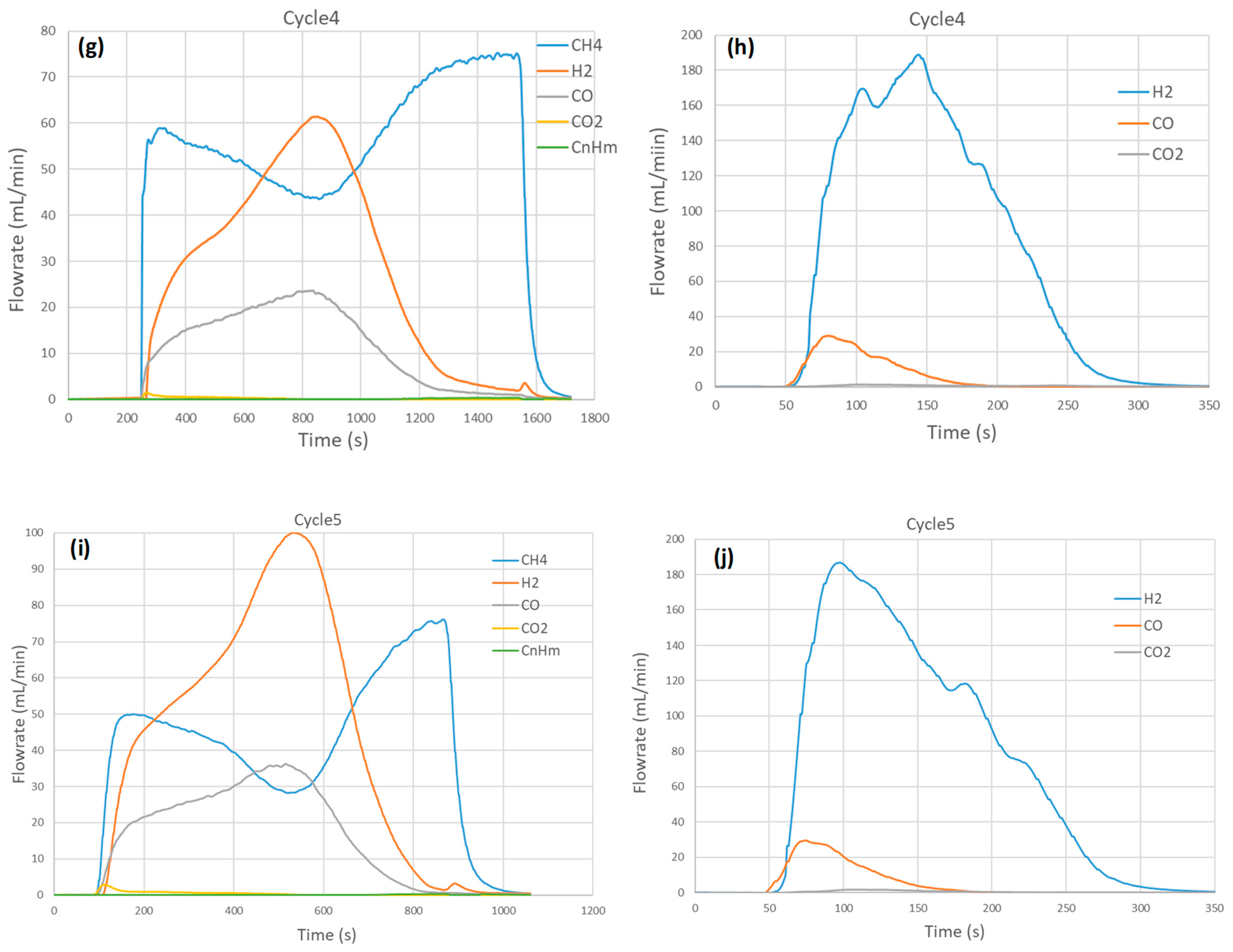
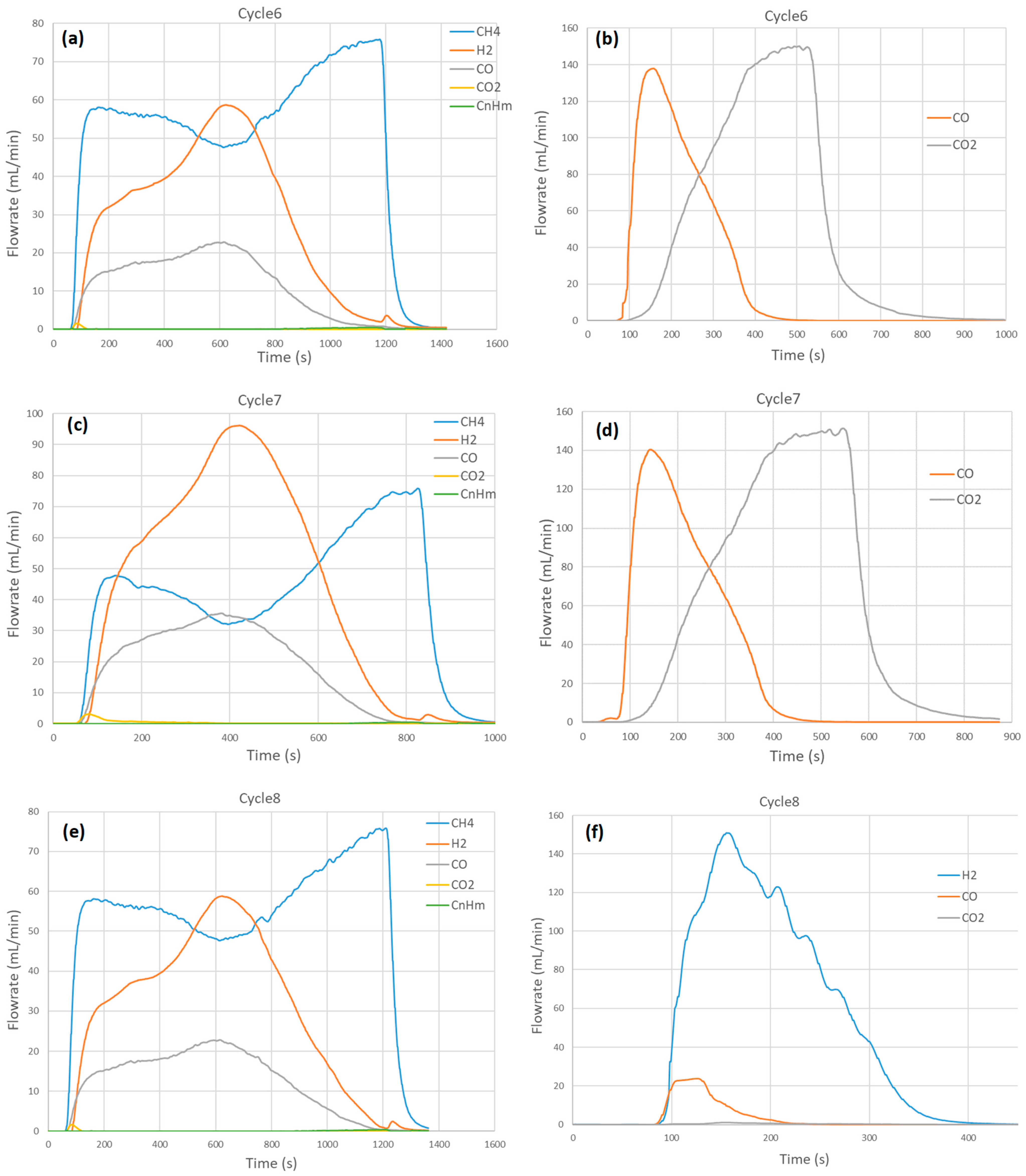
2.4. Thermochemical Cycling Stability and Materials Characterization
3. Experimental Set-Up, Materials, and Methods
3.1. Principle of the Chemical Looping Process, Involved Reactions, and Quantification of Product Yields
3.2. Materials Synthesis and Characterization
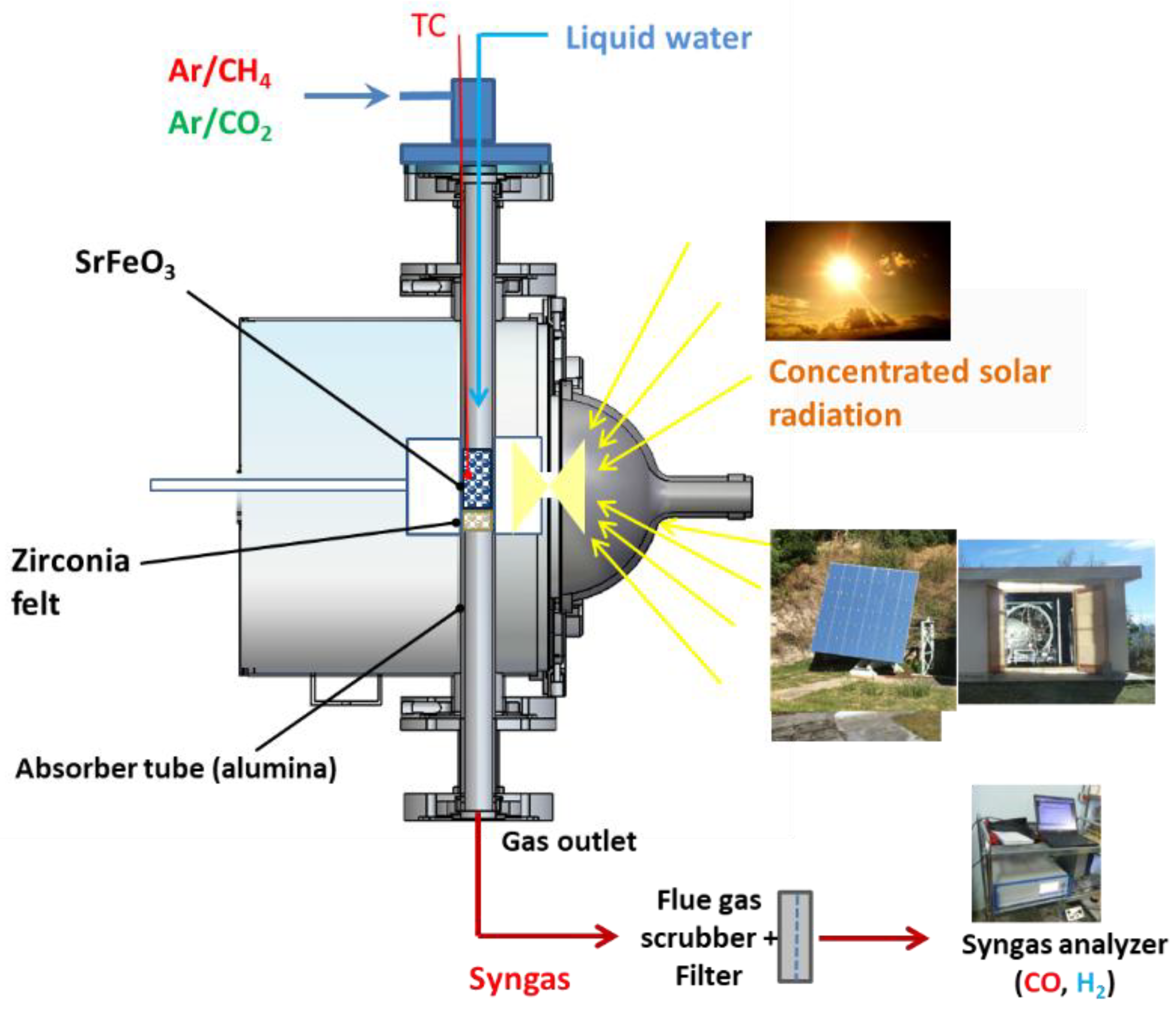
3.3. Solar Tubular Reactor
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kuravi, S.; Trahan, J.; Goswami, D.Y.; Rahman, M.M.; Stefanakos, E.K. Thermal Energy Storage Technologies and Systems for Concentrating Solar Power Plants. Prog. Energy Combust. Sci. 2013, 39, 285–319. [Google Scholar] [CrossRef]
- Tian, Y.; Zhao, C.Y. A Review of Solar Collectors and Thermal Energy Storage in Solar Thermal Applications. Appl. Energy 2013, 104, 538–553. [Google Scholar] [CrossRef]
- Klerk, A. Fischer–Tropsch Process. In Kirk-Othmer Encyclopedia of Chemical Technology; John Wiley & Sons: Hoboken, NJ, USA, 2013. [Google Scholar]
- Graves, C.; Ebbesen, S.D.; Mogensen, M.; Lackner, K.S. Sustainable Hydrocarbon Fuels by Recycling CO2 and H2O with Renewable or Nuclear Energy. Renew. Sustain. Energy Rev. 2011, 15, 1–23. [Google Scholar] [CrossRef]
- Zheng, Q.; Janke, C.; Farrauto, R. Steam Reforming of Sulfur-Containing Dodecane on a Rh–Pt Catalyst: Influence of Process Parameters on Catalyst Stability and Coke Structure. Appl. Catal. B Environ. 2014, 160–161, 525–533. [Google Scholar] [CrossRef]
- Song, Q.; Xiao, R.; Li, Y.; Shen, L. Catalytic Carbon Dioxide Reforming of Methane to Synthesis Gas over Activated Carbon Catalyst. Ind. Eng. Chem. Res. 2008, 47, 4349–4357. [Google Scholar] [CrossRef]
- Zhang, G.; Dong, Y.; Feng, M.; Zhang, Y.; Zhao, W.; Cao, H. CO2 Reforming of CH4 in Coke Oven Gas to Syngas over Coal Char Catalyst. Chem. Eng. J. 2010, 156, 519–523. [Google Scholar] [CrossRef]
- Zhang, G.; Su, A.; Du, Y.; Qu, J.; Xu, Y. Catalytic Performance of Activated Carbon Supported Cobalt Catalyst for CO2 Reforming of CH4. J. Colloid Interface Sci. 2014, 433, 149–155. [Google Scholar] [CrossRef]
- Zhang, G.; Du, Y.; Xu, Y.; Zhang, Y. Effects of Preparation Methods on the Properties of Cobalt/Carbon Catalyst for Methane Reforming with Carbon Dioxide to Syngas. J. Ind. Eng. Chem. 2014, 20, 1677–1683. [Google Scholar] [CrossRef]
- Kathiraser, Y.; Oemar, U.; Saw, E.T.; Li, Z.; Kawi, S. Kinetic and Mechanistic Aspects for CO2 Reforming of Methane over Ni Based Catalysts. Chem. Eng. J. 2015, 278, 62–78. [Google Scholar] [CrossRef]
- Otsuka, K.; Wang, Y.; Nakamura, M. Direct Conversion of Methane to Synthesis Gas through Gas–Solid Reaction Using CeO2–ZrO2 Solid Solution at Moderate Temperature. Appl. Catal. A Gen. 1999, 183, 317–324. [Google Scholar] [CrossRef]
- Otsuka, K.; Ushiyama, T.; Yamanaka, I. Partial Oxidation of Methane Using the Redox of Cerium Oxide. Chem. Lett. 1993, 22, 1517–1520. [Google Scholar] [CrossRef]
- Otsuka, K.; Wang, Y.; Sunada, E.; Yamanaka, I. Direct Partial Oxidation of Methane to Synthesis Gas by Cerium Oxide. J. Catal. 1998, 175, 152–160. [Google Scholar] [CrossRef]
- Zeng, L.; Cheng, Z.; Fan, J.A.; Fan, L.-S.; Gong, J. Metal Oxide Redox Chemistry for Chemical Looping Processes. Nat. Rev. Chem. 2018, 2, 349–364. [Google Scholar] [CrossRef]
- Luo, M.; Yi, Y.; Wang, S.; Wang, Z.; Du, M.; Pan, J.; Wang, Q. Review of Hydrogen Production Using Chemical-Looping Technology. Renew. Sustain. Energy Rev. 2018, 81, 3186–3214. [Google Scholar] [CrossRef]
- Skocypec, R.D.; Hogan, R.E.; Muir, J.F. Solar Reforming of Methane in a Direct Absorption Catalytic Reactor on a Parabolic Dish: II—Modeling and Analysis. Sol. Energy 1994, 52, 479–490. [Google Scholar] [CrossRef]
- Gokon, N.; Yamawaki, Y.; Nakazawa, D.; Kodama, T. Kinetics of Methane Reforming over Ru/γ-Al2O3-Catalyzed Metallic Foam at 650–900 °C for Solar Receiver-Absorbers. Int. J. Hydrogen Energy 2011, 36, 203–215. [Google Scholar] [CrossRef]
- Lu, J.; Chen, Y.; Ding, J.; Wang, W. High Temperature Energy Storage Performances of Methane Reforming with Carbon Dioxide in a Tubular Packed Reactor. Appl. Energy 2016, 162, 1473–1482. [Google Scholar] [CrossRef]
- Yu, T.; Yuan, Q.; Lu, J.; Ding, J.; Lu, Y. Thermochemical Storage Performances of Methane Reforming with Carbon Dioxide in Tubular and Semi-Cavity Reactors Heated by a Solar Dish System. Appl. Energy 2017, 185, 1994–2004. [Google Scholar] [CrossRef]
- Welte, M.; Warren, K.; Scheffe, J.R.; Steinfeld, A. Combined Ceria Reduction and Methane Reforming in a Solar-Driven Particle-Transport Reactor. Ind. Eng. Chem. Res. 2017, 56, 10300–10308. [Google Scholar] [CrossRef]
- Furler, P.; Scheffe, J.; Marxer, D.; Gorbar, M.; Bonk, A.; Vogt, U.; Steinfeld, A. Thermochemical CO2 Splitting via Redox Cycling of Ceria Reticulated Foam Structures with Dual-Scale Porosities. Phys. Chem. Chem. Phys. 2014, 16, 10503–10511. [Google Scholar] [CrossRef]
- Haeussler, A.; Abanades, S.; Julbe, A.; Jouannaux, J.; Drobek, M.; Ayral, A.; Cartoixa, B. Remarkable Performance of Microstructured Ceria Foams for Thermochemical Splitting of H2O and CO2 in a Novel High–Temperature Solar Reactor. Chem. Eng. Res. Des. 2020, 156, 311–323. [Google Scholar] [CrossRef]
- Wieckert, C.; Steinfeld, A. Solar Thermal Reduction of ZnO Using CH4:ZnO and C:ZnO Molar Ratios Less Than 1. J. Sol. Energy Eng. 2001, 124, 55–62. [Google Scholar] [CrossRef]
- Steinfeld, A.; Frei, A.; Kuhn, P.; Wuillemin, D. Solar Thermal Production of Zinc and Syngas via Combined ZnO-Reduction and CH4-Reforming Processes. Int. J. Hydrogen Energy 1995, 20, 793–804. [Google Scholar] [CrossRef]
- Chuayboon, S.; Abanades, S. Combined ZnO Reduction and Methane Reforming for Co-Production of Pure Zn and Syngas in a Prototype Solar Thermochemical Reactor. Fuel Process. Technol. 2021, 211, 106572. [Google Scholar] [CrossRef]
- Chuayboon, S.; Abanades, S. Thermochemical Performance Assessment of Solar Continuous Methane-Driven ZnO Reduction for Co-Production of Pure Zinc and Hydrogen-Rich Syngas. Chem. Eng. J. 2022, 429, 132356. [Google Scholar] [CrossRef]
- Chuayboon, S.; Abanades, S. Solar Carbo-Thermal and Methano-Thermal Reduction of MgO and ZnO for Metallic Powder and Syngas Production by Green Extractive Metallurgy. Processes 2022, 10, 154. [Google Scholar] [CrossRef]
- Shimizu, T.; Kitayama, Y.; Kodama, T. Thermochemical Conversion of CH4 to C2-Hydrocarbons and H2 over SnO2/Fe3O4/SiO2 in Methane−Water Co-Feed System. Energy Fuels 2001, 15, 463–469. [Google Scholar] [CrossRef]
- Villafán-Vidales, H.I.; Abanades, S.; Montiel-González, M.; Romero-Paredes-Rubio, H. Carbo- and Methanothermal Reduction of Tungsten Trioxide into Metallic Tungsten for Thermochemical Production of Solar Fuels. Energy Technol. 2017, 5, 692–702. [Google Scholar] [CrossRef]
- Otsuka, K.; Hatano, M.; Morikawa, A. Hydrogen from Water by Reduced Cerium Oxide. J. Catal. 1983, 79, 493–496. [Google Scholar] [CrossRef]
- Li, K.; Wang, H.; Wei, Y.; Yan, D. Syngas Production from Methane and Air via a Redox Process Using Ce–Fe Mixed Oxides as Oxygen Carriers. Appl. Catal. B Environ. 2010, 97, 361–372. [Google Scholar] [CrossRef]
- Li, K.; Wang, H.; Wei, Y.; Yan, D. Direct Conversion of Methane to Synthesis Gas Using Lattice Oxygen of CeO2–Fe2O3 Complex Oxides. Chem. Eng. J. 2010, 156, 512–518. [Google Scholar] [CrossRef]
- Chuayboon, S.; Abanades, S.; Rodat, S. Stepwise Solar Methane Reforming and Water-Splitting via Lattice Oxygen Transfer in Iron and Cerium Oxides. Energy Technol. 2020, 8, 1900415. [Google Scholar] [CrossRef]
- Chuayboon, S.; Abanades, S.; Rodat, S. Solar Chemical Looping Reforming of Methane Combined with Isothermal H2O/CO2 Splitting Using Ceria Oxygen Carrier for Syngas Production. J. Energy Chem. 2020, 41, 60–72. [Google Scholar] [CrossRef]
- Haeussler, A.; Abanades, S.; Jouannaux, J.; Drobek, M.; Ayral, A.; Julbe, A. Recent Progress on Ceria Doping and Shaping Strategies for Solar Thermochemical Water and CO2 Splitting Cycles. AIMS Mater. Sci. 2019, 6, 657–684. [Google Scholar] [CrossRef]
- Furler, P.; Scheffe, J.; Gorbar, M.; Moes, L.; Vogt, U.; Steinfeld, A. Solar Thermochemical CO2 Splitting Utilizing a Reticulated Porous Ceria Redox System. Energy Fuels 2012, 26, 7051–7059. [Google Scholar] [CrossRef]
- Nair, M.M.; Abanades, S. Tailoring Hybrid Nonstoichiometric Ceria Redox Cycle for Combined Solar Methane Reforming and Thermochemical Conversion of H2O/CO2. Energy Fuels 2016, 30, 6050–6058. [Google Scholar] [CrossRef]
- Sørensen, O.T. Thermodynamic Studies of the Phase Relationships of Nonstoichiometric Cerium Oxides at Higher Temperatures. J. Solid State Chem. 1976, 18, 217–233. [Google Scholar] [CrossRef]
- Pantu, P.; Kim, K.; Gavalas, G.R. Methane Partial Oxidation on Pt/CeO2–ZrO2 in the Absence of Gaseous Oxygen. Appl. Catal. A Gen. 2000, 193, 203–214. [Google Scholar] [CrossRef]
- Li, K.; Wang, H.; Wei, Y.; Yan, D. Transformation of Methane into Synthesis Gas Using the Redox Property of Ce–Fe Mixed Oxides: Effect of Calcination Temperature. Int. J. Hydrogen Energy 2011, 36, 3471–3482. [Google Scholar] [CrossRef]
- Li, K.; Wang, H.; Wei, Y.; Yan, D. Partial Oxidation of Methane to Syngas with Air by Lattice Oxygen Transfer over ZrO2-Modified Ce–Fe Mixed Oxides. Chem. Eng. J. 2011, 173, 574–582. [Google Scholar] [CrossRef]
- Warren, K.J.; Reim, J.; Randhir, K.; Greek, B.; Carrillo, R.; Hahn, D.W.; Scheffe, J.R. Theoretical and Experimental Investigation of Solar Methane Reforming through the Nonstoichiometric Ceria Redox Cycle. Energy Technol. 2017, 5, 2138–2149. [Google Scholar] [CrossRef]
- Krenzke, P.T.; Fosheim, J.R.; Zheng, J.; Davidson, J.H. Synthesis Gas Production via the Solar Partial Oxidation of Methane-Ceria Redox Cycle: Conversion, Selectivity, and Efficiency. Int. J. Hydrogen Energy 2016, 41, 12799–12811. [Google Scholar] [CrossRef]
- Cho, P.; Mattisson, T.; Lyngfelt, A. Comparison of Iron-, Nickel-, Copper- and Manganese-Based Oxygen Carriers for Chemical-Looping Combustion. Fuel 2004, 83, 1215–1225. [Google Scholar] [CrossRef]
- Zhu, X.; Wang, H.; Wei, Y.; Li, K.; Cheng, X. Hydrogen and Syngas Production from Two-Step Steam Reforming of Methane Using CeO2 as Oxygen Carrier. J. Nat. Gas Chem. 2011, 20, 281–286. [Google Scholar] [CrossRef]
- Johansson, M.; Mattisson, T.; Lyngfelt, A. Investigation of Mn3O4 With Stabilized ZrO2 for Chemical-Looping Combustion. Chem. Eng. Res. Des. 2006, 84, 807–818. [Google Scholar] [CrossRef]
- Neal, L.M.; Shafiefarhood, A.; Li, F. Dynamic Methane Partial Oxidation Using a Fe2O3 @La0.8Sr0.2FeO3-δ Core–Shell Redox Catalyst in the Absence of Gaseous Oxygen. ACS Catal. 2014, 4, 3560–3569. [Google Scholar] [CrossRef]
- Zhu, X.; Wei, Y.; Wang, H.; Li, K. Ce–Fe Oxygen Carriers for Chemical-Looping Steam Methane Reforming. Int. J. Hydrogen Energy 2013, 38, 4492–4501. [Google Scholar] [CrossRef]
- Qin, L.; Guo, M.; Liu, Y.; Cheng, Z.; Fan, J.A.; Fan, L.-S. Enhanced Methane Conversion in Chemical Looping Partial Oxidation Systems Using a Copper Doping Modification. Appl. Catal. B Environ. 2018, 235, 143–149. [Google Scholar] [CrossRef]
- Zheng, Y.; Li, K.; Wang, H.; Tian, D.; Wang, Y.; Zhu, X.; Wei, Y.; Zheng, M.; Luo, Y. Designed Oxygen Carriers from Macroporous LaFeO3 Supported CeO2 for Chemical-Looping Reforming of Methane. Appl. Catal. B Environ. 2017, 202, 51–63. [Google Scholar] [CrossRef]
- Haeussler, A.; Julbe, A.; Abanades, S. Investigation of Reactive Perovskite Materials for Solar Fuel Production via Two-Step Redox Cycles: Thermochemical Activity, Thermodynamic Properties and Reduction Kinetics. Mater. Chem. Phys. 2022, 276, 125358. [Google Scholar] [CrossRef]
- Chuayboon, S.; Abanades, S.; Rodat, S. Syngas Production via Solar-Driven Chemical Looping Methane Reforming from Redox Cycling of Ceria Porous Foam in a Volumetric Solar Reactor. Chem. Eng. J. 2019, 356, 756–770. [Google Scholar] [CrossRef]
- Haeussler, A.; Abanades, S.; Costa Oliveira, F.A.; Barreiros, M.A.; Caetano, A.P.F.; Novais, R.M.; Pullar, R.C. Solar Redox Cycling of Ceria Structures Based on Fiber Boards, Foams, and Biomimetic Cork-Derived Ecoceramics for Two-Step Thermochemical H2O and CO2 Splitting. Energy Fuels 2020, 34, 9037–9049. [Google Scholar] [CrossRef]
- Abanades, S.; Haeussler, A. Two-Step Thermochemical Cycles Using Fibrous Ceria Pellets for H2 Production and CO2 Reduction in Packed-Bed Solar Reactors. Sustain. Mater. Technol. 2021, 29, e00328. [Google Scholar] [CrossRef]
- Haeussler, A.; Abanades, S.; Julbe, A.; Jouannaux, J.; Cartoixa, B. Two-Step CO2 and H2O Splitting Using Perovskite-Coated Ceria Foam for Enhanced Green Fuel Production in a Porous Volumetric Solar Reactor. J. CO2 Util. 2020, 41, 101257. [Google Scholar] [CrossRef]
- Zhang, X.; Pei, C.; Chang, X.; Chen, S.; Liu, R.; Zhao, Z.-J.; Mu, R.; Gong, J. FeO6 Octahedral Distortion Activates Lattice Oxygen in Perovskite Ferrite for Methane Partial Oxidation Coupled with CO2 Splitting. J. Am. Chem. Soc. 2020, 142, 11540–11549. [Google Scholar] [CrossRef]
- Royer, S.; Duprez, D.; Can, F.; Courtois, X.; Batiot-Dupeyrat, C.; Laassiri, S.; Alamdari, H. Perovskites as Substitutes of Noble Metals for Heterogeneous Catalysis: Dream or Reality. Chem. Rev. 2014, 114, 10292–10368. [Google Scholar] [CrossRef]
- Hwang, J.; Rao, R.R.; Giordano, L.; Katayama, Y.; Yu, Y.; Shao-Horn, Y. Perovskites in Catalysis and Electrocatalysis. Science 2017, 358, 751–756. [Google Scholar] [CrossRef]
- Zhao, K.; He, F.; Huang, Z.; Wei, G.; Zheng, A.; Li, H.; Zhao, Z. Perovskite-Type Oxides LaFe1−xCoxO3 for Chemical Looping Steam Methane Reforming to Syngas and Hydrogen Co-Production. Appl. Energy 2016, 168, 193–203. [Google Scholar] [CrossRef]
- Haribal, V.P.; He, F.; Mishra, A.; Li, F. Iron-Doped BaMnO3 for Hybrid Water Splitting and Syngas Generation. ChemSusChem 2017, 10, 3402–3408. [Google Scholar] [CrossRef]
- Wang, X.; Du, X.; Yu, W.; Zhang, J.; Wei, J. Coproduction of Hydrogen and Methanol from Methane by Chemical Looping Reforming. Ind. Eng. Chem. Res. 2019, 58, 10296–10306. [Google Scholar] [CrossRef]
- Heifets, E.; Kotomin, E.A.; Bagaturyants, A.A.; Maier, J. Thermodynamic Stability of Non-Stoichiometric SrFeO3−δ: A Hybrid DFT Study. Phys. Chem. Chem. Phys. 2019, 21, 3918–3931. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Haribal, V.; Li, F. Perovskite Nanocomposites as Effective CO2-Splitting Agents in a Cyclic Redox Scheme. Sci. Adv. 2017, 3, e1701184. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Wang, X.; Liu, Y.; Wei, J.; Zhang, J. Effect of Composition on the Redox Performance of Strontium Ferrite Nanocomposite. Energy Fuels 2020, 34, 8644–8652. [Google Scholar] [CrossRef]
- Wang, X.; Yang, L.; Ji, X.; Gao, Y.; Li, F.; Zhang, J.; Wei, J. Reduction Kinetics of SrFeO3−δ/CaO·MnO Nanocomposite as Effective Oxygen Carrier for Chemical Looping Partial Oxidation of Methane. Front. Chem. Sci. Eng. 2022, 16, 1726–1734. [Google Scholar] [CrossRef]
- Wang, X.; Abanades, S.; Chuayboon, S.; Zhang, J.; Wei, J. Solar-Driven Chemical Looping Reforming of Methane over SrFeO3-δ-Ca0.5Mn0.5O Nanocomposite Foam. Int. J. Hydrogen Energy 2022, 47, 33664–33676. [Google Scholar] [CrossRef]
- Jia, T.; Popczun, E.J.; Lekse, J.W.; Duan, Y. The Optimal Co-Doping of SrFe1−xCoxO3−δ Oxygen Carriers in Redox Applications. Phys. Chem. Chem. Phys. 2020, 22, 16721–16726. [Google Scholar] [CrossRef]
- Zhang, R.; Cao, Y.; Li, H.; Zhao, Z.; Zhao, K.; Jiang, L. The Role of CuO Modified La0.7Sr0.3FeO3 Perovskite on Intermediate-Temperature Partial Oxidation of Methane via Chemical Looping Scheme. Int. J. Hydrogen Energy 2020, 45, 4073–4083. [Google Scholar] [CrossRef]
- Zhu, A.; Li, D.; Zhu, T.; Zhu, X. Tailored SrFeO3-δ for Chemical Looping Dry Reforming of Methane. Green Chem. Eng. 2025, 6, 102–115. [Google Scholar] [CrossRef]
- Sastre, D.; Galván, C.Á.; Pizarro, P.; Coronado, J.M. Enhanced Performance of CH4 Dry Reforming over La0.9Sr0.1FeO3/YSZ under Chemical Looping Conditions. Fuel 2022, 309, 122122. [Google Scholar] [CrossRef]
- Yang, L.; Zhao, Z.; Cui, C.; Zhang, J.; Wei, J. Effect of Nickel and Cobalt Doping on the Redox Performance of SrFeO3−δ toward Chemical Looping Dry Reforming of Methane. Energy Fuels 2023, 37, 12045–12057. [Google Scholar] [CrossRef]
- Oliveira, F.A.C.; Barreiros, M.A.; Haeussler, A.; Caetano, A.P.F.; Mouquinho, A.I.; Oliveira e Silva, P.M.; Novais, R.M.; Pullar, R.C.; Abanades, S. High Performance Cork-Templated Ceria for Solar Thermochemical Hydrogen Production via Two-Step Water-Splitting Cycles. Sustain. Energy Fuels 2020, 4, 3077–3089. [Google Scholar] [CrossRef]
- Levêque, G.; Abanades, S. Design and Operation of a Solar-Driven Thermogravimeter for High Temperature Kinetic Analysis of Solid–Gas Thermochemical Reactions in Controlled Atmosphere. Sol. Energy 2014, 105, 225–235. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cycle No. | Ar in 1st Step (mL/min) | CH4 in 1st Step (mL/min) | Ar in 2nd Step (mL/min) | H2O (g/min)/CO2 (mL/min) in 2nd Step | T (°C) |
|---|---|---|---|---|---|
| 1 | 425 | 75 | 500 | 0.19 g/min | 1000 |
| 2 | 425 | 75 | 500 | 0.19 g/min | 1000 |
| 3 | 425 | 75 | 500 | 0.19 g/min | 950 |
| 4 | 425 | 75 | 500 | 0.19 g/min | 1000 |
| 5 | 425 | 75 | 500 | 0.19 g/min | 1050 |
| 6 | 425 | 75 | 350 | 150 mL/min | 1000 |
| 7 | 425 | 75 | 350 | 150 mL/min | 1050 |
| 8 | 425 | 75 | 500 | 0.19 g/min | 1000 |
| 9 | 475 | 25 | 500 | 0.19 g/min | 1000 |
| 10 | 350 | 150 | 500 | 0.19 g/min | 1000 |
| 11 | 350 | 150 | 500 | 0.19 g/min | 1000 |
| 12 | 425 | 75 | 500 | 0.19 g/min | 1000 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abanades, S.; Wang, X.; Chuayboon, S. Chemical Looping CH4 Reforming Through Isothermal Two-Step Redox Cycling of SrFeO3 Oxygen Carrier in a Tubular Solar Reactor. Molecules 2025, 30, 1076. https://doi.org/10.3390/molecules30051076
Abanades S, Wang X, Chuayboon S. Chemical Looping CH4 Reforming Through Isothermal Two-Step Redox Cycling of SrFeO3 Oxygen Carrier in a Tubular Solar Reactor. Molecules. 2025; 30(5):1076. https://doi.org/10.3390/molecules30051076
Chicago/Turabian StyleAbanades, Stéphane, Xinhe Wang, and Srirat Chuayboon. 2025. "Chemical Looping CH4 Reforming Through Isothermal Two-Step Redox Cycling of SrFeO3 Oxygen Carrier in a Tubular Solar Reactor" Molecules 30, no. 5: 1076. https://doi.org/10.3390/molecules30051076
APA StyleAbanades, S., Wang, X., & Chuayboon, S. (2025). Chemical Looping CH4 Reforming Through Isothermal Two-Step Redox Cycling of SrFeO3 Oxygen Carrier in a Tubular Solar Reactor. Molecules, 30(5), 1076. https://doi.org/10.3390/molecules30051076