
Indium Imidazo[4,5,-b]porphyrins as Photocatalysts for Oxidation of Sulfides

 ,
, 
Abstract

1. Introduction
2. Results and Discussion
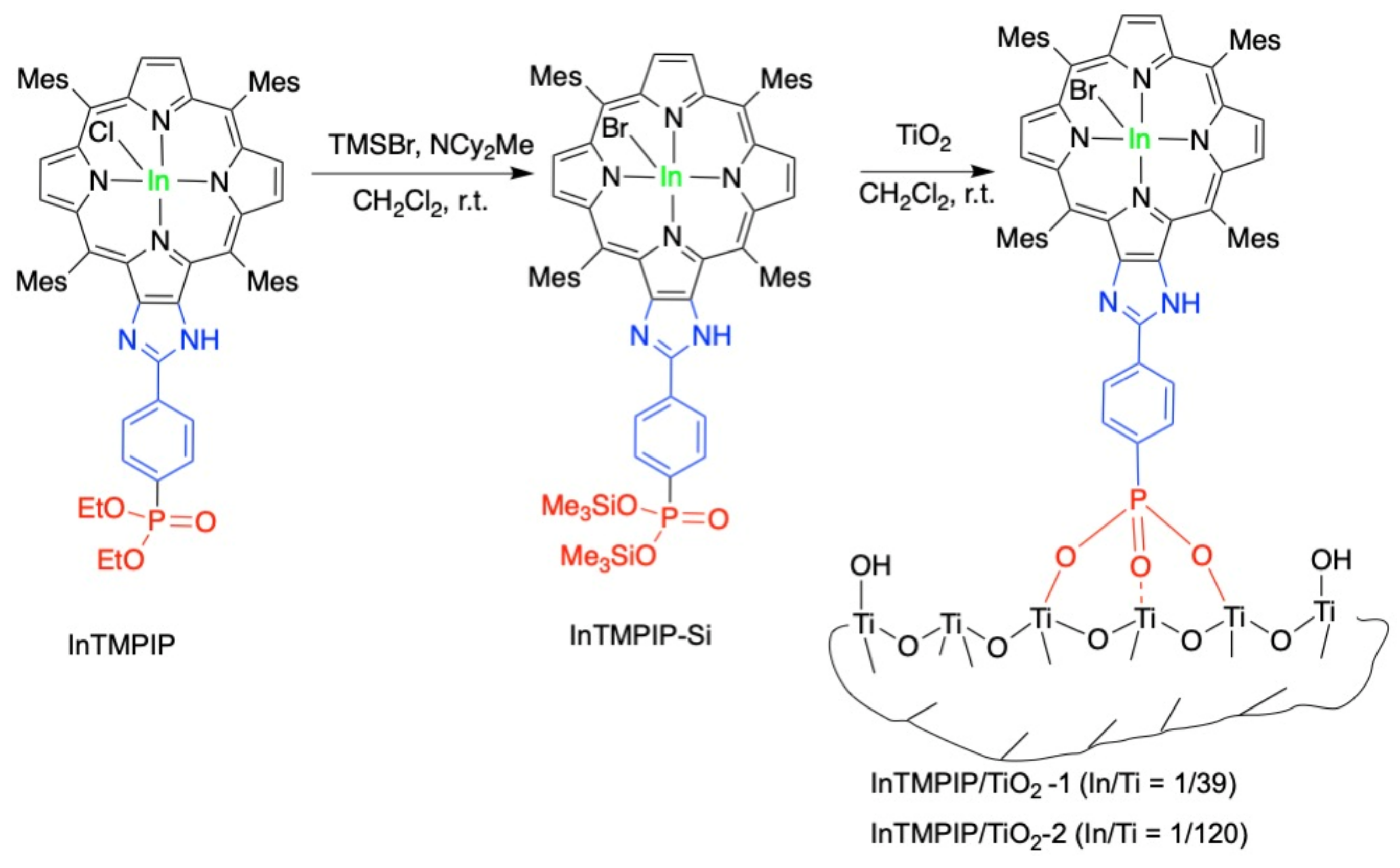
2.1. Synthesis of InTMPIP
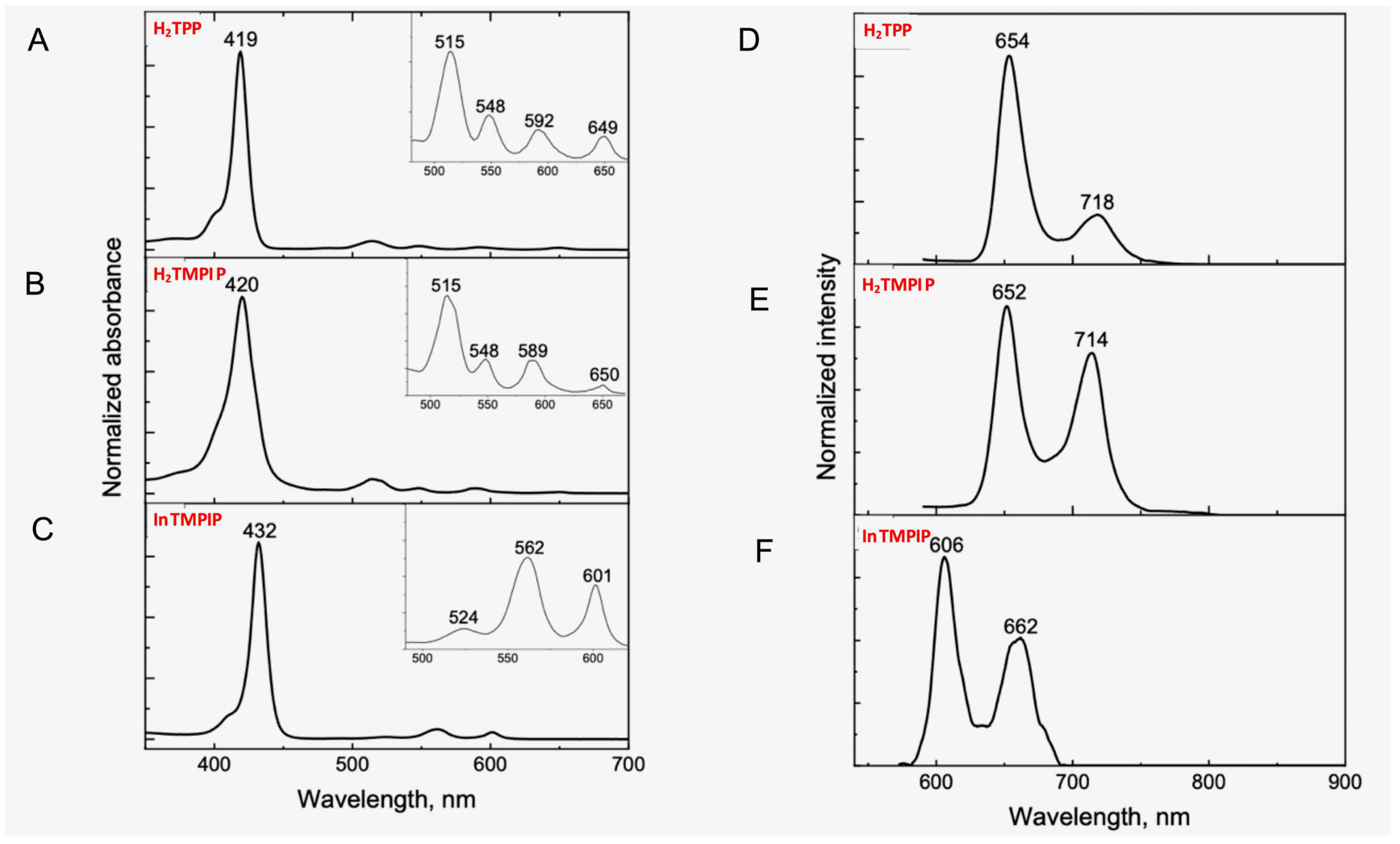
2.2. Optical Properties of H2TMPIP and InTMPIP, and Singlet Oxygen Generation
2.3. Homogeneous Photooxidation of Sulfides
2.4. Photostability Studies
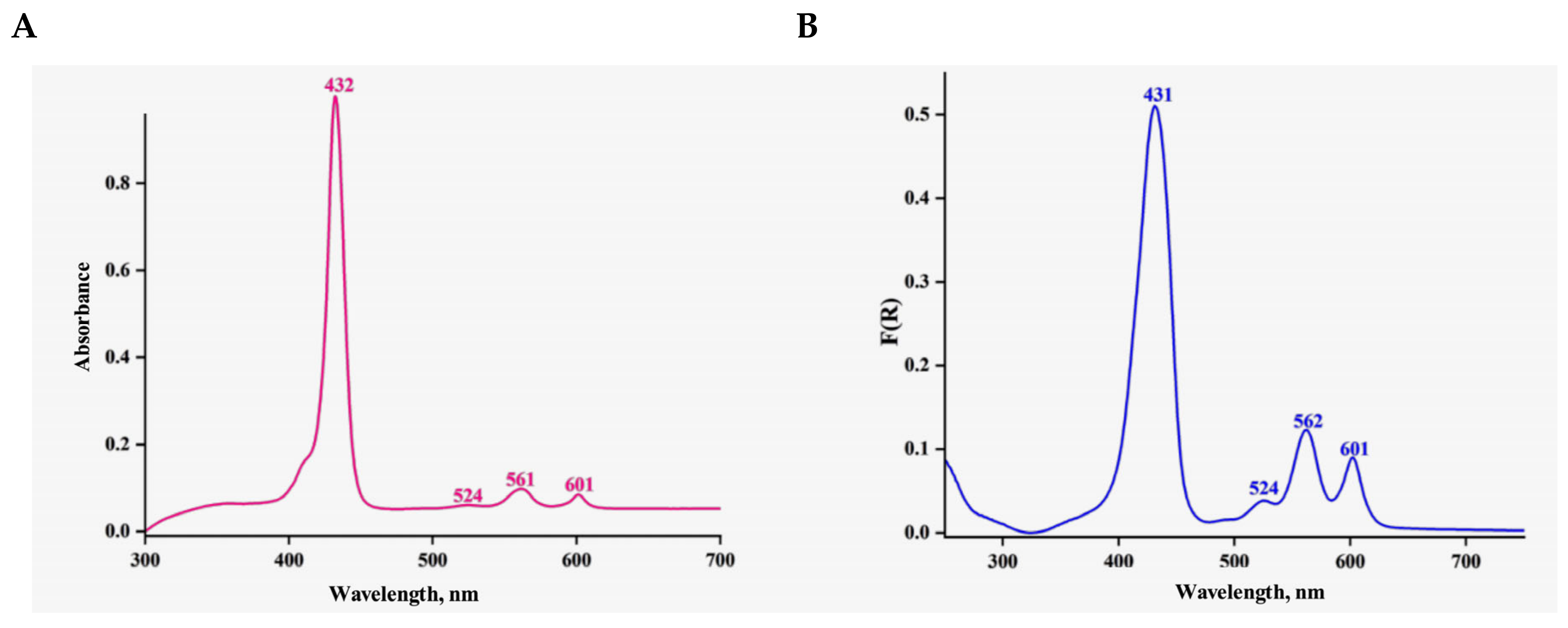
2.5. Grafting of InTMPIP on Mesoporous Titania
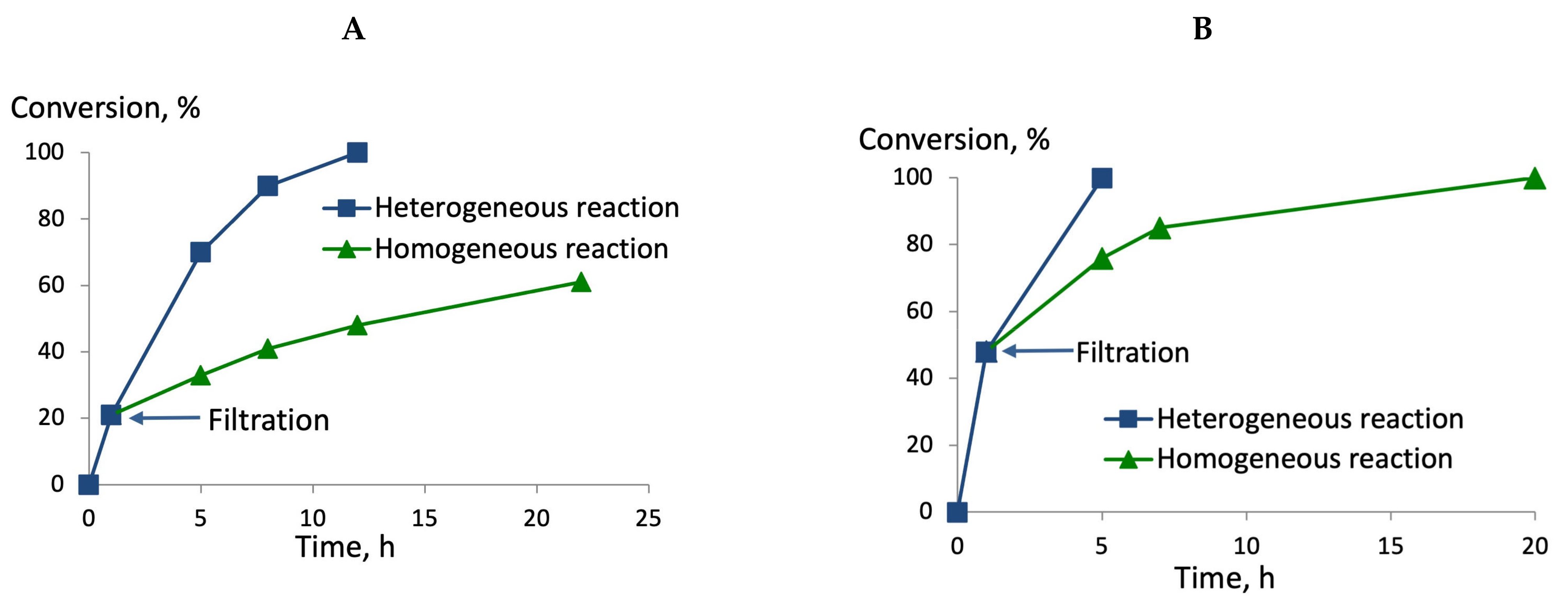
2.6. Photocatalytic Properties of InTMPIP/TiO2-2
3. Materials and Methods
3.1. Materials
3.2. Methods
3.3. Synthesis of {2-[4-(Diethoxyphosphoryl)phenyl]-5,10,15,20-tetramesityl-1H-imidazo[4,5-b]porphyrinato(2−)}(chloro)indium(III)
3.4. Determination of Emission Quantum Yields and Singlet Oxygen Quantum Yields
3.5. Photocatalytic Oxidation of Sulfides
3.6. Synthesis of Heterogenized Catalysts
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chao, H.; Li, R.-H.; Ye, B.-H.; Li, H.; Feng, X.-L.; Cai, J.-W.; Zhou, J.-Y.; Ji, L.-N. Syntheses, characterization and third order non-linear optical properties of the ruthenium(II) complexes containing 2-phenylimidazo[4,5-f][1,10]phenanthroline derivatives. J. Chem. Soc. Dalton Trans. 1999, 3711–3717. [Google Scholar] [CrossRef]
- Wu, J.-Z.; Li, L.; Zeng, T.-X.; Ji, L.-N.; Zhou, J.-Y.; Luo, T.; Li, R.-H. Synthesis, characterization and luminiscent DNA-binding study of a series of ruthenium complexes containing 2-arylimidazo[f]1,10-phenanthroline. Polyhedron 1997, 16, 103–107. [Google Scholar] [CrossRef]
- Han, M.-J.; Gao, L.-H.; Wang, K.-Z. Ruthenium(II) complex of 2-(9-anthryl)-1H-imidazo[4,5-f][1,10]phenanthroline: Synthesis, spectrophotometric pH titrations and DNA interaction. New J. Chem. 2006, 30, 208–214. [Google Scholar] [CrossRef]
- Herrero, C.; Quaranta, A.; Fallahpour, R.-A.; Leibl, W.; Aukauloo, A. Identification of the different mechanisms of activation of a [RuII(tpy)(bpy)(OH2)]2+ catalyst by modified ruthenium sensitizers in supramolecular complexes. J. Phys. Chem. C 2013, 117, 9605–9612. [Google Scholar] [CrossRef]
- Luan, T.-X.; Du, L.; Wang, J.-R.; Li, K.; Zhang, Q.; Li, P.-Z.; Zhao, Y. Highly Effective generation of singlet oxygen by an imidazole-linked robust photosensitizing covalent organic framework. ACS Nano 2022, 16, 21565–21575. [Google Scholar] [CrossRef]
- Crossley, M.J.; McDonald, J.A. Fused porphyrin-imidazole systems: New building blocks for synthesis of porphyrin arrays. J. Chem. Soc. Perkin Trans. 1 1999, 2429–2431. [Google Scholar] [CrossRef]
- Crossley, M.J.; Thordarson, P.; Wu, R.A.S. Efficient formation of lipophilic dihydroxotin(IV) porphyrins and bis-porphyrins. J. Chem. Soc. Perkin Trans. 1 2001, 2294–2302. [Google Scholar] [CrossRef]
- Curiel, D.; Ohkubo, K.; Reimers, J.R.; Fukuzumi, S.; Crossley, M.J. Photoinduced electron transfer in a β,β′-pyrrolic fused ferrocene-(zinc porphyrin)-fullerene. Phys. Chem. Chem. Phys. 2007, 9, 5260–5266. [Google Scholar] [CrossRef]
- Kashiwagi, Y.; Ohkubo, K.; McDonald, J.A.; Blake, I.M.; Crossley, M.J.; Araki, Y.; Ito, O.; Imahori, H.; Fukuzumi, S. Long-lived charge-separated state produced by photoinduced electron transfer in a zinc imidazoporphyrin-C60 dyad. Org. Lett. 2003, 5, 2719–2721. [Google Scholar] [CrossRef]
- Hayashi, H.; Touchy, A.S.; Kinjo, Y.; Kurotobi, K.; Toude, Y.; Ito, S.; Saarenpaeae, H.; Tkachenko, N.V.; Lemmetyinen, H.; Imahori, H. Triarylamine-substituted imidazole- and quinoxaline-fused push-pull porphyrins for dye-sensitized solar cells. ChemSusChem 2013, 6, 508–517. [Google Scholar] [CrossRef]
- Reimers, J.R.; McKemmish, L.K.; McKenzie, R.H.; Hush, N.S. Non-adiabatic effects in thermochemistry, spectroscopy and kinetics: The general importance of all three Born-Oppenheimer breakdown corrections. Phys. Chem. Chem. Phys. 2015, 17, 24641–24665. [Google Scholar] [CrossRef] [PubMed]
- Birin, K.P.; Abdulaeva, I.A.; Polivanovskaia, D.A.; Martynov, A.G.; Shokurov, A.V.; Gorbunova, Y.G.; Tsivadze, A.Y. Imidazoporphyrins with appended polycyclic aromatic hydrocarbons: To conjugate or not to conjugate? Dyes Pigm. 2021, 186, 109042. [Google Scholar] [CrossRef]
- Shepeleva, I.I.; Birin, K.P.; Polivanovskaia, D.A.; Martynov, A.G.; Shokurov, A.V.; Tsivadze, A.Y.; Selektor, S.L.; Gorbunova, Y.G. Unusual ‘Turn-on’ ratiometric response of fluorescent porphyrin-pyrene dyads to the nitroaromatic compounds. Chemosensors 2023, 11, 43. [Google Scholar] [CrossRef]
- Longevial, J.-F.; Rose, C.; Poyac, L.; Clément, S.; Richeter, S. Molecular systems combining porphyrinoids and N-heterocyclic carbenes. Eur. J. Inorg. Chem. 2021, 2021, 776–791. [Google Scholar] [CrossRef]
- Poyac, L.; Rose, C.; Wahiduzzaman, M.; Lebrun, A.; Cazals, G.; Devillers, C.H.; Yot, P.G.; Clément, S.; Richeter, S. Synthesis, characterization, and encapsulation properties of rigid and flexible porphyrin cages assembled from N-heterocyclic carbene–Metal Bonds. Inorg. Chem. 2021, 60, 19009–19021. [Google Scholar] [CrossRef]
- Poyac, L.; Scoditti, S.; Dumail, X.; Granier, M.; Clément, S.; Gramage-Doria, R.; Devillers, C.H.; Richeter, S. Electronic, steric and catalytic properties of N-heterocyclic carbene rhodium(I) complexes linked to (metallo)porphyrins. Chem. Commun. 2022, 58, 13270–13273. [Google Scholar] [CrossRef]
- Kechiche, A.; Al Shehimy, S.; Khrouz, L.; Monnereau, C.; Bucher, C.; Parola, S.; Bessmertnykh-Lemeune, A.; Rousselin, Y.; Cheprakov, A.V.; Nasri, H. Phosphonate-substituted porphyrins as efficient, cost-effective and reusable photocatalysts. Dalton Trans. 2024, 53, 7498–7516. [Google Scholar] [CrossRef]
- Sheldon, A. Metalloporphyrins in Catalytic Oxidations. Marcel Dekker: New York, NY, USA, 1994; p. 408. [Google Scholar]
- Stephenson, N.A.; Bell, A.T. Effects of porphyrin composition on the activity and selectivity of the iron(III) porphyrin catalysts for the epoxidation of cyclooctene by hydrogen peroxide. J. Mol. Catal. A Chem. 2007, 272, 108–117. [Google Scholar] [CrossRef]
- Da Silva, G.; Pires, S.M.G.; Silva, V.L.M.; Simões, M.M.Q.; Neves, M.G.P.M.S.; Rebelo, S.L.H.; Silva, A.M.S.; Cavaleiro, J.A.S. A green and sustainable method for the oxidation of 1,3-dihydrobenzo[c]thiophenes to sulfones using metalloporphyrin complexes. Catal. Commun. 2014, 56, 68–71. [Google Scholar] [CrossRef]
- Abdulaeva, I.A.; Birin, K.P.; Michalak, J.; Romieu, A.; Stern, C.; Bessmertnykh-Lemeune, A.; Guilard, R.; Gorbunova, Y.G.; Tsivadze, A.Y. On the synthesis of functionalized porphyrins and porphyrin conjugates via β-aminoporphyrins. New J. Chem. 2016, 40, 5758–5774. [Google Scholar] [CrossRef]
- Abdulaeva, I.A.; Birin, K.P.; Chassagnon, R.; Bessmertnykh-Lemeune, A. Hybrid materials based on imidazo[4,5-b]porphyrins for catalytic oxidation of sulfides. Catalysts 2023, 13, 402. [Google Scholar] [CrossRef]
- Hoffmann, N. Photochemical reactions as key steps in organic synthesis. Chem. Rev. 2008, 108, 1052–1103. [Google Scholar] [CrossRef] [PubMed]
- Prier, C.K.; Rankic, D.A.; MacMillan, D.W.C. Visible light photoredox catalysis with transition metal complexes: Applications in organic synthesis. Chem. Rev. 2013, 113, 5322–5363. [Google Scholar] [CrossRef] [PubMed]
- Schultz, D.M.; Yoon, T.P. Solar synthesis: Prospects in visible light photocatalysis. Science 2014, 343, 1239176. [Google Scholar] [CrossRef] [PubMed]
- Ghogare, A.A.; Greer, A. Using singlet oxygen to synthesize natural products and drugs. Chem. Rev. 2016, 116, 9994–10034. [Google Scholar] [CrossRef]
- Lu, W.; Zhou, L. Oxidation of C–H Bonds; John Wiley & Sons, Inc.: Oxford, UK, 2017; p. 496. [Google Scholar]
- Clerici, M.G.; Kholdeeva, O.A. (Eds.) Liquid Phase Oxidation via Heterogeneous Catalysis: Organic Synthesis and Industrial Applications; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013; p. 546. [Google Scholar]
- Teles, J.H.; Hermans, I.; Franz, G.; Sheldon, R.A. Oxidation. In Ullmann’s Encyclopedia of Industrial Chemistry; Willey-VCH: Hoboken, NJ, USA, 2015. [Google Scholar] [CrossRef]
- Mejía, E. (Ed.) Catalytic Aerobic Oxidations; Catalysis Series; The Royal Society of Chemistry: Cambridge, UK, 2020; Volume 39, p. 334. [Google Scholar]
- Wang, D.; Weinstein, A.B.; White, P.B.; Stahl, S.S. Ligand-promoted palladium-catalyzed aerobic oxidation reactions. Chem. Rev. 2018, 118, 2636–2679. [Google Scholar] [CrossRef]
- Strukul, G.; Scarso, A. Environmentally benign oxidants. In Liquid Phase Oxidation via Heterogeneous Catalysis; Clerici, M.G., Kholdeeva, O.A., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013; pp. 1–20. [Google Scholar]
- De Rosa, M.C.; Crutchley, R.J. Photosensitized singlet oxygen and its applications. Coord. Chem. Rev. 2002, 233–234, 351–371. [Google Scholar] [CrossRef]
- Al-Nu’airat, J.; Oluwoye, I.; Zeinali, N.; Altarawneh, M.; Dlugogorski, B.Z. Review of chemical reactivity of singlet oxygen with organic fuels and contaminants. Chem. Rec. 2021, 21, 315–342. [Google Scholar] [CrossRef]
- Quaranta, M.; Borisov, S.M.; Klimant, I. Indicators for optical oxygen sensors. Bioananal. Rev. 2012, 4, 115–157. [Google Scholar] [CrossRef]
- To, W.-P.; Liu, Y.; Lau, T.-C.; Che, C.-M. A robust palladium(II)–porphyrin complex as catalyst for visible light induced oxidative C–H Functionalization. Chem.-Eur. J. 2013, 19, 5654–5664. [Google Scholar] [CrossRef]
- Ciulla, T.A.; Criswell, M.H.; Snyder, W.J.; Small, W. Photodynamic therapy with PhotoPoint photosensitiser MV6401, indium chloride methyl pyropheophorbide, achieves selective closure of rat corneal neovascularisation and rabbit choriocapillaris. Br. J. Ophthalmol. 2005, 89, 113. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zheng, X.; Dobhal, M.P.; Gryshuk, A.; Morgan, J.; Dougherty, T.J.; Oseroff, A.; Pandey, R.K. Methyl pyropheophorbide-a analogues: Potential fluorescent probes for the peripheral-type benzodiazepine receptor. Effect of central metal in photosensitizing efficacy. J. Med. Chem. 2005, 48, 3692–3695. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.A.; Zhang, X.; Reeson, T.C.; Chen, Y.-S.; Zhang, J. Facile control of the charge density and photocatalytic activity of an anionic indium porphyrin framework via in situ metalation. J. Am. Chem. Soc. 2014, 136, 15881–15884. [Google Scholar] [CrossRef]
- Mitrofanov, A.; Brandes, S.; Herbst, F.; Rigolet, S.; Bessmertnykh-Lemeune, A.; Beletskaya, I. Immobilization of copper complexes with (1,10-phenanthrolinyl)phosphonates on titania supports for sustainable catalysis. J. Mater. Chem. A 2017, 5, 12216–12235. [Google Scholar] [CrossRef]
- Kee, H.L.; Bhaumik, J.; Diers, J.R.; Mroz, P.; Hamblin, M.R.; Bocian, D.F.; Lindsey, J.S.; Holten, D. Photophysical characterization of imidazolium-substituted Pd(II), In(III), and Zn(II) porphyrins as photosensitizers for photodynamic therapy. Photochem. Photobiol. A 2008, 200, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Gouterman, M. Spectra of porphyrins. J. Mol. Spectrosc. 1961, 6, 138–163. [Google Scholar] [CrossRef]
- Teixeira, R.; Serra, V.V.; Botequim, D.; Paulo, P.M.R.; Andrade, S.M.; Costa, S.M.B. Fluorescence spectroscopy of porphyrins and phthalocyanines: Some insights into supramolecular self-assembly, microencapsulation, and imaging microscopy. Molecules 2021, 26, 4264. [Google Scholar] [CrossRef]
- Wilkinson, F.; Helman, W.P.; Ross, A.B. Quantum yields for the photosensitized formation of the lowest electronically excited singlet state of molecular oxygen in solution. J. Phys. Chem. Ref. Data 1993, 22, 113–262. [Google Scholar] [CrossRef]
- Stingl, K.A.; Tsogoeva, S.B. Recent advances in sulfoxidation reactions: A metal-free approach. Tetrahedron Asymmetry 2010, 21, 1055–1074. [Google Scholar] [CrossRef]
- Liu, Y.; Howarth, A.J.; Hupp, J.T.; Farha, O.K. Selective photooxidation of a mustard-gas simulant catalyzed by a porphyrinic metal–organic framework. Angew. Chem. Int. Ed. 2015, 54, 9001–9005. [Google Scholar] [CrossRef]
- Han, J.; Soloshonok, V.A.; Klika, K.D.; Drabowicz, J.; Wzorek, A. Chiral sulfoxides: Advances in asymmetric synthesis and problems with the accurate determination of the stereochemical outcome. Chem. Soc. Rev. 2018, 47, 1307–1350. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Li, Z.; Bolm, C. Iron(II)-catalyzed direct synthesis of NH sulfoximines from sulfoxides. Angew. Chem. Int. Ed. 2018, 57, 324–327. [Google Scholar] [CrossRef]
- Ahmadian, M.; Anbia, M. Oxidative desulfurization of liquid fuels using polyoxometalate-based catalysts: A review. Energy Fuels 2021, 35, 10347–10373. [Google Scholar] [CrossRef]
- Barona-Castaño, J.C.; Carmona-Vargas, C.C.; Brocksom, T.J.; De Oliveira, K.T. Porphyrins as catalysts in scalable organic reactions. Molecules 2016, 21, 310. [Google Scholar] [CrossRef] [PubMed]
- Baciocchi, E.; Crescenzi, C.; Lanzalunga, O. Photoinduced electron transfer reactions of benzyl phenyl sulfides promoted by 9,10-dicyanoanthracene. Tetrahedron 1997, 53, 4469–4478. [Google Scholar] [CrossRef]
- Baciocchi, E.; Del Giacco, T.; Lanzalunga, O.; Mencarelli, P.; Procacci, B. Photosensitized oxidation of alkyl phenyl sulfoxides. C−S bond cleavage in alkyl phenyl sulfoxide radical cations. J. Org. Chem. 2008, 73, 5675–5682. [Google Scholar] [CrossRef]
- Kupwade, R.V. A Concise review on synthesis of sulfoxides and sulfones with special reference to oxidation of sulfides. J. Chem. Rev. 2019, 1, 99–113. [Google Scholar]
- Matavos-Aramyan, S.; Soukhakian, S.; Jazebizadeh, M.H. Selected methods for the synthesis of sulfoxides and sulfones with emphasis on oxidative protocols. Phosphorus Sulfur Silicon Relat. Elem. 2020, 195, 181–193. [Google Scholar] [CrossRef]
- Caron, S.; Dugger, R.W.; Ruggeri, S.G.; Ragan, J.A.; Ripin, D.H.B. Large-scale oxidations in the pharmaceutical industry. Chem. Rev. 2006, 106, 2943–2989. [Google Scholar] [CrossRef]
- Skolia, E.; Gkizis, P.L.; Kokotos, C.G. Aerobic photocatalysis: Oxidation of sulfides to sulfoxides. ChemPlusChem 2022, 87, e202200008. [Google Scholar] [CrossRef]
- Hikita, T.; Tamaru, K.; Yamagishi, A.; Iwamoto, T. Production of an optically active sulfoxide by use of colloidally dispersed. Λ-tris(2,2′-bipyridyl)ruthenium(II) montmorillonite as a photosensitizer. Inorg. Chem. 1989, 28, 2221–2223. [Google Scholar] [CrossRef]
- Baciocchi, E.; Giacco, T.D.; Elisei, F.; Gerini, M.F.; Guerra, M.; Lapi, A.; Liberali, P. Electron transfer and singlet oxygen mechanisms in the photooxygenation of dibutyl sulfide and thioanisole in MeCN sensitized by N-methylquinolinium tetrafluoborate and 9,10-dicyanoanthracene. The probable involvement of a thiadioxirane intermediate in electron transfer photooxygenations. J. Am. Chem. Soc. 2003, 125, 16444–16454. [Google Scholar] [PubMed]
- Bonesi, S.M.; Manet, I.; Freccero, M.; Fagnoni, M.; Albini, A. Photosensitized oxidation of sulfides: Discriminating between the singlet-oxygen mechanism and electron transfer involving superoxide anion or molecular oxygen. Chem.-Eur. J. 2006, 12, 4844–4857. [Google Scholar] [CrossRef]
- Clennan, E.L. Persulfoxide: Key intermediate in reactions of singlet oxygen with sulfides. Acc. Chem. Res. 2001, 34, 875–884. [Google Scholar] [CrossRef]
- Clennan, E.L.; Greer, A. Effect of alcohols on the photooxidative behavior of diethyl sulfide. J. Org. Chem. 1996, 61, 4793–4797. [Google Scholar] [CrossRef]
- Bonesi, S.M.; Albini, A. Effect of protic cosolvents on the photooxygenation of diethyl sulfide. J. Org. Chem. 2000, 65, 4532–4536. [Google Scholar] [CrossRef]
- Abdulaeva, I.A. Functionalised Imidazoporphyrins and Their Application in Catalysis. Ph.D. Thesis, Université de Bourgogne, Dijon, France, Lomonosov Moscow State University, Moscow, Russia, 2017. [Google Scholar]
- Neveselý, T.; Svobodová, E.; Chudoba, J.; Sikorski, M.; Cibulka, R. Efficient metal-free aerobic photooxidation of sulfides to sulfoxides mediated by a vitamin B2 derivative and visible light. Adv. Synth. Catal. 2016, 358, 1654–1663. [Google Scholar] [CrossRef]
- Bonesi, S.M.; Fagnoni, M.; Monti, S.; Albini, A. Photosensitized oxidation of phenyl and tert-butyl sulfides. Photochem. Photobiol. Sci. 2004, 3, 489–493. [Google Scholar] [CrossRef]
- Ashford, D.L.; Gish, M.K.; Vannucci, A.K.; Brennaman, M.K.; Templeton, J.L.; Papanikolas, J.M.; Meyer, T.J. Molecular chromophore–catalyst assemblies for solar fuel applications. Chem. Rev. 2015, 115, 13006–13049. [Google Scholar] [CrossRef]
- Queffélec, C.; Petit, M.; Janvier, P.; Knight, D.A.; Bujoli, B. Surface modification using phosphonic acids and esters. Chem. Rev. 2012, 112, 3777–3807. [Google Scholar] [CrossRef]
- Guerrero, G.; Alauzun, J.G.; Granier, M.; Laurencin, D.; Mutin, P.H. Phosphonate coupling molecules for the control of surface/interface properties and the synthesis of nanomaterials. Dalton Trans. 2013, 42, 12569–12585. [Google Scholar] [CrossRef] [PubMed]
- DiGiacomo, P.M.; Dines, M.B. Nonaqueous Preparation of Layered or Amorphous Organometallic Inorganic Polymers. U.S. Patent No. 4,299,943, 10 November 1981. [Google Scholar]
- Guerrero, G.; Mutin, P.H.; Vioux, A. Anchoring of phosphonate and phosphinate coupling molecules on titania particles. Chem. Mater. 2001, 13, 4367–4373. [Google Scholar] [CrossRef]
- Kamm, J.M.; Iverson, C.P.; Lau, W.-Y.; Hopkins, M.D. Axial ligand effects on the structures of self-assembled gallium–porphyrin monolayers on highly oriented pyrolytic graphite. Langmuir 2016, 32, 487–495. [Google Scholar] [CrossRef]
- Enakieva, Y.Y.; Volostnykh, M.V.; Nefedov, S.E.; Kirakosyan, G.A.; Gorbunova, Y.G.; Tsivadze, A.Y.; Bessmertnykh-Lemeune, A.G.; Stern, C.; Guilard, R. Gallium(III) and indium(III) complexes with meso-monophosphorylated porphyrins: Synthesis and structure. A first example of dimers formed by the self-assembly of meso-porphyrinylphosphonic acid monoester. Inorg. Chem. 2017, 56, 3055–3070. [Google Scholar] [CrossRef]
- Seybold, P.G.; Gouterman, M. Porphyrins: XIII: Fluorescence spectra and quantum yields. J. Mol. Spectrosc. 1969, 31, 1–10. [Google Scholar] [CrossRef]
- Levitus, M. Tutorial: Measurement of fluorescence spectra and determination of relative fluorescence quantum yields of transparent samples. Methods Appl. Fluoresc. 2020, 8, 033001. [Google Scholar] [CrossRef]
- Gou, L.; Coretsopoulos, C.N.; Scranton, A.B. Measurement of the dissolved oxygen concentration in acrylate monomers with a novel photochemical method. J. Polym. Sci. Part A Polym. Chem. 2004, 42, 1285–1292. [Google Scholar] [CrossRef]
- Hosseini-Eshbala, F.; Sedrpoushan, A.; Dehdashti, M.N.; Breit, B.; Mohanazadeh, F.; Veisi, H. Needle ball-like nanostructured mixed Cu–Ni–Co oxides: Synthesis, characterization and application to the selective oxidation of sulfides to sulfoxides. Mater. Sci. Eng. Proc. Conf. 2019, 103, 109814. [Google Scholar] [CrossRef]
- Gao, Y.; Lam, Y. Polymer-supported N-phenylsulfonyloxaziridine (Davis reagent): A versatile oxidant. Adv. Synth. Catal. 2008, 350, 2937–2946. [Google Scholar] [CrossRef]
- Amri, N.; Wirth, T. Flow electrosynthesis of sulfoxides, sulfones, and sulfoximines without supporting electrolytes. J. Org. Chem. 2021, 86, 15961–15972. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
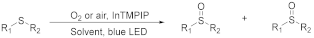 | ||||||||
| Entry | Sulfide | Photocatalyst (mol%) | Solvent | Reaction Conditions 1 | Time (h) | Conversion2 (%) | Yield 2 (%) | |
|---|---|---|---|---|---|---|---|---|
| Sulfoxide | Sulfone | |||||||
| 1 2 3 4 5 6 7 | 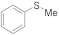 | 0.05 0.005 0.005 0.1 0.0005 0.005 0.0005 | MeCN/H2O MeCN/H2O MeOH/CHCl3 MeOH/CHCl3 MeOH/CHCl3 MeOH/CHCl3 MeOH/CHCl3 | A A A B B C C | 1 1 1 5 24 2 7 2 | 100 100 100 100 97 38 50 43 45 | 98 98 99 92 97 100 100 100 100 | 2 2 1 8 3 0 0 0 0 |
| 8 9 | 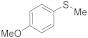 | 0.005 0.0005 | MeCN/H2O MeOH/CHCl3 | A B | 1 24 48 | 100 66 100 | 98 95 91 | 2 5 9 |
| 10 11 12 13 | 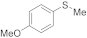 | 0.005 0.005 0.005 0.0005 | MeCN/H2O MeOH/CHCl3 MeOH/CHCl3 MeOH/CHCl3 | A A B B | 1 1 24 24 48 | 100 100 100 57 100 | 99.5 97 97 96 97 | 0.5 3 3 4 3 |
| 14 |  | 0.005 | MeCN/H2O | A | 1 | 100 | 98 | 2 |
| 15 16 17 | 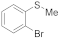 | 0.005 0.005 0.0005 | MeCN/H2O MeOH/CHCl3 MeOH/CHCl3 | A B B | 1 24 24 48 | 100 100 23 55 | 100 100 100 100 | 0 0 0 0 |
| 18 19 20 21 | 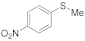 | 0.005 0.05 0.005 0.05 | MeCN/H2O MeCN/H2O MeOH/CHCl3 MeOH/CHCl3 | A A C B | 20 12 20 20 24 | 40 55 100 68 3 93 | 100 100 98 37 4 100 | 0 0 2 0 0 |
| 22 23 | 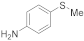 | 0.005 0.005 | MeCN/H2O MeOH/CHCl3 | A A | 5 10 5 | 45 77 100 | 100 100 100 | 0 0 0 |
| 24 25 26 | 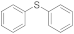 | 0.005 0.005 0.1 | MeCN/H2O MeOH/CHCl3 MeOH/CHCl3 | A A B | 3 6 3 16 24 | 53 100 63 82 24 | 98 98 93 | 2 2 7 |
| 27 28 | 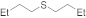 | 0.005 0.005 | MeCN/H2O MeOH/CHCl3 | A B | 1 24 | 100 100 | 95 98 | 5 2 |
| 29 30 | 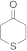 | 0.005 0.005 | MeCN/H2O MeOH/CHCl3 | A B | 1 24 | 100 83 | 100 98 | 0 2 |
| 31 32 | 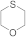 | 0.0005 0.00005 | MeOH/CHCl3 MeOH/CHCl3 | B B | 1 24 | 100 100 | 100 98 | 0 2 |
| 33 | 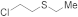 | 0.00005 | MeOH/CHCl3 | B | 48 | 100 | 91 | 9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abdulaeva, I.A.; Filatov, M.A.; Kechiche, A.; Bessmertnykh-Lemeune, A. Indium Imidazo[4,5,-b]porphyrins as Photocatalysts for Oxidation of Sulfides. Molecules 2025, 30, 864. https://doi.org/10.3390/molecules30040864
Abdulaeva IA, Filatov MA, Kechiche A, Bessmertnykh-Lemeune A. Indium Imidazo[4,5,-b]porphyrins as Photocatalysts for Oxidation of Sulfides. Molecules. 2025; 30(4):864. https://doi.org/10.3390/molecules30040864
Chicago/Turabian StyleAbdulaeva, Inna A., Mikhail A. Filatov, Azhar Kechiche, and Alla Bessmertnykh-Lemeune. 2025. "Indium Imidazo[4,5,-b]porphyrins as Photocatalysts for Oxidation of Sulfides" Molecules 30, no. 4: 864. https://doi.org/10.3390/molecules30040864
APA StyleAbdulaeva, I. A., Filatov, M. A., Kechiche, A., & Bessmertnykh-Lemeune, A. (2025). Indium Imidazo[4,5,-b]porphyrins as Photocatalysts for Oxidation of Sulfides. Molecules, 30(4), 864. https://doi.org/10.3390/molecules30040864