
Understanding the Role of Active Lattice Oxygen in CO Oxidation Catalyzed by Copper-Doped Mn2O3@MnO2

Abstract

1. Introduction
2. Results and Discussion
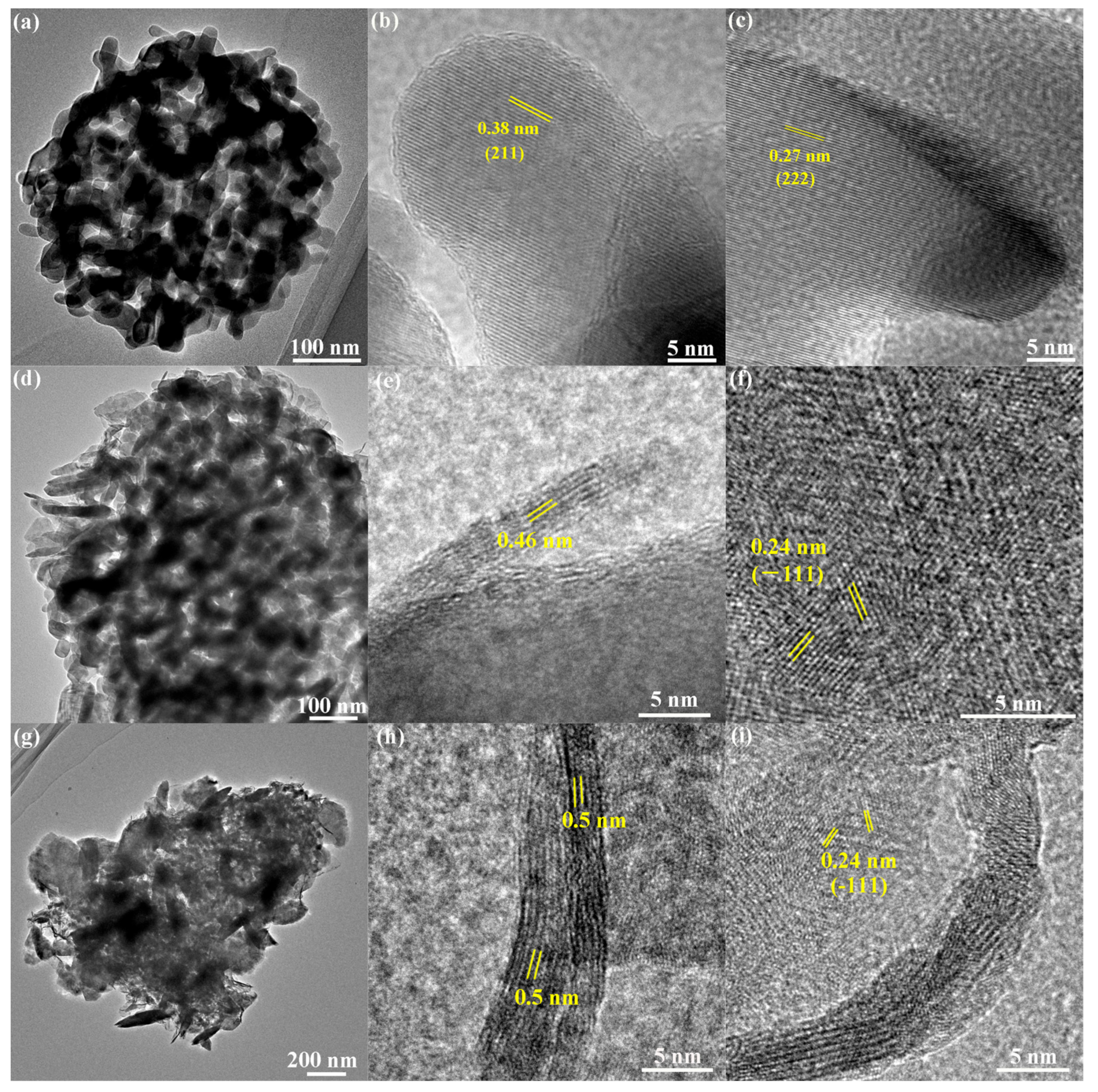
2.1. Crystal Structure and Morphology
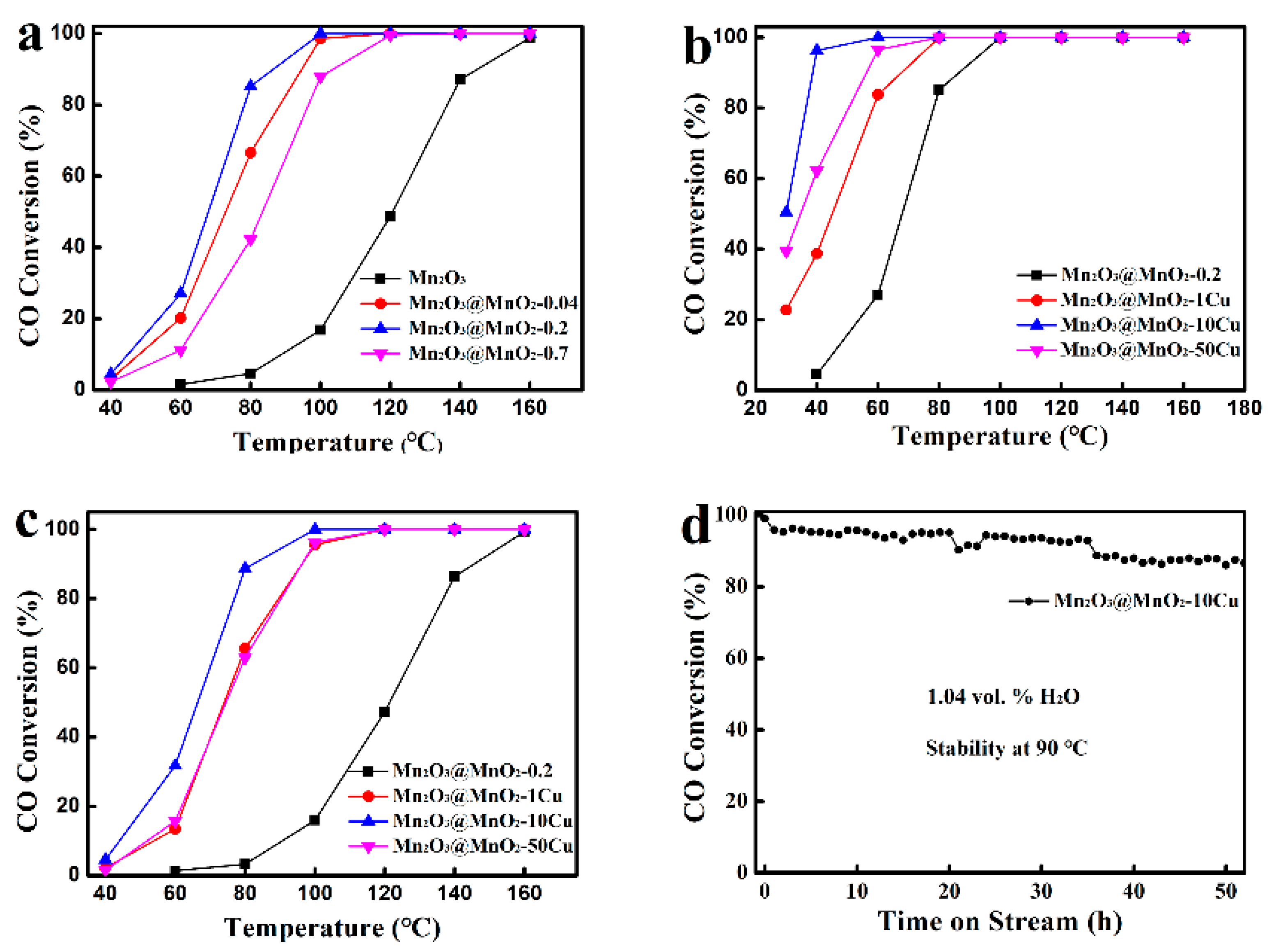
2.2. Catalytic Activity for CO Oxidation
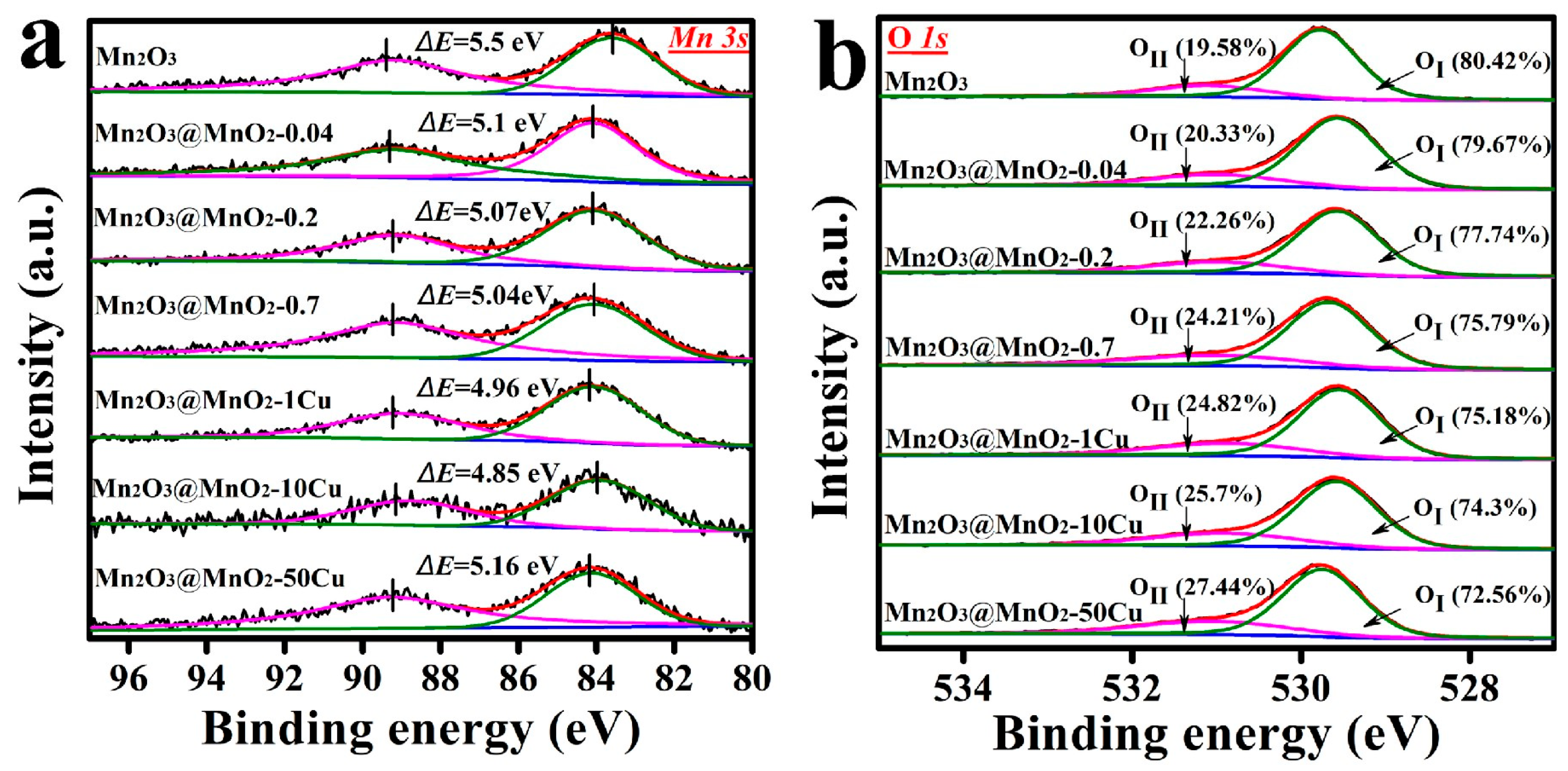
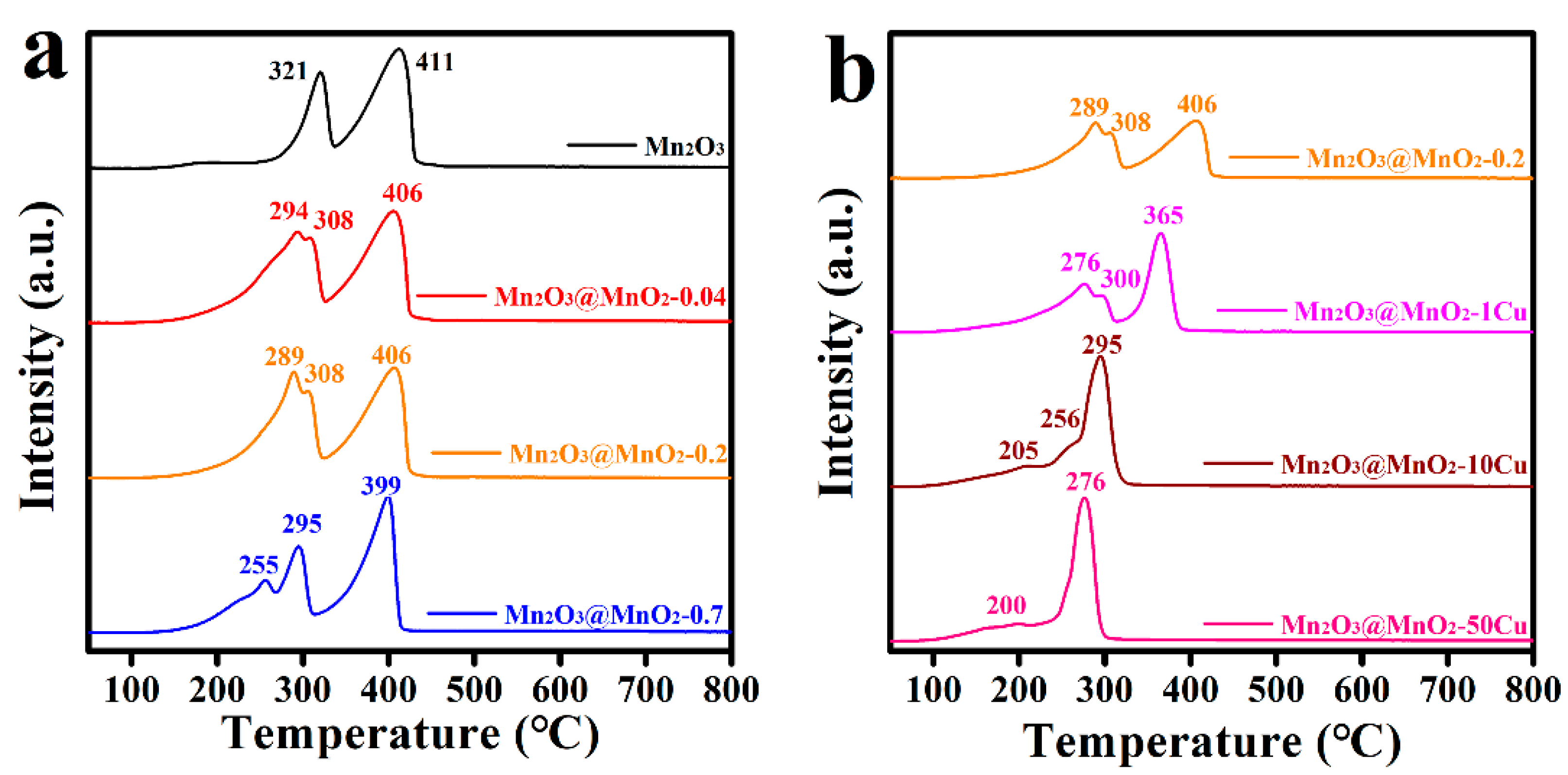
2.3. Physicochemical Properties of As-Prepared Catalysts
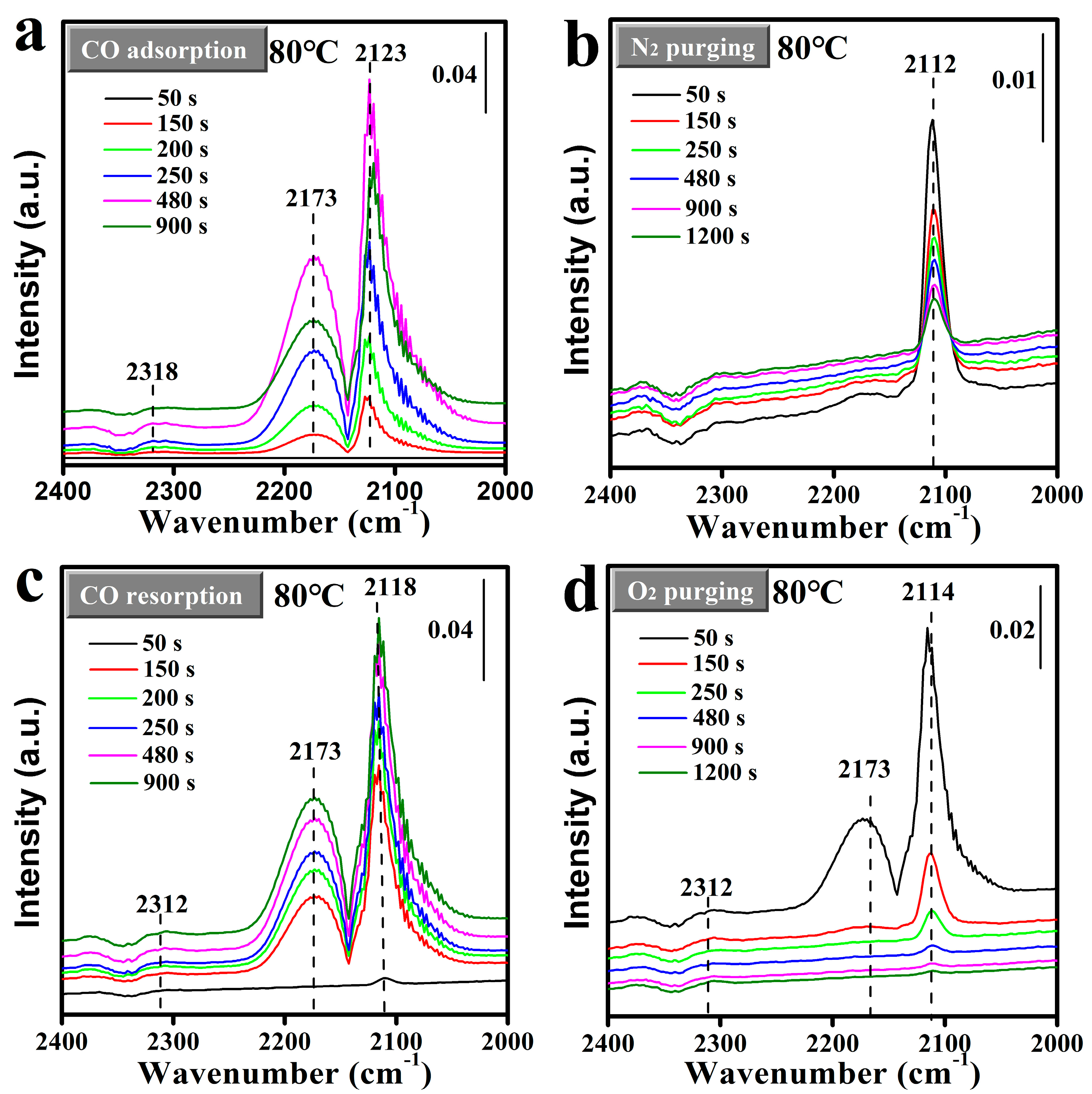
2.4. Analysis of Surface Reaction Process
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. Catalyst Preparation
3.2.1. Mn2O3 Nanosphere
3.2.2. Mn2O3@MnO2
3.2.3. Cu-Doped Mn2O3@MnO2
3.3. Materials Characterization and Catalytic Activity Tests
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pope, D.P.; Mishra, V.; Thompson, L.; Siddiqui, A.R.; Rehfuess, E.A.; Weber, M.; Bruce, N.G. Risk of low birth weight and stillbirth associated with indoor air pollution from solid fuel use in developing countries. Epidemiol. Rev. 2010, 32, 70–81. [Google Scholar] [CrossRef] [PubMed]
- Saavedra, J.; Doan, H.; Pursell, C.; Grabow, L.; Chandler, B. The critical role of water at the gold-titania interface in catalytic CO oxidation. Science 2014, 345, 6204. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Li, X.M.; Lai, C.Y.; Zhang, Y.P.; Lin, X.; Ding, S.P. Recent advances in noble metal-based catalysts for CO oxidation. RSC Adv. 2024, 14, 30566. [Google Scholar] [CrossRef] [PubMed]
- Soubaihi, R.M.A.; Saoud, K.M.; Dutta, J. Low-temperature CO oxidation by silver nanoparticles in silica aerogel mesoreactors. Chem. Eng. J. 2023, 455, 140576. [Google Scholar] [CrossRef]
- Wu, C.H.; Liu, C.; Su, D.; Xin, H.L.; Fang, H.T.; Eren, B.; Zhang, S.; Murray, C.B.; Salmeron, M.B. Bimetallic synergy in cobalt–palladium nanocatalysts for CO oxidation. Nat. Catal. 2018, 2, 78–85. [Google Scholar] [CrossRef]
- Zhang, J.Y.; Shu, M.; Niu, Y.X.; Yi, L.; Yi, H.H.; Zhou, Y.S.; Zhao, S.Z.; Tang, X.L.; Gao, F.Y. Advances in CO catalytic oxidation on typical noble metal catalysts: Mechanism, performance and optimization. Chem. Eng. J. 2024, 495, 153523. [Google Scholar] [CrossRef]
- Zhang, Y.N.; Qin, G.Z.; Zheng, J.F.; Li, Y.F.; Huang, Z.G.; Han, X.J. Promotion effect of CO oxidation via activation of surface lattice oxygen by single atom Cu/MnO2 catalyst. Mol. Catal. 2023, 540, 113057. [Google Scholar] [CrossRef]
- Dasireddy, V.D.; Likozar, B. Cu-Mn-O nano-particle/nano-sheet spinel-type materials as catalysts in methanol steam reforming (MSR) and preferential oxidation (PROX) reaction for purified hydrogen production. Renew. Energy 2022, 182, 713–724. [Google Scholar] [CrossRef]
- Ma, Y.M.; Li, Y.H.; Liang, P.Y.; Min, X.B.; Sun, T.J. SnO2-modiffed CuMnOx catalysts for humid CO oxidation at low temperature. J. Ind. Eng. Chem. 2024, 138, 300–310. [Google Scholar] [CrossRef]
- He, Z.Y.; Zheng, B. Microscopic investigation of CO oxidation reaction by copper–manganese oxide catalysts. Catal. Lett. 2025, 155, 4. [Google Scholar] [CrossRef]
- Hu, Z.; Liu, X.; Meng, D.; Guo, Y.; Guo, Y.; Lu, G. Effect of ceria crystal plane on the physicochemical and catalytic properties of Pd/ceria for CO and propane oxidation. ACS Catal. 2016, 6, 2265–2279. [Google Scholar] [CrossRef]
- Xie, X.; Li, Y.; Liu, Z.Q.; Haruta, M.; Shen, W. Low-temperature oxidation of CO catalysed by Co3O4 nanorods. Nature 2009, 458, 746–749. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.; Teng, F.; Bulgan, G.; Zong, R.; Zhu, Y. Effect of phase structure of MnO2 nanorod catalyst on the activity for CO oxidation. J. Phys. Chem. C 2008, 112, 5307–5315. [Google Scholar] [CrossRef]
- Chen, B.; Wu, B.; Yu, L.; Crocker, M.; Shi, C. Investigation into the catalytic roles of various oxygen species over different crystal phases of MnO2 for C6H6 and HCHO Oxidation. ACS Catal. 2020, 10, 6176–6187. [Google Scholar] [CrossRef]
- Song, W.; Chen, J.; Li, Z.; Fang, X. Self-powered MXene/GaN van der waals heterojunction ultraviolet photodiodes with superhigh efficiency and stable current outputs. Adv. Mater. 2021, 33, e2101059. [Google Scholar] [CrossRef]
- Khan, A.; Wang, H.; Liu, Y.; Jawad, A.; Ifthikar, J.; Liao, Z.; Wang, T.; Chen, Z. Highly efficient α-Mn2O3@α-MnO2-500 nanocomposite for peroxymonosulfate activation: Comprehensive investigation of manganese oxides. J. Mater. Chem. A 2018, 6, 1590–1600. [Google Scholar] [CrossRef]
- Zeng, Y.; Meng, Y.; Lai, Z.; Zhang, X.; Yu, M.; Fang, P.; Wu, M.; Tong, Y.; Lu, X. An ultrastable and high-performance flexible fiber-shaped Ni–Zn battery based on a Ni–NiO heterostructured nanosheet cathode. Adv. Mater. 2017, 29, 1702698. [Google Scholar] [CrossRef]
- Chen, J.; Ouyang, W.; Yang, W.; He, J.H.; Fang, X. Recent progress of heterojunction ultraviolet photodetectors: Materials, integrations, and applications. Adv. Funct. Mater. 2020, 30, 1909909. [Google Scholar] [CrossRef]
- Yang, W.; Peng, Y.; Wang, Y.; Wang, Y.; Liu, H.; Su, Z.A.; Yang, W.; Chen, J.; Si, W.; Li, J. Controllable redox-induced in-situ growth of MnO2 over Mn2O3 for toluene oxidation: Active heterostructure interfaces. Appl. Catal. B Environ. 2020, 278, 119279. [Google Scholar] [CrossRef]
- Yang, R.; Fan, Y.; Ye, R.; Tang, Y.; Cao, X.; Yin, Z.; Zeng, Z. MnO2-based materials for environmental applications. Adv. Mater. 2021, 33, e2004862. [Google Scholar] [CrossRef]
- An, K.; Alayoglu, S.; Musselwhite, N.; Plamthottam, S.; Melaet, G.; Lindeman, A.E.; Somorjai, G.A. Enhanced CO oxidation rates at the interface of mesoporous oxides and Pt nanoparticles. J. Am. Chem. Soc. 2013, 135, 16689–16696. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.; Yu, X.; Zhou, Y.; Miao, S.; Li, Y.; Kuld, S.; Sehested, J.; Liu, J.; Aoki, T.; Hong, S.; et al. Structure of the catalytically active copper–ceria interfacial perimeter. Nat. Catal. 2019, 2, 334–341. [Google Scholar] [CrossRef]
- May, Y.A.; Wei, S.; Yu, W.Z.; Wang, W.W.; Jia, C.J. Highly efficient CuO/α-MnO2 catalyst for low-temperature CO oxidation. Langmuir 2020, 36, 11196–11206. [Google Scholar] [CrossRef]
- Elmhamdi, A.; Pascual, L.; Retuerto, M.; Martínez-Arias, A. Catalysts of ceria supported on copper-chromium oxide: Ceria promotion of the CO-PROX activity. Int. J. Hydrog. Energ. 2021, 46, 38712–38723. [Google Scholar] [CrossRef]
- Chen, C.S.; You, J.H.; Lin, J.H.; Chen, Y.Y. Effect of highly dispersed active sites of Cu/TiO2 catalyst on CO oxidation. Catal. Commun. 2008, 9, 2381–2385. [Google Scholar] [CrossRef]
- Chen, C.S.; Chen, T.C.; Chen, C.C.; Lai, Y.T.; You, J.H.; Chou, T.M.; Chen, C.H.; Lee, J.F. Effect of Ti3+ on TiO2-supported Cu catalysts used for CO oxidation. Langmuir 2012, 28, 9996–10006. [Google Scholar] [CrossRef]
- Polster, C.S.; Nair, H.; Baertsch, C.D. Study of active sites and mechanism responsible for highly selective CO oxidation in H2 rich atmospheres on a mixed Cu and Ce oxide catalyst. J. Catal. 2009, 266, 308–319. [Google Scholar] [CrossRef]
- Pakharukova, V.P.; Moroz, E.M.; Zyuzin, D.A.; Ishchenko, A.V.; Dolgikh, L.Y.; Strizhak, P.E. Structure of copper oxide species supported on monoclinic zirconia. J. Phys. Chem. C 2015, 119, 28828–28835. [Google Scholar] [CrossRef]
- Wang, W.W.; Du, P.P.; Zou, S.H.; He, H.Y.; Wang, R.X.; Jin, Z.; Shi, S.; Huang, Y.Y.; Si, R.; Song, Q.S.; et al. Highly dispersed copper oxide clusters as active species in copper-ceria catalyst for preferential oxidation of carbon monoxide. ACS Catal. 2015, 5, 2088–2099. [Google Scholar] [CrossRef]
- Rong, S.; He, T.; Zhang, P. Self-assembly of MnO2 nanostructures into high purity three-dimensional framework for high efficiency formaldehyde mineralization. Appl. Catal. B Environ. 2020, 267, 118375. [Google Scholar] [CrossRef]
- Liu, Y.; Zhou, H.; Cao, R.; Liu, X.; Zhang, P.; Zhan, J.; Liu, L. Facile and green synthetic strategy of birnessite-type MnO2 with high efficiency for airborne benzene removal at low temperatures. Appl. Catal. B Environ. 2019, 245, 569–582. [Google Scholar] [CrossRef]
- Zhu, L.; Wang, J.; Rong, S.; Wang, H.; Zhang, P. Cerium modified birnessite-type MnO2 for gaseous formaldehyde oxidation at low temperature. Appl. Catal. B Environ. 2017, 211, 212–221. [Google Scholar] [CrossRef]
- Cao, R.; Zhang, P.; Liu, Y.; Zheng, X. Ammonium-treated birnessite-type MnO2 to increase oxygen vacancies and surface acidity for stably decomposing ozone in humid condition. Appl. Surf. Sci. 2019, 495, 143607. [Google Scholar] [CrossRef]
- Rong, S.; Li, K.; Zhang, P.; Liu, F.; Zhang, J. Potassium associated manganese vacancy in birnessite-type manganese dioxide for airborne formaldehyde oxidation. Catal. Sci. Technol. 2018, 8, 1799–1812. [Google Scholar] [CrossRef]
- Wang, J.; Li, J.; Zhang, P.; Zhang, G. Understanding the “seesaw effect” of interlayered K+ with different structure in manganese oxides for the enhanced formaldehyde oxidation. Appl. Catal. B Environ. 2018, 224, 863–870. [Google Scholar] [CrossRef]
- Aschauer, U.; Chen, J.; Selloni, A. Peroxide and superoxide states of adsorbed O2 on anatase TiO2 (101) with subsurface defects. Phys. Chem. Chem. Phys. 2010, 12, 12956–12960. [Google Scholar] [CrossRef]
- Feng, N.; Zhu, Z.; Zhao, P.; Wang, L.; Wan, H.; Guan, G. Facile fabrication of trepang-like CeO2@MnO2 nanocomposite with high catalytic activity for soot removal. Appl. Surf. Sci. 2020, 515, 146013. [Google Scholar] [CrossRef]
- Mo, S.; Zhang, Q.; Li, J.; Sun, Y.; Ren, Q.; Zou, S.; Zhang, Q.; Lu, J.; Fu, M.; Mo, D.; et al. Highly efficient mesoporous MnO2 catalysts for the total toluene oxidation: Oxygen-vacancy defect engineering and involved intermediates using in situ DRIFTS. Appl. Catal. B Environ. 2020, 264, 118464. [Google Scholar] [CrossRef]
- Wang, Z.; Chen, B.; Crocker, M.; Yu, L.; Shi, C. New insights into alkaline metal modified CoMn-oxide catalysts for formaldehyde oxidation at low temperatures. Appl. Catal. A Gen. 2020, 596, 117512. [Google Scholar] [CrossRef]
- Yang, Y.J.; Zhang, P.Y.; Jia, J.B. Vanadium-doped MnO2 for efficient room-temperature catalytic decomposition of ozone in air. Appl. Surf. Sci. 2019, 484, 45–53. [Google Scholar] [CrossRef]
- Zhang, H.; Li, H.N.; Zhang, P.Y.; Hu, T.X.; Wang, X.J. Highly active copper-intercalated weakly crystallized δ-MnO2 for low-temperature oxidation of CO in dry and humid air. Front. Environ. Sci. Eng. 2024, 18, 62. [Google Scholar] [CrossRef]
- Yang, W.H.; Wang, Y.; Yang, W.N.; Liu, H.; Li, Z.G.; Peng, Y.; Li, J.H. Surface In situ doping modification over Mn2O3 for toluene and propene catalytic oxidation: The effect of isolated Cuδ+ insertion into the mezzanine of surface MnO2 cladding. ACS Appl. Mater. Interfaces 2021, 13, 2753–2764. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.W.; Yu, W.Z.; Du, P.P.; Xu, H.; Jin, Z.; Si, R.; Ma, C.; Shi, S.; Jia, C.J.; Yan, C.H. Crystal plane effect of ceria on supported copper oxide cluster catalyst for CO oxidation: Importance of metal–support interaction. ACS Catal. 2017, 7, 1313–1329. [Google Scholar] [CrossRef]
- Zhang, H.; Sui, S.; Zheng, X.; Cao, R.; Zhang, P. One-pot synthesis of atomically dispersed Pt on MnO2 for efficient catalytic decomposition of toluene at low temperatures. Appl. Catal. B Environ. 2019, 257, 117878. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Mn (ICP, wt%) | Cu (ICP, wt%) | K (ICP, wt%) | BET (m2·g−1) | Pore Diameter (nm) | AOS of Mn | XPSO II/OI |
|---|---|---|---|---|---|---|---|
| Mn2O3 | 73.7 | _ | _ | 51.5 | 4.9/26.4 | 2.763 | 0.24 |
| Mn2O3@MnO2-0.04 | 69.2 | _ | 0.010 | 59.7 | 4.9/26.4 | 3.219 | 0.26 |
| Mn2O3@MnO2-0.2 | 69.45 | _ | 0.057 | 80.2 | 4.9/26.4 | 3.247 | 0.29 |
| Mn2O3@MnO2-0.7 | 71.51 | _ | 0.34 | 110.9 | 4.9/13.9 | 3.281 | 0.32 |
| Mn2O3@MnO2-1Cu | 70.93 | 0.29 | 0.071 | 67.0 | 4.9/26.4 | 3.371 | 0.33 |
| Mn2O3@MnO2-10Cu | 68.03 | 1.33 | 0.066 | 93.0 | 4.9/17.3 | 3.495 | 0.35 |
| Mn2O3@MnO2-50Cu | 68.25 | 2.17 | 0.025 | 107.8 | 4.9/13.9 | 3.146 | 0.38 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, H.; Meng, T.; Zhang, M.; Zhang, P.; Sun, P.; Li, H.; Yu, Y. Understanding the Role of Active Lattice Oxygen in CO Oxidation Catalyzed by Copper-Doped Mn2O3@MnO2. Molecules 2025, 30, 865. https://doi.org/10.3390/molecules30040865
Zhang H, Meng T, Zhang M, Zhang P, Sun P, Li H, Yu Y. Understanding the Role of Active Lattice Oxygen in CO Oxidation Catalyzed by Copper-Doped Mn2O3@MnO2. Molecules. 2025; 30(4):865. https://doi.org/10.3390/molecules30040865
Chicago/Turabian StyleZhang, Hao, Tan Meng, Min Zhang, Pengyi Zhang, Peizhe Sun, Huinan Li, and Yangyang Yu. 2025. "Understanding the Role of Active Lattice Oxygen in CO Oxidation Catalyzed by Copper-Doped Mn2O3@MnO2" Molecules 30, no. 4: 865. https://doi.org/10.3390/molecules30040865
APA StyleZhang, H., Meng, T., Zhang, M., Zhang, P., Sun, P., Li, H., & Yu, Y. (2025). Understanding the Role of Active Lattice Oxygen in CO Oxidation Catalyzed by Copper-Doped Mn2O3@MnO2. Molecules, 30(4), 865. https://doi.org/10.3390/molecules30040865