

Ultrasound-Assisted Extraction of Polysaccharides from Lyophyllum decastes: Structural Analysis and Bioactivity Assessment

Abstract
1. Introduction
2. Results and Discussion
2.1. Isolation and Purification of Polysaccharides
2.2. Chemical Composition of Different LDP Components
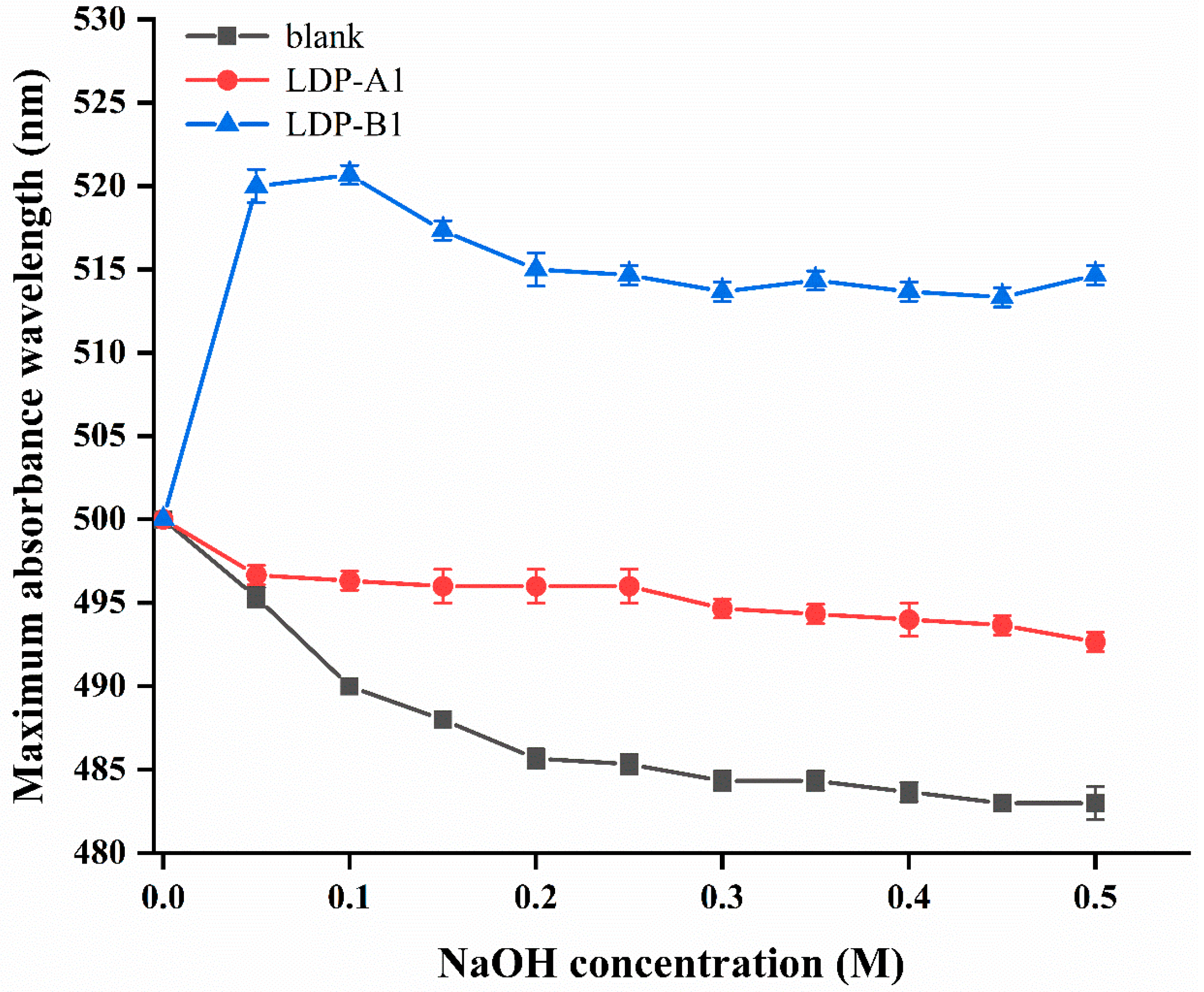
2.3. UV Spectrum Analysis of Different LDP Components
2.4. Monosaccharide Composition and Molecular Weight Analysis
2.5. Fourier-Transform Infrared (FTIR) Spectroscopy Analysis
2.6. Triple-Helix Determination
2.7. Scanning Electron Microscopy (SEM)
2.8. X-Ray Diffraction (XRD) Analysis
2.9. Antioxidant Activity Analysis
2.9.1. ABTS Radical Scavenging Activity
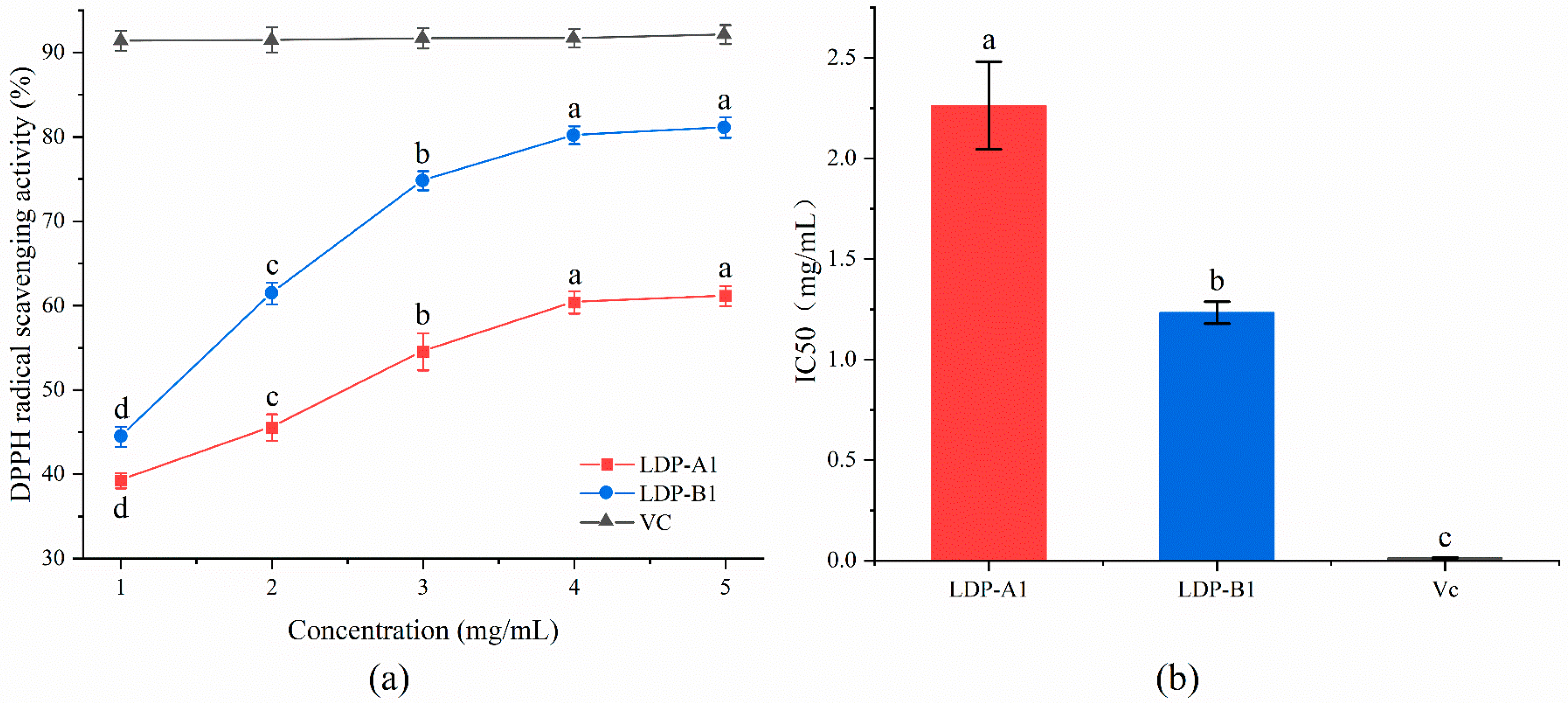
2.9.2. DPPH Radical Scavenging Activity
2.9.3. Hydroxyl Radical Scavenging Activity
2.9.4. Reducing Power
2.10. Hypoglycemic Activity Analysis
2.10.1. α-Amylase Inhibitory Activity
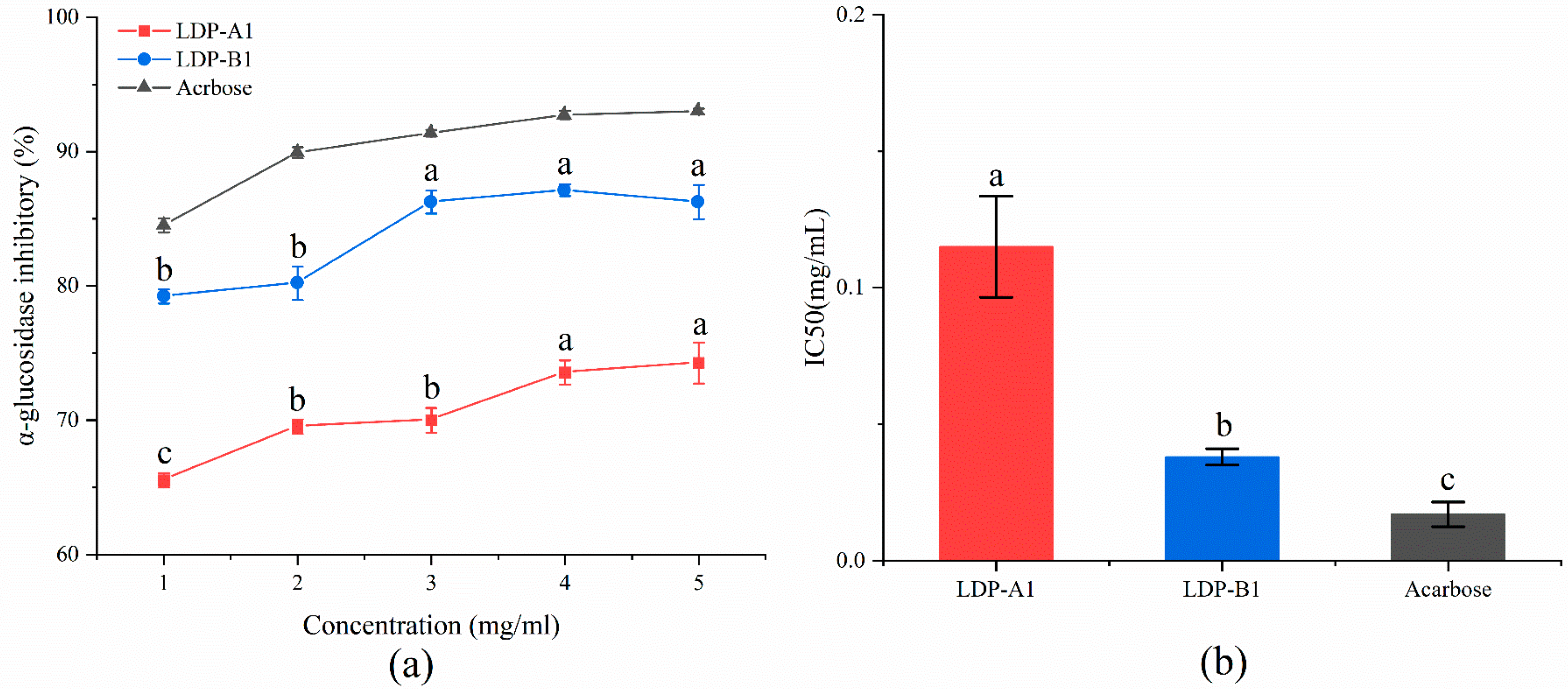
2.10.2. α-Glucosidase Inhibitory Activity
2.11. Anti-Inflammatory Activities
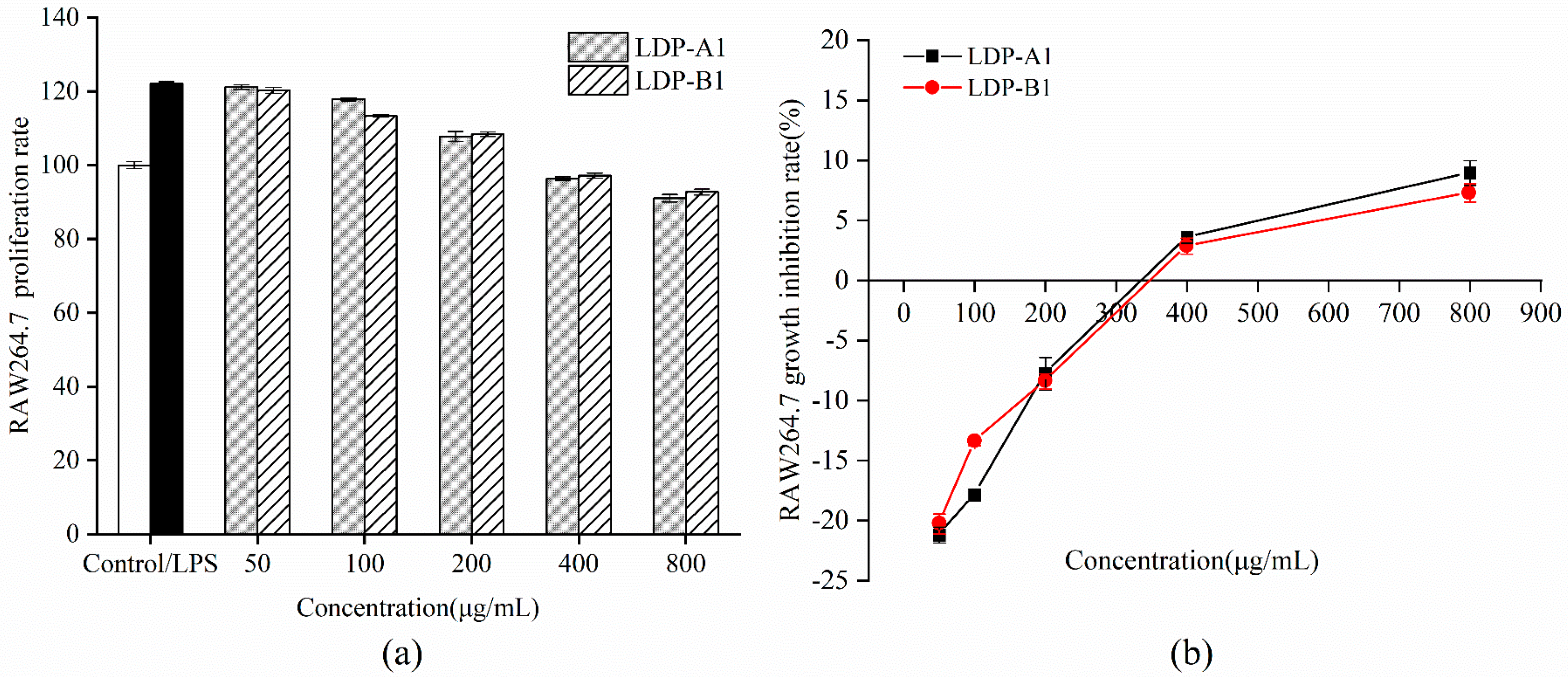
2.11.1. Cell Proliferation Rate
2.11.2. Effects of Polysaccharide Samples on LPS-Induced NO and Cytokine Secretion in RAW264.7 Cells
2.12. Anti-Tumor Activity
3. Materials and Methods
3.1. Materials and Reagents
3.2. Preparation of Polysaccharides from L. decastes
3.3. Isolation and Purification of Polysaccharides from LDPs
3.4. Determination of Chemical Composition
3.5. UV Full-Wavelength Scan Analysis
3.6. Monosaccharide Composition and Molecular Weight
3.7. Fourier-Transform Infrared (FTIR) Spectroscopy Analysis
3.8. Triple-Helix Determination
3.9. Scanning Electron Microscopy (SEM)
3.10. X-Ray Diffraction (XRD) Analysis
3.11. Antioxidant Activity Analysis
3.11.1. ABTS Radical Scavenging Activity
3.11.2. DPPH Radical Scavenging Activity
3.11.3. Hydroxyl Radical Scavenging Activity
3.11.4. Reducing Power
3.12. Hypoglycemic Activity Analysis
3.12.1. Inhibition Rate of α-Amylase
3.12.2. Inhibition Rate of α-Glucosidase
3.13. Anti-Inflammatory Activities
3.13.1. Determination of Cell Proliferation Rate
3.13.2. Assay of NO Release
3.13.3. Effect of Polysaccharide Sample on the Secretion of Cytokines in RAW264.7 Cells
3.14. Anti-Tumor Activity
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhang, F.F.; Xiao, Y.; Pan, L.; Yu, L.; Liu, Y.F.; Li, D.S.; Liu, X.J. Effects of polysaccharides from Lyophyllum decastes (Fr.) Singer on gut microbiota via in vitro-simulated digestion and fermentation. Front. Microbiol. 2023, 14, 1083917. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.P.; Wang, Y.N.; Qin, C.Q.; Ye, S.M.; Zhang, F.M.; Linhardt, R.J.; Zhang, A.Q. Structural characterization of an antioxidant polysaccharide isolated from the fruiting bodies of Lyophyllum decastes. J. Mol. Struct. 2023, 1285, 135507. [Google Scholar] [CrossRef]
- Li, X.J.; Xiao, S.J.; Xie, Y.H.; Chen, J.; Xu, H.R.; Yin, Y.; Zhang, R.; Yang, T.; Zhou, T.Y.; Zhang, S.Y.; et al. Structural characterization and immune activity evaluation of a polysaccharide from Lyophyllum Decastes. Int. J. Biol. Macromol. 2024, 278, 134628. [Google Scholar] [CrossRef] [PubMed]
- Arana-Gabriel, Y.; Burrola-Aguilar, C.; Garibay-Orijel, R.; Matías-Ferrer, N.; Franco-Maass, S.; Mata, G. Genetic characterization, evaluation of growth and production of biomass of strains from wild edible mushrooms of Lyophyllum of Central Mexico. Braz. J. Microbiol. 2018, 49, 632–640. [Google Scholar] [CrossRef]
- Tel, G.; Ozturk, M.; Duru, M.E.; Turkoglu, A. Antioxidant and anticholinesterase activities of five wild mushroom species with total bioactive contents. Pharm. Biol. 2015, 53, 824–830. [Google Scholar] [CrossRef] [PubMed]
- Ukawa, Y.; Furuichi, Y.; Kokean, Y.; Nishii, T.; Hisamatsu, M. EfEffect of Hatakeshimeji (Lyophyllum decastes Sing.) mushroom on serum lipid levels in rats. J. Nutr. Sci. Vitaminol. 2002, 48, 73–76. [Google Scholar] [CrossRef]
- Fu, Y.; Li, F.; Ding, Y.; Li, H.Y.; Xiang, X.R.; Ye, Q.; Zhang, J.; Zhao, L.; Qin, W.; Gan, R.Y.; et al. Polysaccharides from loquat (Eriobotrya japonica) leaves: Impacts of extraction methods on their physicochemical characteristics and biological activities. Int. J. Biol. Macromol. 2020, 146, 508–517. [Google Scholar] [CrossRef]
- Huang, F.; Liu, H.J.; Zhang, R.F.; Dong, L.H.; Liu, L.; Ma, Y.X.; Jia, X.C.; Wang, G.J.; Zhang, M.W. Physicochemical properties and prebiotic activities of polysaccharides from longan pulp based on different extraction techniques. Carbohydr. Polym. 2019, 206, 344–351. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Ji, H.Y.; Yang, Z.Z.; Liu, A.J. Relationship between structural properties and antitumor activity of Astragalus polysaccharides extracted with different temperatures. Int. J. Biol. Macromol. 2019, 124, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.Z.; Li, S.Z.; Liu, C.M.; Yang, S.M.; Li, X.L. Ultrasound complex enzymes assisted extraction and biochemical activities of polysaccharides from Epimedium leaves. Process Biochem. 2012, 47, 2040–2050. [Google Scholar] [CrossRef]
- Leong, Y.K.; Yang, F.C.; Chang, J.S. Extraction of polysaccharides from edible mushrooms: Emerging technologies and recent advances. Carbohydr. Polym. 2021, 251, 117006. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.J.; He, H.Q.; Wang, Q.; Yang, X.Y.; Jiang, S.J.; Wang, D.B. A Review of Development and Utilization for Edible Fungal Polysaccharides: Extraction, Chemical Characteristics, and Bioactivities. Polymers 2022, 14, 4454. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.L.; Guo, J.H.; Tian, J.Y.; Ma, K.; Liu, Y.Q. Research progress on degradation methods and product properties of plant polysaccharides. J. Light Ind. 2023, 38, 55–62. [Google Scholar]
- Tang, Z.Z.; Zhou, C.X.; Cai, Y.; Tang, Y.J.; Sun, W.J.; Yao, H.P.; Zheng, T.R.; Chen, H.; Xiao, Y.R.; Shan, Z.; et al. Purification, characterization and antioxidant activities in vitro of polysaccharides from Amaranthus hybridus L. PeerJ 2020, 8, e9077. [Google Scholar] [CrossRef]
- Ji, X.L.; Cheng, Y.Q.; Tian, J.Y.; Zhang, S.Q.; Jing, Y.S.; Shi, M.M. Structural characterization of polysaccharide from jujube (Ziziphus jujuba Mill.) fruit. Chem. Biol. Technol. Agric. 2021, 8, 54. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, J.; Meng, Z. Purification, characterization and anti-tumor activities of polysaccharides extracted from wild Russula griseocarnosa. Int. J. Biol. Macromol. 2018, 109, 1054–1060. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.H.; Chen, G.J.; Wang, Z.R.; Kan, J.Q. A comparison of a polysaccharide extracted from ginger (Zingiber officinale) stems and leaves using different methods: Preparation, structure characteristics, and biological activities. Int. J. Biol. Macromol. 2020, 151, 635–649. [Google Scholar] [CrossRef]
- Zhang, A.L.; Shen, Y.W.; Cen, M.P.; Hong, X.Z.; Shao, Q.S.; Chen, Y.B.; Zheng, B.S. Polysaccharide and crocin contents, and antioxidant activity of saffron from different origins. Ind. Crops Prod. 2019, 133, 111–117. [Google Scholar] [CrossRef]
- Wang, Y.P.; Wang, C.N.; Guo, M.R. Effects of Ultrasound Treatment on Extraction and Rheological Properties of Polysaccharides from Auricularia Cornea var. Li. Molecules 2019, 24, 939. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Li, S.; Xiong, Q.; Jiang, C.; Lai, X. Extraction, characterization and biological activities of polysaccharides from Amomum villosum. Carbohydr. Polym. 2013, 95, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zeng, Y.; Men, Y.; Zhang, J.; Liu, H.; Sun, Y. Structural characterization and immunomodulatory activity of exopolysaccharides from submerged culture of Auricularia auricula-judae. Int. J. Biol. Macromol. 2018, 115, 978–984. [Google Scholar] [CrossRef] [PubMed]
- Luo, Q.-L.; Tang, Z.-H.; Zhang, X.-F.; Zhong, Y.-H.; Yao, S.-Z.; Wang, L.-S.; Lin, C.-W.; Luo, X. Chemical properties and antioxidant activity of a water-soluble polysaccharide from Dendrobium officinale. Int. J. Biol. Macromol. 2016, 89, 219–227. [Google Scholar] [CrossRef]
- Pu, X.; Ma, X.; Liu, L.; Ren, J.; Li, H.; Li, X.; Yu, S.; Zhang, W.; Fan, W. Structural characterization and antioxidant activity in vitro of polysaccharides from angelica and astragalus. Carbohydr. Polym. 2016, 137, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Dammak, M.I.; Ben Salem, Y.; Belaid, A.; Ben Mansour, H.; Hammami, S.; Le Cerf, D.; Majdoub, H. Partial characterization and antitumor activity of a polysaccharide isolated from watermelon rinds. Int. J. Biol. Macromol. 2019, 136, 632–641. [Google Scholar] [CrossRef] [PubMed]
- Tu, W.; Zhu, J.; Bi, S.; Chen, D.; Song, L.; Wang, L.; Zi, J.; Yu, R. Isolation, characterization and bioactivities of a new polysaccharide from Annona squamosa and its sulfated derivative. Carbohydr. Polym. 2016, 152, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Vasilieva, T.; Sigarev, A.; Kosyakov, D.; Ul’yanovskii, N.; Anikeenko, E.; Chuhchin, D.; Ladesov, A.; Hein, A.M.; Miasnikov, V. Formation of low molecular weight oligomers from chitin and chitosan stimulated by plasma-assisted processes. Carbohydr. Polym. 2017, 163, 54–61. [Google Scholar] [CrossRef]
- Zhao, P.; Zhou, H.; Zhao, C.; Li, X.; Wang, Y.; Wang, Y.; Huan, L.; Gao, W. Purification, characterization and immunomodulatory activity of fructans from Polygonatum odoratum and P. cyrtonema. Carbohydr. Polym. 2019, 214, 44–52. [Google Scholar] [CrossRef]
- Jing, Y.S.; Huang, L.J.; Lv, W.J.; Tong, H.; Song, L.Y.; Hu, X.Q.; Yu, R.M. Structural Characterization of a Novel Polysaccharide from Pulp Tissues of Litchi chinensis and Its Immunomodulatory Activity. J. Agric. Food Chem. 2014, 62, 902–911. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Zhu, C.P.; Huang, G.Q.; Xiao, J.X. Fractionation and structural characterization of polysaccharides derived from red grape pomace. Process Biochem. 2021, 109, 37–45. [Google Scholar] [CrossRef]
- Palacios, I.; García-Lafuente, A.; Guillamón, E.; Villares, A. Novel isolation of water-soluble polysaccharides from the fruiting bodies of Pleurotus ostreatus mushrooms. Carbohydr. Res. 2012, 358, 72–77. [Google Scholar] [CrossRef]
- Guo, X.Y.; Kang, J.; Xu, Z.Y.; Guo, Q.B.; Zhang, L.F.; Ning, H.F.; Cui, S.W. Triple-helix polysaccharides: Formation mechanisms and analytical methods. Carbohydr. Polym. 2021, 262, 117962. [Google Scholar] [CrossRef]
- Xu, S.Q.; Xu, X.J.; Zhang, L.N. Branching Structure and Chain Conformation of Water-Soluble Glucan Extracted from Auricularia auricula-judae. J. Agric. Food Chem. 2012, 60, 3498–3506. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Kang, Y.H. Synergistic interaction of Auricularia auricula-judae polysaccharide with yam starch: Effects on physicochemical properties and in vitro starch digestibility. Food Sci. Biotechnol. 2018, 27, 1579–1588. [Google Scholar] [CrossRef]
- Movahedian, M.; Thomas, J.; Rahmani, J.; Clark, C.C.T.; Rashidkhani, B.; Ghanavati, M. Association between dietary glycemic index and glycemic load, insulin index and load with incidence of age-related cataract: Results from a case-control study. Diabetes Metab. Syndr. Clin. Res. Rev. 2020, 14, 199–204. [Google Scholar] [CrossRef]
- Wang, S.Q.; Li, G.; Zhang, X.F.; Wang, Y.Q.; Qiang, Y.; Wang, B.L.; Zou, J.B.; Niu, J.F.; Wang, Z.Z. Structural characterization and antioxidant activity of Polygonatum sibiricum polysaccharides. Carbohydr. Polym. 2022, 291, 119524. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.P.; Guo, M.R. Purification and structural characterization of polysaccharides isolated from Auricularia cornea var. Li. Carbohydr. Polym. 2020, 230, 115680. [Google Scholar] [CrossRef] [PubMed]
- Xiong, F.; Li, X.; Zheng, L.; Hu, N.; Cui, M.; Li, H. Characterization and antioxidant activities of polysaccharides from Passiflora edulis Sims peel under different degradation methods. Carbohydr. Polym. 2019, 218, 46–52. [Google Scholar] [CrossRef]
- Wu, Y.; Zheng, Y.L.; Yang, W.L.; Wang, C.C.; Hu, J.H.; Fu, S.K. Synthesis and characterization of a novel amphiphilic chitosan-polylactide graft copolymer. Carbohydr. Polym. 2005, 59, 165–171. [Google Scholar] [CrossRef]
- Ben Jeddou, K.; Chaari, F.; Maktouf, S.; Nouri-Ellouz, O.; Helbert, C.B.; Ghorbel, R.E. Structural, functional, and antioxidant properties of water-soluble polysaccharides from potatoes peels. Food Chem. 2016, 205, 97–105. [Google Scholar] [CrossRef]
- Ktari, N.; Feki, A.; Trabelsi, I.; Triki, M.; Maalej, H.; Ben Slima, S.; Nasri, M.; Ben Amara, I.; Ben Salah, R. Structure, functional and antioxidant properties in Tunisian beef sausage of a novel polysaccharide from Trigonella foenum-graecum seeds. Int. J. Biol. Macromol. 2017, 98, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.J.; Li, H.Z.; Netala, V.R.; Hou, T.Y.; Zhang, Z.J. Optimization of complex enzyme-ultrasonic synergistic extraction of water-soluble polysaccharides from Perilla frutescens seed meal: Purification, characterization and in vitro antioxidant activity. J. Food Process. Preserv. 2022, 46, e16201. [Google Scholar] [CrossRef]
- Ren, Y.P.; Liu, S.X. Effects of separation and purification on structural characteristics of polysaccharide from quinoa (Chenopodium quinoa willd). Biochem. Biophys. Res. Commun. 2020, 522, 286–291. [Google Scholar] [CrossRef] [PubMed]
- Song, L.J.; Liu, S.Q.; Zhang, L.; Pan, L.Q.; Xu, L. Polysaccharides from Nitraria retusa Fruit: Extraction, Purification, Structural Characterization, and Antioxidant Activities. Molecules 2023, 28, 1266. [Google Scholar] [CrossRef]
- Mei, X.Y.; Yang, W.J.; Huang, G.; Huang, H.L. The antioxidant activities of balsam pear polysaccharide. Int. J. Biol. Macromol. 2020, 142, 232–236. [Google Scholar] [CrossRef]
- Liu, S.; Li, M.; Liu, W.W.; Zhang, Z.; Wang, X.; Dong, H.J. Structure and properties of acidic polysaccharides isolated from Massa Medicata Fermentata: Neuroprotective and antioxidant activity. Int. J. Biol. Macromol. 2024, 259, 129128. [Google Scholar] [CrossRef] [PubMed]
- Qi, H.M.; Zhao, T.T.; Zhang, Q.B.; Li, Z.; Zhao, Z.Q.; Xing, R. Antioxidant activity of different molecular weight sulfated polysaccharides from Ulva pertusa Kjellm (Chlorophyta). J. Appl. Phycol. 2005, 17, 527–534. [Google Scholar] [CrossRef]
- Mutailifu, P.; Nuerxiati, R.; Lu, C.F.; Huojiaaihemaiti, H.; Abuduwaili, A.; Yili, A. Extraction, purification, and characterization of polysaccharides from Alhagi pseudoalhagi with antioxidant and hypoglycemic activities. Process Biochem. 2022, 121, 339–348. [Google Scholar] [CrossRef]
- Mirzadeh, M.; Lelekami, A.K.; Khedmat, L. Plant/algal polysaccharides extracted by microwave: A review on hypoglycemic, hypolipidemic, prebiotic, and immune-stimulatory effect. Carbohydr. Polym. 2021, 266, 118134. [Google Scholar] [CrossRef] [PubMed]
- Hsu, W.-K.; Hsu, T.-H.; Lin, F.-Y.; Cheng, Y.-K.; Yang, J.P.-W. Separation, purification, and α-glucosidase inhibition of polysaccharides from Coriolus versicolor LH1 mycelia. Carbohydr. Polym. 2013, 92, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.L.; Guo, J.H.; Cao, T.Z.; Zhang, T.T.; Liu, Y.Q.; Yan, Y.Z. Review on mechanisms and structure-activity relationship of hypoglycemic effects of polysaccharides from natural resources. Food Sci. Hum. Wellness 2023, 12, 1969–1980. [Google Scholar] [CrossRef]
- Wan, P.; Yang, X.; Cai, B.; Chen, H.; Sun, H.; Chen, D.; Pan, J. Ultrasonic extraction of polysaccharides from Laminaria japonica and their antioxidative and glycosidase inhibitory activities. J. Ocean Univ. China 2015, 14, 651–662. [Google Scholar] [CrossRef]
- Wang, Y.F.; Yang, Z.W.; Wei, X.L. SSugar compositions, α-glucosidase inhibitory and amylase inhibitory activities of polysaccharides from leaves and flowers of Camellia sinensis obtained by different extraction methods. Int. J. Biol. Macromol. 2010, 47, 534–539. [Google Scholar] [CrossRef] [PubMed]
- Cho, M.; Han, J.H.; You, S. Inhibitory effects of fucan sulfates on enzymatic hydrolysis of starch. LWT Food Sci. Technol. 2011, 44, 1164–1171. [Google Scholar] [CrossRef]
- Xue, H.K.; Hao, Z.T.; Gao, Y.C.; Cai, X.; Tang, J.T.; Liao, X.J.; Tan, J.Q. Research progress on the hypoglycemic activity and mechanisms of natural polysaccharides. Int. J. Biol. Macromol. 2023, 252, 126199. [Google Scholar] [CrossRef]
- Zhong, Q.W.; Zhou, T.S.; Qiu, W.H.; Wang, Y.K.; Xu, Q.L.; Ke, S.Z.; Wang, S.J.; Jin, W.H.; Chen, J.W.; Zhang, H.W.; et al. Characterization and hypoglycemic effects of sulfated polysaccharides derived from brown seaweed Undaria pinnatifida. Food Chem. 2021, 341, 128148. [Google Scholar] [CrossRef]
- Zeng, A.Q.; Yang, R.J.; Yu, S.H.; Zhao, W. A novel hypoglycemic agent: Polysaccharides from laver (Porphyra spp.). Food Funct. 2020, 11, 9048–9056. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.F.; Fu, Y.P.; Chen, X.F.; Austarheim, I.; Inngjerdingen, K.T.; Huang, C.; Lei, F.Y.; Song, X.; Li, L.X.; Ye, G.; et al. Polysaccharides with immunomodulating activity from roots of Gentiana crassicaulis. Carbohydr. Polym. 2017, 172, 306–314. [Google Scholar] [CrossRef]
- Zhu, Y.T.; Wang, X.J.; Pan, W.S.; Shen, X.F.; He, Y.J.; Yin, H.L.; Zhou, K.; Zou, L.K.; Chen, S.J.; Liu, S.L. Exopolysaccharides produced by yogurt-texture improving Lactobacillus plantarum RS20D and the immunoregulatory activity. Int. J. Biol. Macromol. 2019, 121, 342–349. [Google Scholar] [CrossRef]
- Shen, Y.; Wu, S.H.; Song, M.M.; Zhang, H.M.; Zhao, H.; Wu, L.L.; Zhao, H.B.; Qiu, H.B.; Zhang, Y. The Isolation, Structural Characterization and Anti-Inflammatory Potentials of Neutral Polysaccharides from the Roots of Isatis indigotica Fort. Molecules 2024, 29, 4987. [Google Scholar] [CrossRef] [PubMed]
- He, L.A.; Yan, X.T.; Liang, J.; Li, S.J.; He, H.R.; Xiong, Q.P.; Lai, X.P.; Hou, S.Z.; Huang, S. Comparison of different extraction methods for polysaccharides from Dendrobium officinale stem. Carbohydr. Polym. 2018, 198, 101–108. [Google Scholar] [CrossRef]
- Shi, F.L.; Yan, X.L.; Cheong, K.L.; Liu, Y. Extraction, purification, and characterization of polysaccharides from marine algae Gracilaria lemaneiformis with anti-tumor activity. Process Biochem. 2018, 73, 197–203. [Google Scholar] [CrossRef]
- Zhang, L.; Li, X.L.; Xu, X.J.; Zeng, F.B. Correlation between antitumor activity, molecular weight, and conformation of lentinan. Carbohydr. Res. 2005, 340, 1515–1521. [Google Scholar] [CrossRef]
- Chen, L.; Huang, G.L.; Hu, J.C. Preparation, deproteinization, characterisation, and antioxidant activity of polysaccharide from cucumber (Cucumis saticus L.). Int. J. Biol. Macromol. 2018, 108, 408–411. [Google Scholar] [CrossRef]
- Chen, H.; Sun, J.; Liu, J.; Gou, Y.R.; Zhang, X.; Wu, X.N.; Sun, R.; Tang, S.X.; Kan, J.; Qian, C.L.; et al. Structural characterization and anti-inflammatory activity of alkali-soluble polysaccharides from purple sweet potato. Int. J. Biol. Macromol. 2019, 131, 484–494. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.T.; Xu, F.R.; Li, J.L.; Li, J.; Mo, C.; Zhao, M.; Wang, L.F. Ultrasonic-assisted extraction of polysaccharides from coix seeds: Optimization, purification, and in vitro digestibility. Food Chem. 2022, 374, 131636. [Google Scholar] [CrossRef]
- Bradford, M.M. Rapid and Sensitive Method for Quantitation of Microgram Quantities of Protein Utilizing Principle of Protein-Dye Binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yu, L.; Bi, H.T.; Li, X.H.; Ni, W.H.; Han, H.; Li, N.; Wang, B.Q.; Zhou, Y.F.; Tai, G.H. Total fractionation and characterization of the water-soluble polysaccharides isolated from Panax ginseng C. A. Meyer. Carbohydr. Polym. 2009, 77, 544–552. [Google Scholar] [CrossRef]
- Cui, L.N.; Wang, J.Y.; Huang, R.; Tan, Y.; Zhang, F.; Zhou, Y.F.; Sun, L. Analysis of pectin from Panax ginseng flower buds and their binding activities to galectin-3. Int. J. Biol. Macromol. 2019, 128, 459–467. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.Y.; Su, Y.Y.; Su, J.Q.; Xue, J.J.; Zhang, R.; Li, X.L.; Li, Y.; Ding, Y.; Chu, X.L. Optimization of Enzyme-Assisted Aqueous Extraction of Polysaccharide from Acanthopanax senticosus and Comparison of Physicochemical Properties and Bioactivities of Polysaccharides with Different Molecular Weights. Molecules 2023, 28, 6585. [Google Scholar] [CrossRef]
- Lavudi, H.N.; Kottapalli, S.; Goycoolea, F.M. Extraction and physicochemical characterization of galactomannans from Dichrostachys cinerea seeds. Food Hydrocoll. 2018, 82, 451–456. [Google Scholar] [CrossRef]
- Wang, L.; Luo, Y.; Wu, Y.N.; Liu, Y.; Wu, Z.Q. Fermentation and complex enzyme hydrolysis for improving the total soluble phenolic contents, flavonoid aglycones contents and bio-activities of guava leaves tea. Food Chem. 2018, 264, 189–198. [Google Scholar] [CrossRef]
- Shimada, K.; Fujikawa, K.; Yahara, K.; Nakamura, T. Antioxidative Properties of Xanthan on the Autoxidation of Soybean Oil in Cyclodextrin Emulsion. J. Agric. Food Chem. 1992, 40, 945–948. [Google Scholar] [CrossRef]
- Yin, C.M.; Fan, X.Z.; Fan, Z.; Shi, D.F.; Gao, H. Optimization of enzymes-microwave-ultrasound assisted extraction of Lentinus edodes polysaccharides and determination of its antioxidant activity. Int. J. Biol. Macromol. 2018, 111, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Meng, Y.H.; Su, A.P.; Yuan, S.; Zhao, H.G.; Tan, S.Y.; Hu, C.Y.; Deng, H.; Guo, Y.R. Evaluation of Total Flavonoids, Myricetin, and Quercetin from Hovenia dulcis Thunb. As Inhibitors of α-Amylase and α-Glucosidase. Plant Foods Hum. Nutr. 2016, 71, 444–449. [Google Scholar] [CrossRef]
- Tong, T.; Li, J.; Ko, D.O.; Kim, B.S.; Zhang, C.; Ham, K.S.; Kang, S.G. In vitro antioxidant potential and inhibitory effect of seaweed on enzymes relevant for hyperglycemia. Food Sci. Biotechnol. 2014, 23, 2037–2044. [Google Scholar] [CrossRef]
- Zhao, X.L.; Yu, L.; Zhang, S.D.; Ping, K.; Ni, H.Y.; Qin, X.Y.; Zhao, C.J.; Wang, W.; Efferth, T.; Fu, Y.J. Cryptochlorogenic acid attenuates LPS-induced inflammatory response and oxidative stress via upregulation of the Nrf2/HO-1 signaling pathway in RAW 264.7 macrophages. Int. Immunopharmacol. 2020, 83, 106436. [Google Scholar] [CrossRef] [PubMed]
- Cui, M.X.; Wang, Y.; Liu, K.H. Structure Analysis and Anti-Inflammatory Effect of a Polysaccharide from Hippocampus mohnikei. Chem. Biodivers. 2022, 19, e202100739. [Google Scholar] [CrossRef]
- Zhang, H.; Guo, Q.; Liang, Z.; Wang, M.; Wang, B.; Sun-Waterhouse, D.; Waterhouse, G.I.N.; Wang, J.; Ma, C.; Kang, W. Anti-inflammatory and antioxidant effects of Chaetoglobosin Vb in LPS-induced RAW264.7 cells: Achieved via the MAPK and NF-κB signaling pathways. Food Chem. Toxicol. 2021, 147, 111915. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Liu, M.; Liu, Z.K.; Cheng, L.; Li, M.S.; Li, C.W. Purification, Structural Characterization, and Antitumor Activity of a Polysaccharide from Perilla Seeds. Int. J. Mol. Sci. 2023, 24, 15904. [Google Scholar] [CrossRef] [PubMed]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polysaccharide Components | Total Sugar Content | Protein Content |
|---|---|---|
| LDP-A1 | 87.21 ± 0.57% | N.D. |
| LDP-B1 | 86.47 ± 0.29% | N.D. |
| Monosaccharide Composition and Content (mol %) | Different Components of Polysaccharides | |
|---|---|---|
| LDP-A1 | LDP-B1 | |
| Man | 15.1% | 8.9% |
| GlcA | N.D. | 2.1% |
| Rha | 0.2% | 0.2% |
| GalA | N.D. | 0.2% |
| Glc | 40.7% | 53.3% |
| Gal | 41.5% | 27.8% |
| Xyl | 0.3% | 0.5% |
| Ara | 0.1% | 0.3% |
| Fuc | 2.1% | 6.8% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, Q.; Liang, B.; Wang, J.; Dai, Y. Ultrasound-Assisted Extraction of Polysaccharides from Lyophyllum decastes: Structural Analysis and Bioactivity Assessment. Molecules 2025, 30, 961. https://doi.org/10.3390/molecules30040961
Wu Q, Liang B, Wang J, Dai Y. Ultrasound-Assisted Extraction of Polysaccharides from Lyophyllum decastes: Structural Analysis and Bioactivity Assessment. Molecules. 2025; 30(4):961. https://doi.org/10.3390/molecules30040961
Chicago/Turabian StyleWu, Qiong, Bin Liang, Jiaming Wang, and Yonggang Dai. 2025. "Ultrasound-Assisted Extraction of Polysaccharides from Lyophyllum decastes: Structural Analysis and Bioactivity Assessment" Molecules 30, no. 4: 961. https://doi.org/10.3390/molecules30040961
APA StyleWu, Q., Liang, B., Wang, J., & Dai, Y. (2025). Ultrasound-Assisted Extraction of Polysaccharides from Lyophyllum decastes: Structural Analysis and Bioactivity Assessment. Molecules, 30(4), 961. https://doi.org/10.3390/molecules30040961