
Effect of the Positioning of Metal Centers on a Cavitand in the Ruthenium-Catalyzed N-Alkylation of Amines


Abstract

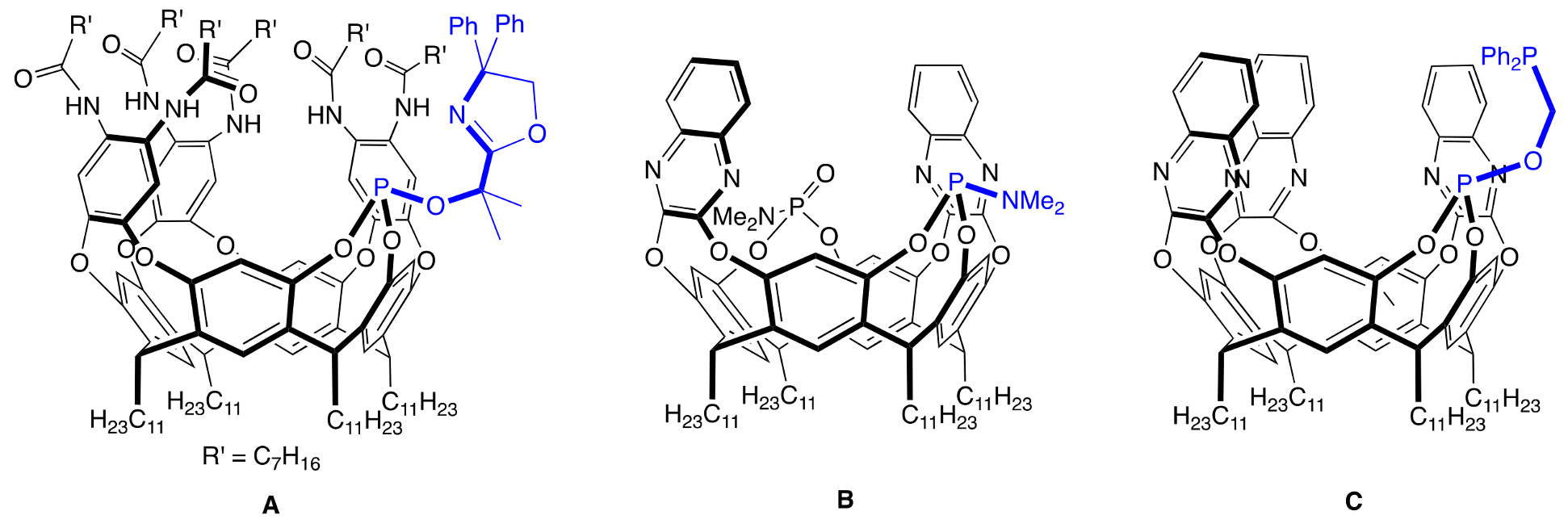
1. Introduction
2. Results and Discussion
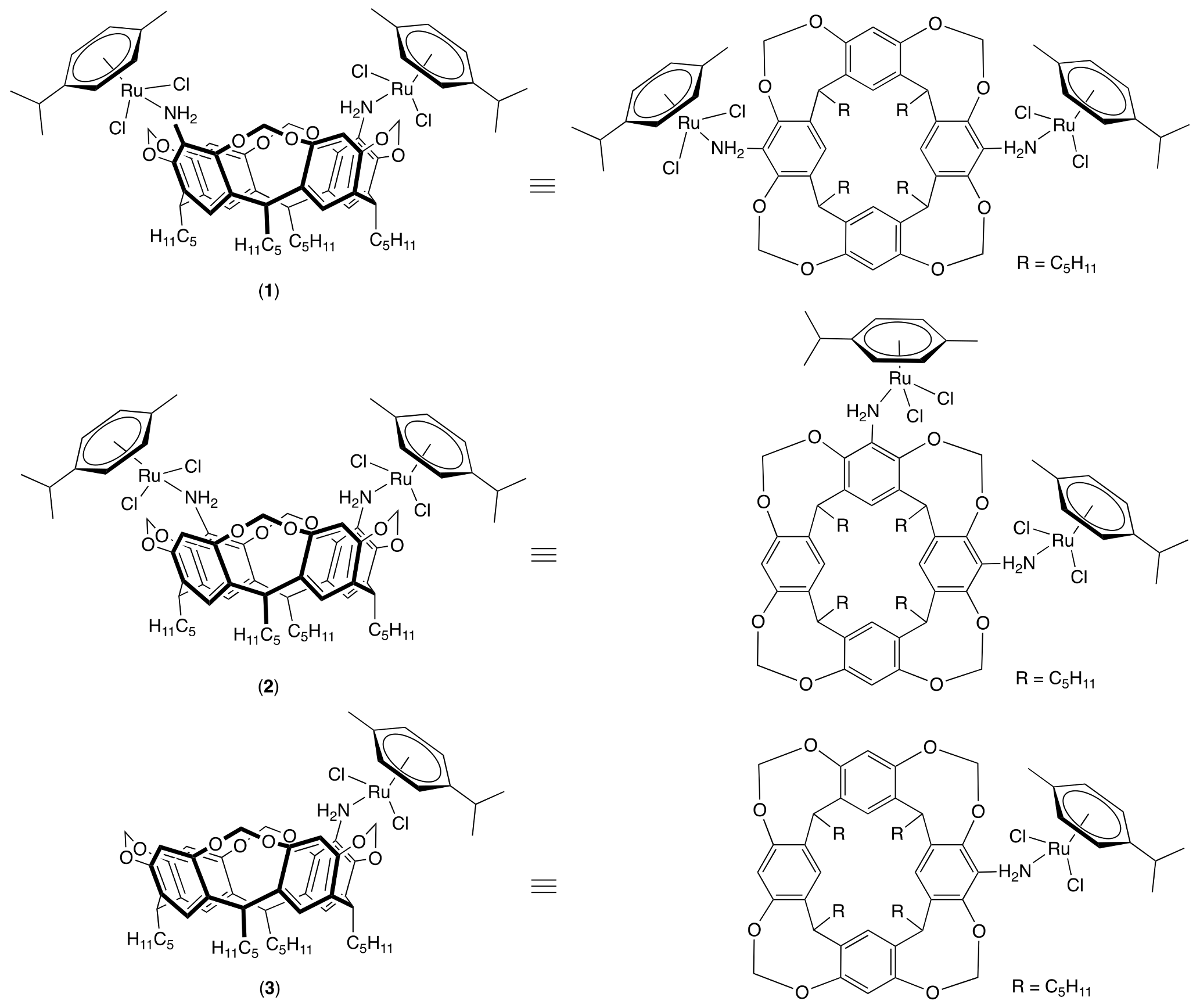
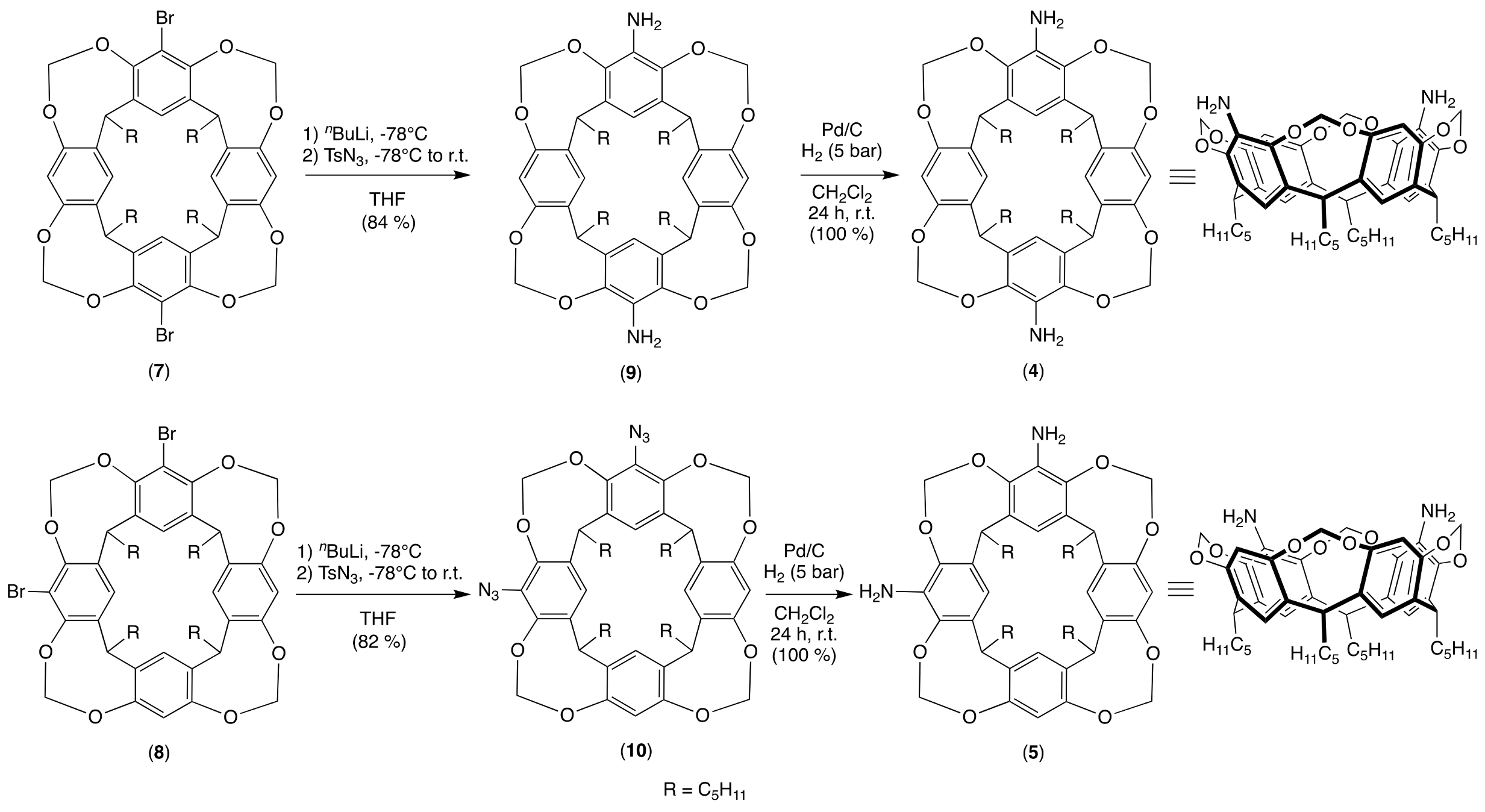
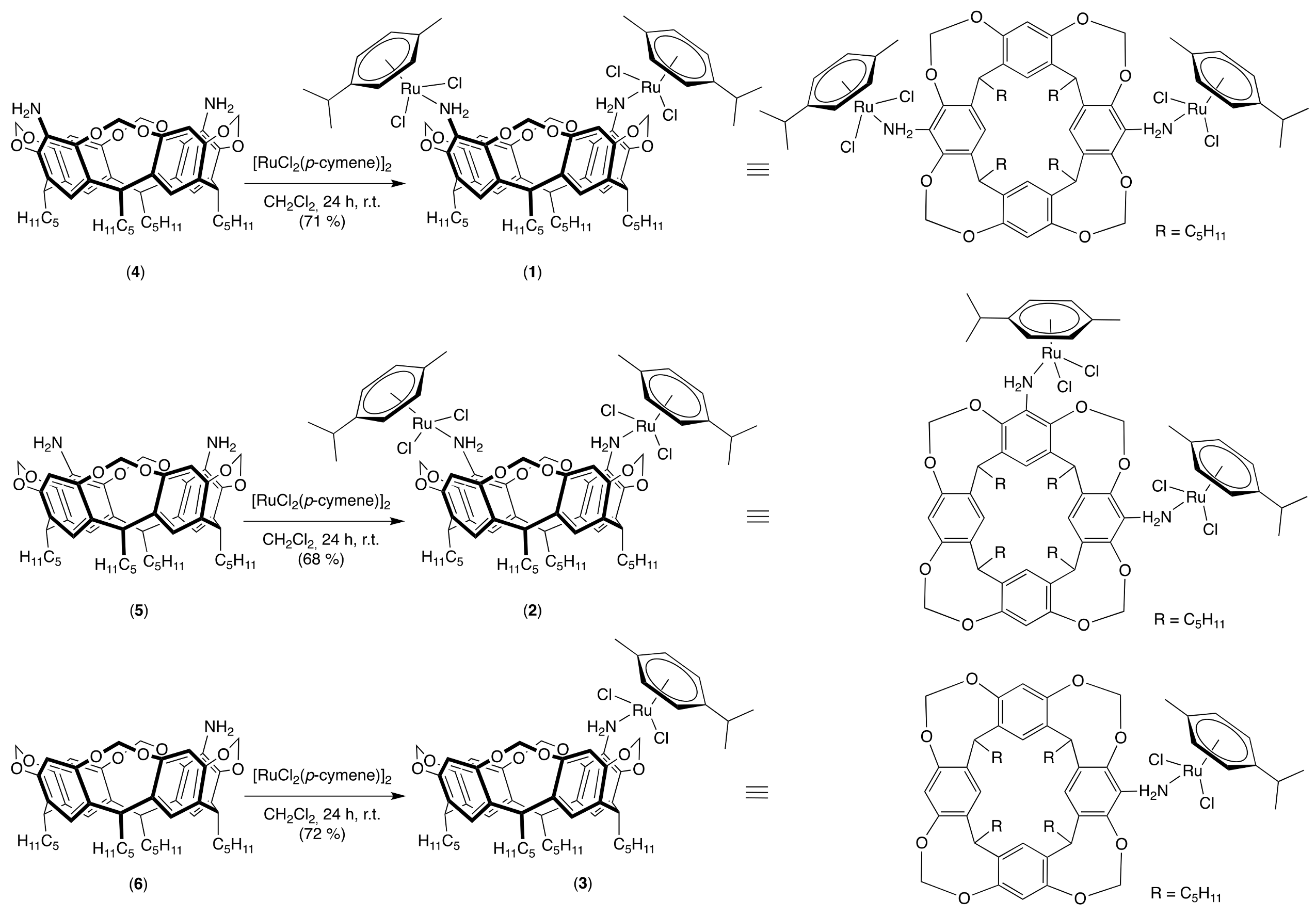
2.1. Synthesis of the Cavitands and Their Ruthenium Complexes
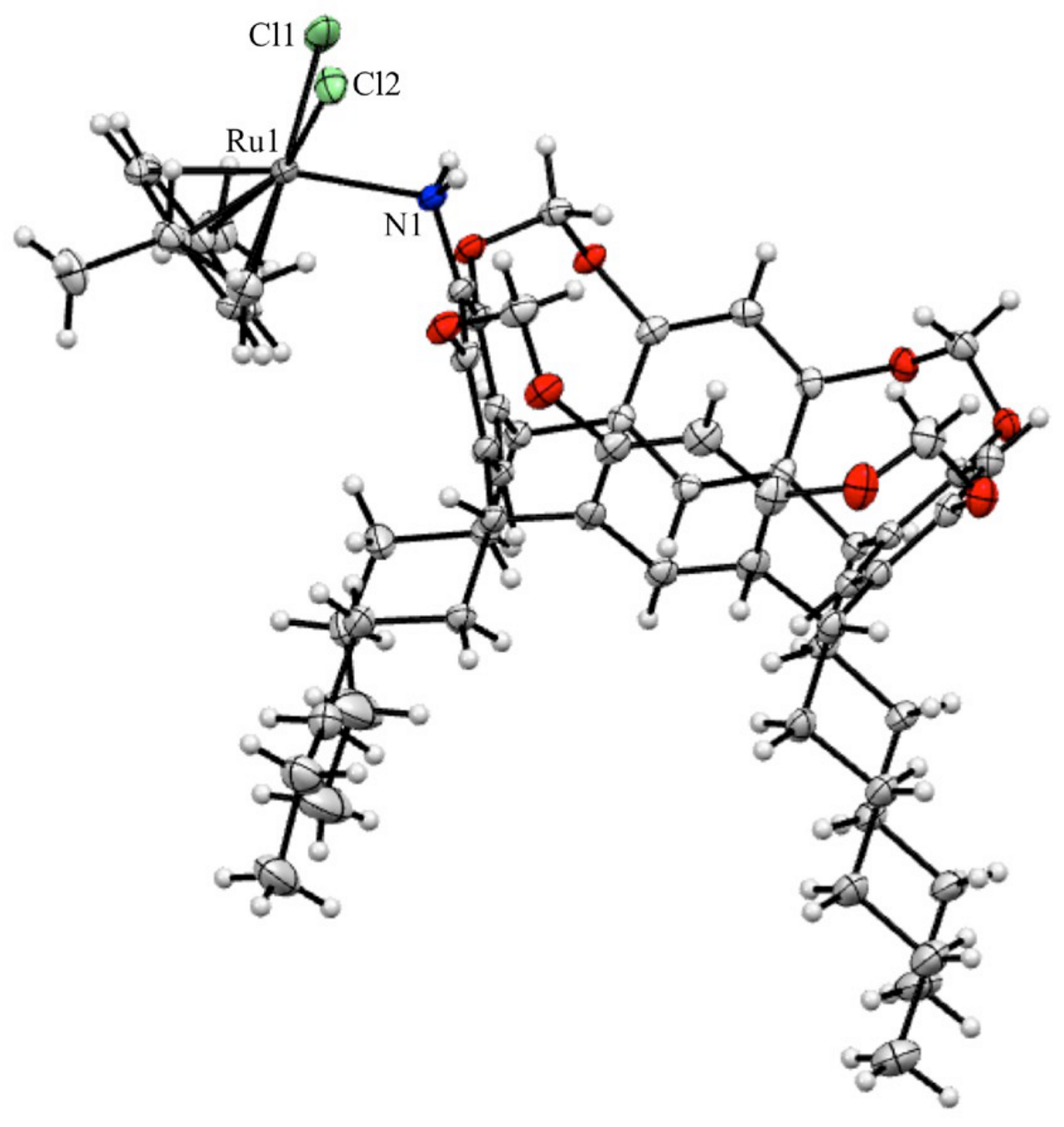
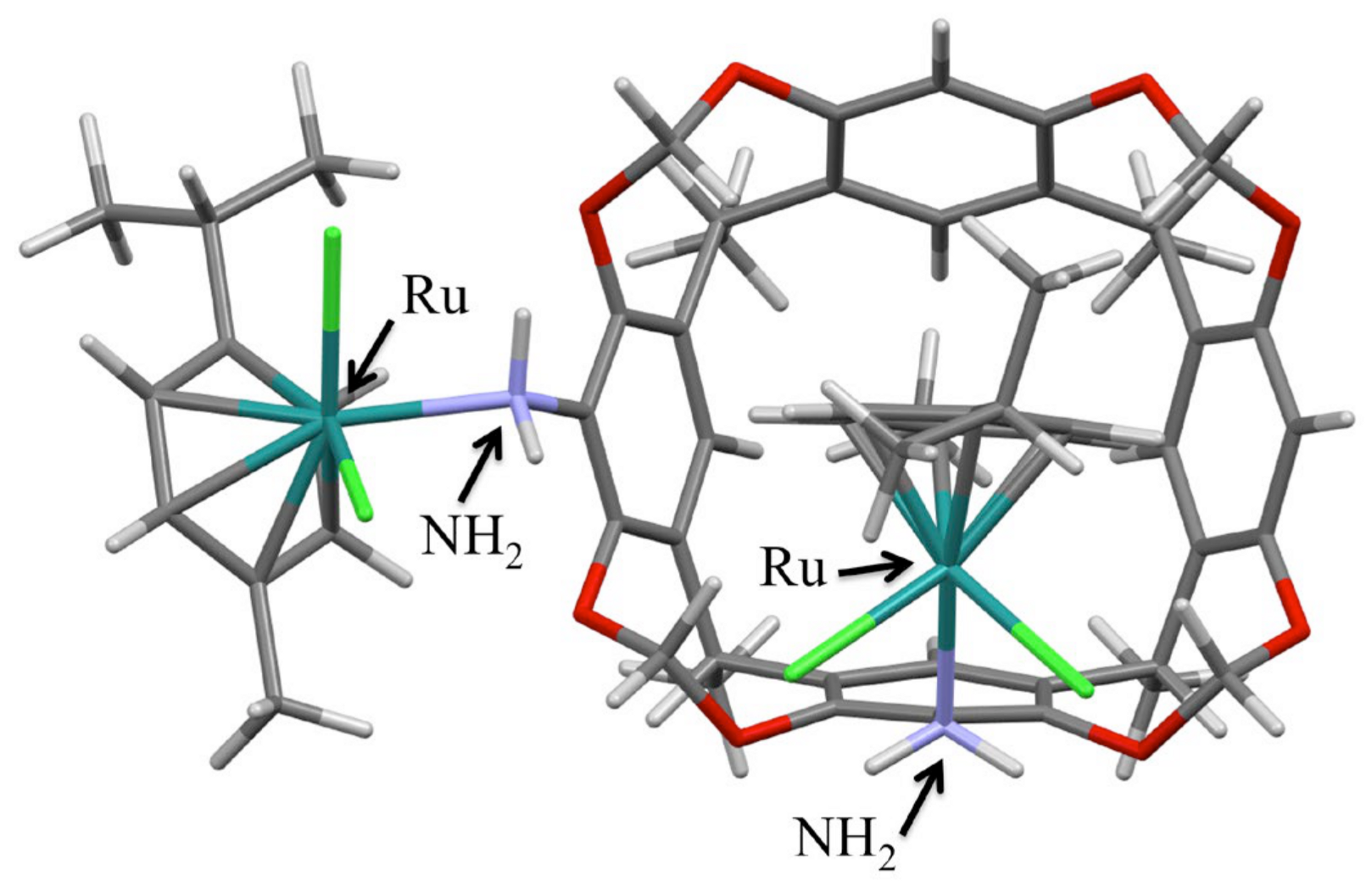
2.2. X-Ray Crystal Structure Analysis
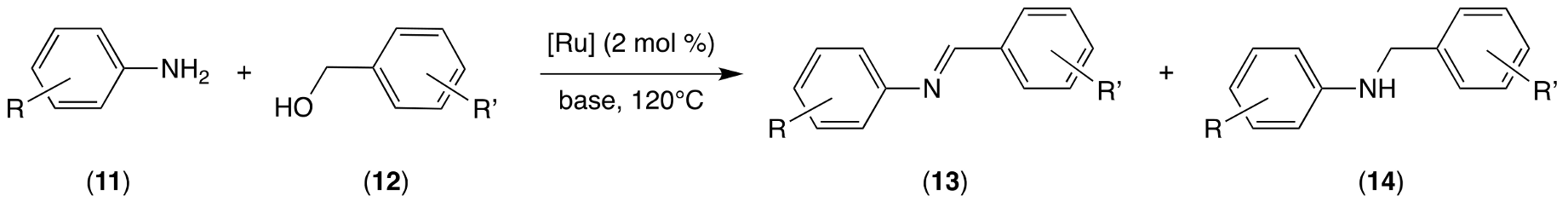
2.3. Catalytic Tests
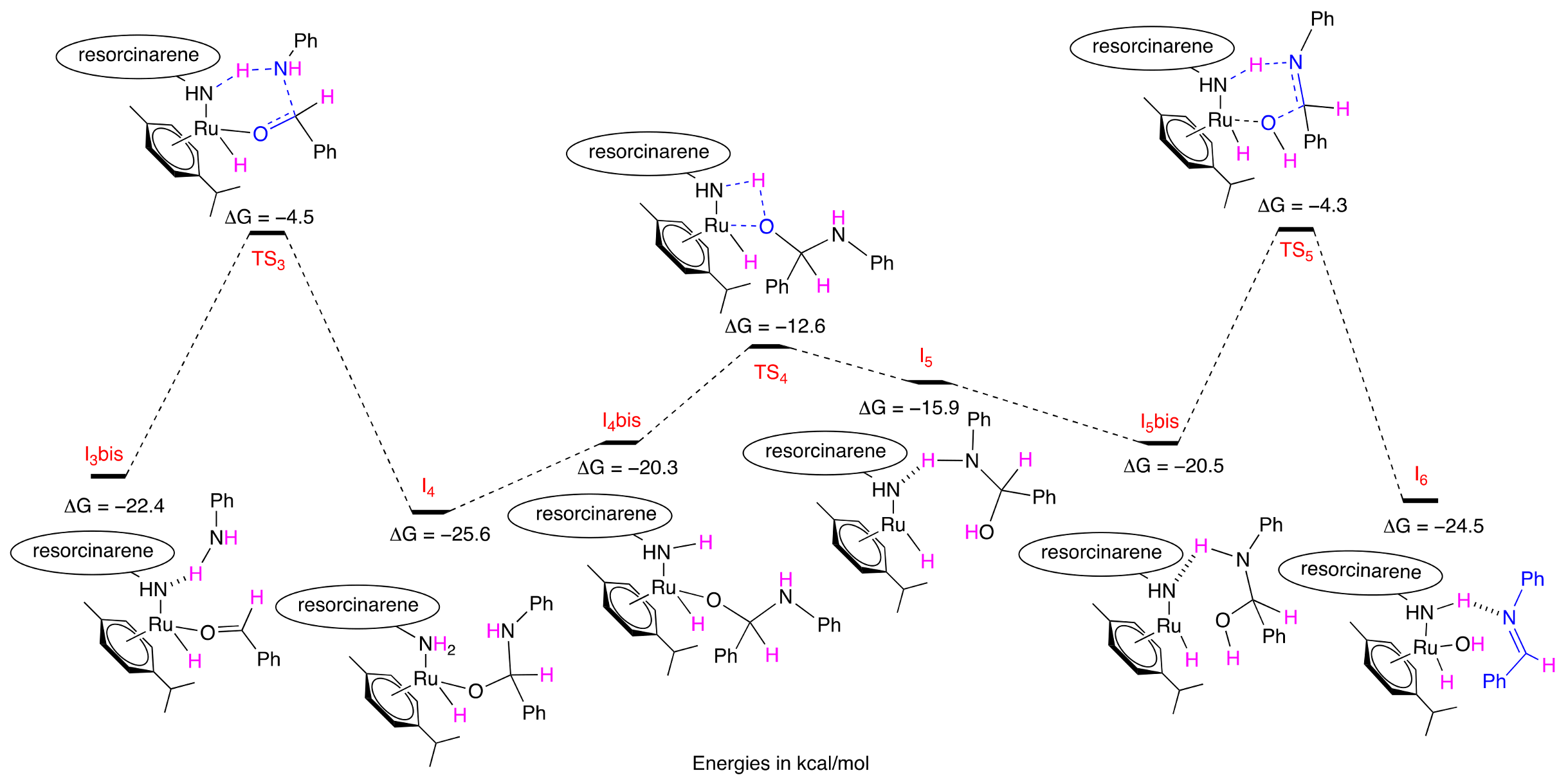
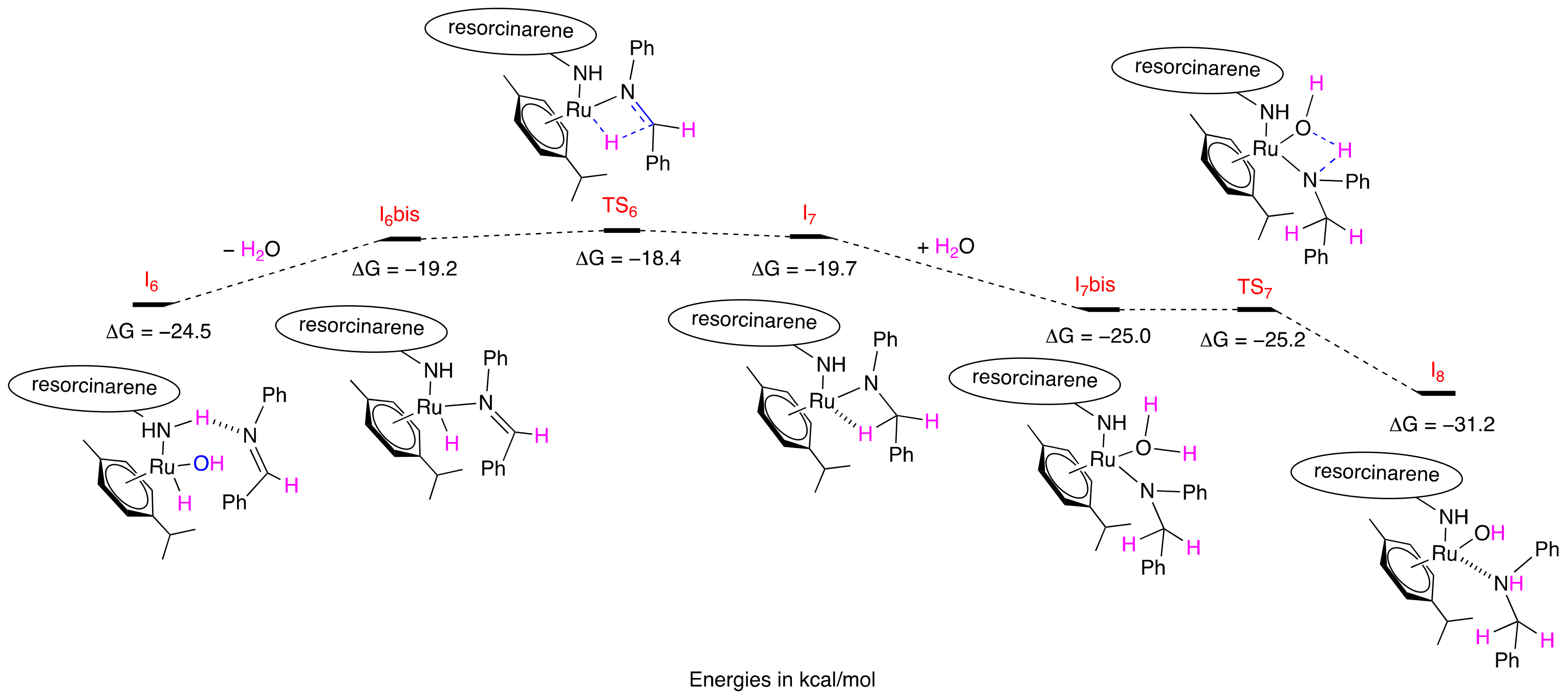
2.4. Mechanistic Computation
3. Materials and Methods
3.1. Synthesis
3.1.1. General Procedure for the Synthesis of 5,X-Diazido-4(24),6(10),12(16),18(22)-tetra-methyl-enedioxy-2,8,14,20-tetrapentyl-resorcin[4]arene (X = 11 or 17)
3.1.2. General Procedure for the Synthesis of 5,X-Diamino-4(24),6(10),12(16),18(22)-te-tramethylenedioxy-2,8,14,20-tetrapentyl-resorcin[4]arene (X = 11 or 17)
3.1.3. General Procedure for the Synthesis of Ruthenium Complexes (1–3)
3.2. X-Ray Crystal Structure Analysis
3.3. Catalytic Tests
3.4. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Baeyer, A. Ueber die Verbindungen der Aldehyde mit den Phenolen. Ber. Dtsch. Chem. Ges. 1872, 5, 280–282. [Google Scholar] [CrossRef]
- Jain, V.K.; Kanaiya, P.H. Chemistry of calix[4]resorcinarenes. Russ. Chem. Rev. 2011, 80, 75–102. [Google Scholar] [CrossRef]
- Moran, J.R.; Karbach, S.; Cram, D.J. Cavitands: Synthetic molecular vessels. J. Am. Chem. Soc. 1982, 104, 5826–5828. [Google Scholar] [CrossRef]
- Cram, D.J. Cavitands: Organic hosts with enforced cavities. Science 1983, 219, 1177–1183. [Google Scholar] [CrossRef]
- Gajjar, J.A.; Vekariya, R.H.; Parekh, H.M. Recent advances in upper rim functionalization of resorcin[4]arene derivatives: Synthesis and applications. Synth. Commun. 2020, 50, 2545–2571. [Google Scholar] [CrossRef]
- Liu, J.-L.; Sun, M.; Shi, Y.-H.; Zhou, X.-M.; Zhang, P.-Z.; Jia, A.-Q.; Zhang, Q.-F. Functional modification, self-assembly and application of calix[4]resorcinarenes. J. Incl. Phenom. Macrocycl. Chem. 2022, 102, 201–233. [Google Scholar] [CrossRef]
- Natarajan, N.; Brenner, E.; Sémeril, D.; Matt, D.; Harrowfield, J. The use of resorcinarene cavitands in metal-based catalysis. J. Eur. J. Org. Chem. 2017, 6100–6113. [Google Scholar] [CrossRef]
- Wang, K.; Liu, Q.; Zhou, L.; Sun, H.; Yao, X.; Hu, X.-Y. State-of-the-art and recent progress in resorcinarene-based cavitand. Chin. Chem. Lett. 2023, 34, 108559. [Google Scholar] [CrossRef]
- Eisler, D.J.; Puddephatt, R.J. Structures and conformational dynamics of gold(I) halide complexes of resorcinarene tetraphosphinite ligands. Inorg. Chem. 2003, 42, 6352–6365. [Google Scholar] [CrossRef]
- El Moll, H.; Sémeril, D.; Matt, D.; Toupet, L. Regioselective grafting of two -CH2P(X)Ph2 units (X = O, lone pair) onto a resorcin[4]arene-derived cavitand. Eur. J. Org. Chem. 2010, 1158–1168. [Google Scholar] [CrossRef]
- El Moll, H.; Sémeril, D.; Matt, D.; Toupet, L. Resorcin[4]arene-derived mono- and diphosphines in Suzuki cross-coupling. Adv. Synth. Catal. 2010, 352, 901–908. [Google Scholar] [CrossRef]
- Şahin, N.; Sémeril, D.; Brenner, E.; Matt, D.; Özdemir, İ.; Kaya, C. Resorcinarene-functionalised imidazolium salts as ligand precursors for palladium-catalysed Suzuki-Miyaura cross-couplings. ChemCatChem 2013, 5, 1116–1125. [Google Scholar] [CrossRef]
- Ngodwana, L.; Bose, S.; Smith, V.; van Otterlo, W.A.L.; Arnott, G.E. A bidentate resorcinarene-based palladium carbene complex. Eur. J. Inorg. Chem. 2017, 1923–1929. [Google Scholar] [CrossRef]
- Gibson, C.; Rebek, J., Jr. Recognition and catalysis in allylic alkylations. Org. Lett. 2002, 4, 1887–1890. [Google Scholar] [CrossRef] [PubMed]
- Endo, N.; Inoue, M.; Iwasawa, T. Rational design of a metallocatalytic cavitand for regioselective hydration of specific alkynes. Eur. J. Org. Chem. 2018, 1136–1140. [Google Scholar] [CrossRef]
- Smart, J.E.; Emerson-King, J.; Jeans, R.J.; Hood, T.M.; Lau, S.; Bara-Estaún, A.; Hintermair, U.; Pringle, P.G.; Chaplin, A.B. A resorcin[4]arene-based phosphite-phosphine ligand for the branched-selective hydroformylation of alkyl alkenes. ACS Catal. 2024, 14, 11803–11807. [Google Scholar] [CrossRef] [PubMed]
- Hkiri, S.; Sémeril, D. How do positions of phosphito units on a calix[4]arene platform affect the enantioselectivity of a catalytic reaction? Organics 2022, 3, 470–480. [Google Scholar] [CrossRef]
- Inoue, M.; Kamiguchi, S.; Ugawa, K.; Hkiri, S.; Bouffard, J.; Sémeril, D.; Iwasawa, T. Evaluation of the catalytic capability of cis- and trans-diquinoxaline spanned cavitands. Eur. J. Org. Chem. 2019, 6261–6268. [Google Scholar] [CrossRef]
- Nixon, T.D.; Whittlesey, M.K.; Williams, J.M.J. Transition metal catalysed reactions of alcohols using borrowing hydrogen methodology. Dalton Trans. 2009, 753–762. [Google Scholar] [CrossRef]
- Bähn, S.; Imm, S.; Neubert, L.; Zhang, M.; Neumann, H.; Beller, M. The catalytic amination of alcohols. ChemCatChem 2011, 3, 1853–1864. [Google Scholar] [CrossRef]
- Corma, A.; Navas, J.; Sabater, M.J. Advances in one-pot synthesis through borrowing hydrogen catalysis. Chem. Rev. 2018, 118, 1410–1459. [Google Scholar] [CrossRef]
- Reed-Berendt, B.G.; Latham, D.E.; Dambatta, M.B.; Morrill, L.C. Borrowing hydrogen for organic synthesis. ACS Cent. Sci. 2021, 7, 570–585. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, N.; Chavagnan, T.; Sémeril, D.; Brenner, E.; Matt, D.; Ramesh, R.; Toupet, L. Cavitand chemistry: Nickel half-sandwich complexes with imidazolylidene ligands bearing one or two resorcinarenyl substituents. Eur. J. Inorg. Chem. 2018, 890–896. [Google Scholar] [CrossRef]
- Binnani, C.; Tyagi, D.; Rai, R.K.; Mobin, S.M.; Singh, S.K. C-H Bond activation/arylation catalyzed by arene-ruthenium-aniline complexes in water. Chem. Asian J. 2016, 11, 3022–3031. [Google Scholar] [CrossRef]
- Tyagi, D.; Binnani, C.; Rai, R.K.; Dwivedi, A.D.; Gupta, K.; Li, P.-Z.; Zhao, Y.; Singh, S.K. Ruthenium-catalyzed oxidative homocoupling of arylboronic acids in water: Ligand tuned reactivity and mechanistic study. Inorg. Chem. 2016, 55, 6332–6343. [Google Scholar] [CrossRef]
- Martínez-Peña, F.; Pizarro, A.M. Control of reversible activation dynamics of [Ru{η6:κ1-C6H5(C6H4)NH2}(XY)]n+ and the effect of chelating-ligand variation. Chem. Eur. J. 2017, 23, 16231–16241. [Google Scholar] [CrossRef]
- Patel, B.; Ranjan, R.; Chauhan, N.R.; Mukhopadhyay, S.; Choudhuryc, A.R.; Vyas, K.M. N-coordinated Ru(II) catalyzed solvent free N-alkylation of primary amines with alcohols through borrowing hydrogen strategy. New J. Chem. 2023, 47, 8305–8317. [Google Scholar] [CrossRef]
- Şahin, N.; Özdemir, N.; Gürbüz, N.; Özdemir, İ. Novel N-alkylbenzimidazole-ruthenium (II) complexes: Synthesis and catalytic activity of N-alkylating reaction under solvent-free medium. Appl. Organometal. Chem. 2019, 33, e4704. [Google Scholar] [CrossRef]
- Kaloğlu, M.; Gürbüz, N.; Sémeril, D.; Özdemir, İ. Ruthenium(II)-(p-cymene)-N-heterocyclic carbene complexes for the N-alkylation of amine using the green hydrogen borrowing methodology. Eur. J. Inorg. Chem. 2018, 1236–1243. [Google Scholar] [CrossRef]
- Hamid, M.H.S.A.; Slatford, P.A.; Williams, J.M.J. Borrowing hydrogen in the activation of alcohols. Adv. Synth. Catal. 2007, 349, 1555–1575. [Google Scholar] [CrossRef]
- Chakrabarti, K.; Paul, B.; Maji, M.; Roy, B.C.; Shee, S.; Kundu, S. Bifunctional Ru(II) complex catalysed carbon-carbon bond formation: An eco-friendly hydrogen borrowing strategy. Org. Biomol. Chem. 2016, 14, 10988–10997. [Google Scholar] [CrossRef] [PubMed]
- Tucker, J.A.; Knobler, C.B.; Trueblood, K.N.; Cram, D.J. Host-guest complexation. 49. Cavitands containing two binding cavities. J. Am. Chem. Soc. 1989, 111, 3688–3699. [Google Scholar] [CrossRef]
- Hwang, H.; Kim, J.; Jeong, J.; Chang, S. Regioselective introduction of heteroatoms at the C-8 position of quinoline N-oxides: Remote C-H activation using N-oxide as a stepping stone. J. Am. Chem. Soc. 2014, 136, 10770–10776. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal structure determination. Acta Crystallogr. Sect. A 2015, A71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C 2015, C71, 3–8. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Chai, J.-D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef]
- Ditchfield, R.; Hehre, W.J.; Pople, J.A. Self-consistent molecular orbital methods. 9. Extended gaussian-type basis for molecular-orbital studies of organic molecules. J. Chem. Phys. 1971, 54, 724–728. [Google Scholar] [CrossRef]
- Fuentealba, P.; Preuss, H.; Stoll, H.; Szentpály, L.V. A proper account of core-polarization with pseudopotentials—Single valence-electron alkali compounds. Chem. Phys. Lett. 1982, 89, 418–422. [Google Scholar] [CrossRef]
- Miertuš, S.; Scrocco, E.; Tomasi, J. Electrostatic interaction of a solute with a continuum. A direct utilization of ab initio molecular potentials for the prevision of solvent effects. Chem. Phys. 1981, 55, 117–129. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Ruthenium Complex | Base | Time (h) | Solvent | Conversion 2 (%) | Product Distribution 2 (Amine 14/Imine 13) |
|---|---|---|---|---|---|---|
| 1 | 3 | tBuOK | 3 | toluene | 88 | 6/94 |
| 2 | 3 | tBuOK | 3 | DMF | 49 | 28/72 |
| 3 | 3 | tBuOK | 3 | dioxane | 64 | 26/74 |
| 4 | 3 | tBuOK | 3 | / | 40 | 92/8 |
| 5 | 3 | KOH | 6 | / | 41 | 97/3 |
| 6 | 3 | K2CO3 | 6 | / | 75 | 13/87 |
| 7 | 3 | Cs2CO3 | 6 | / | 61 | 70/30 |
| 8 | 3 | NaOAc | 6 | / | 55 | 16/84 |
| 9 | 3 | tBuOK | 6 | / | 60 | 93/7 |
| 10 | 1 | tBuOK | 6 | / | 65 | 80/10 |
| 11 | 2 | tBuOK | 6 | / | 82 | 98/2 |
| 12 | D [27] 3 | KOH | 6 | / | 78 | 100/0 |
| 13 | E [28] 4 | tBuOK | 24 | / | 97 | 99/1 |
| 14 | F [29] 5 | tBuOK | 24 | toluene | 91 | 95/5 |
| Entry | 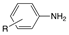 |  | Conversion 2 (%) | Product Distribution 2 (Amine 14/Imine 13) |
|---|---|---|---|---|
| 1 |  |  | 99 | 99/1 |
| 2 | 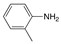 | 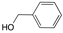 | 17 | 95/5 |
| 3 | 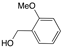 | 24 | 100/0 | |
| 4 | 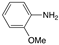 |  | 64 | 100/0 |
| 5 | 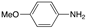 | 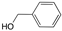 | 95 | 100/0 |
| 6 |  | 94 | 100/0 |
| CCDC depository | 2409059 | chemical formula | C62H80Cl2NO8Ru•(CHCl3)1.15 | |
| color/shape | orange/prism | formula weight (g mol−1) | 1276.51 | |
| crystal system | triclinic | space group | P-1 | |
| unit cell parameters | a (Å) | 11.1926(4) | volume (Å3) | 3566.8(7) |
| b (Å) | 12.0728(4) | Z | 2 | |
| c (Å) | 26.0769(7) | D (g cm−3) | 1.357 | |
| α (°) | 77.7210(10) | μ (mm−1) | 0.537 | |
| β (°) | 82.6390(10) | Tmin, Tmax | 0.929/0.948 | |
| γ (°) | 65.2670(10) | F(000) | 1335 | |
| crystal size (mm) | 0.140 × 0.120 × 0.100 | index ranges | −14 ≤ h ≤ 14 | |
| θ range for data collection (°) | 2.005 ≤ θ ≤ 27.939 | −15 ≤ k ≤ 15 | ||
| reflections collected | 129779 | −34 ≤ l ≤ 32 | ||
| data/restraints/parameters | 14968/9/755 | goodness-of-fit on F2 | 1.029 | |
| final R indices (I > 2.0 σ(I)) | R1 = 0.0411 | R indices (all data) | R1 = 0.0487 | |
| wR2 = 0.0941 | wR2 = 0.0996 | |||
| Δρmax, Δρmin (e Å−3) | 1.989, −1.343 | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Şahin, N.; Gourlaouen, C.; Sémeril, D. Effect of the Positioning of Metal Centers on a Cavitand in the Ruthenium-Catalyzed N-Alkylation of Amines. Molecules 2025, 30, 951. https://doi.org/10.3390/molecules30040951
Şahin N, Gourlaouen C, Sémeril D. Effect of the Positioning of Metal Centers on a Cavitand in the Ruthenium-Catalyzed N-Alkylation of Amines. Molecules. 2025; 30(4):951. https://doi.org/10.3390/molecules30040951
Chicago/Turabian StyleŞahin, Neslihan, Christophe Gourlaouen, and David Sémeril. 2025. "Effect of the Positioning of Metal Centers on a Cavitand in the Ruthenium-Catalyzed N-Alkylation of Amines" Molecules 30, no. 4: 951. https://doi.org/10.3390/molecules30040951
APA StyleŞahin, N., Gourlaouen, C., & Sémeril, D. (2025). Effect of the Positioning of Metal Centers on a Cavitand in the Ruthenium-Catalyzed N-Alkylation of Amines. Molecules, 30(4), 951. https://doi.org/10.3390/molecules30040951