
Anticancer Potential of Azatetracyclic Derivatives: In Vitro Screening and Selective Cytotoxicity of Azide and Monobrominated Compounds

 ,
,  ,
,  ,
,  , and
, and
Abstract
1. Introduction
2. Results and Discussion
2.1. Anticancer Activity
2.2. In Silico ADME and Toxicity Profile
3. Experimental Section
3.1. Synthetic Procedure
3.2. Biological Testing
Cell Proliferation Assay
3.3. In Silico ADMET Predictions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bencini, A.; Lippolis, V. 1,10-Phenanthroline: A versatile building block for the construction of ligands for various purposes. Coord. Chem. Rev. 2010, 254, 2096–2180. [Google Scholar] [CrossRef]
- Accorsi, G.; Listorti, A.; Yoosaf, K.; Armaroli, N. 1,10-Phenanthrolines: Versatile building blocks for luminescent molecules, materials and metal complexes. Chem. Soc. Rev. 2009, 38, 1690–1700. [Google Scholar] [CrossRef]
- Viganor, L.; Howe, O.; McCarron, P.; McCann, M.; Devereux, M. The Antibacterial Activity of Metal Complexes Containing 1,10-phenanthroline: Potential as Alternative Therapeutics in the Era of Antibiotic Resistance. Curr. Top. Med. Chem. 2017, 17, 1280–1302. [Google Scholar] [CrossRef]
- Yoshikawa, N.; Yamazaki, S.; Kakimoto, Y.; Eguchi, S.; Yokoyama, R.; Kanehisa, N.; Tohnai, N.; Nakata, E.; Takashima, H. Emission properties of 1,10-phenanthroline derivatives induced by protonation of a nitrogen atom. J. Mol. Struct. 2021, 1242, 130728. [Google Scholar] [CrossRef]
- Sall, C.; Yapi, A.-D.; Desbois, N.; Chevalley, S.; Chezal, J.-M.; Tan, K.; Teulade, J.-C.; Valentin, A.; Blache, Y. Design, synthesis, and biological activities of conformationally restricted analogs of primaquine with a 1,10-phenanthroline framework. Bioorg. Med. Chem. Lett. 2008, 18, 4666–4669. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, M.C.; Larsen, A.F.; Abdikadir, F.H.; Ulven, T. Phenanthroline-2,9-bistriazoles as selective G-quadruplex ligands. Eur. J. Med. Chem. 2014, 72, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Wesselinova, D.; Neykov, M.; Kaloyanov, N.; Toshkova, R.; Dimitrov, G. Antitumour activity of novel 1,10-phenanthroline and 5-amino-1,10-phenanthroline derivatives. Eur. J. Med. Chem. 2009, 44, 2720–2723. [Google Scholar] [CrossRef]
- Leontie, L.; Druta, I.; Rotaru, A.; Apetroaei, N.; Rusu, G.I. Electronic transport properties of some new monoquaternary salts of 4,4′-bipyridine in thin films. Synth. Met. 2009, 159, 642–648. [Google Scholar] [CrossRef]
- Castedo, L.; Tojo, G. Phenanthrene Alkaloids. In The Alkaloids; Brossi, A., Ed.; Academic Press Inc.: San Diego, CA, USA, 1990; pp. 99–139. [Google Scholar]
- Genès, C.; Lenglet, G.; Depauw, S.; Nhili, R.; Pradoa, S.; David-Cordonnier, M.-H.; Michela, S.; Tillequina, F.; Porée, F.-H. Synthesis and biological evaluation of N-substituted benzo[c]phenanthrolines and benzo[c]phenanthrolinones as antiproliferative agents. Eur. J. Med. Chem. 2011, 46, 2117–2131. [Google Scholar] [CrossRef] [PubMed]
- Al Matarneh, C.M.; Amarandi, R.M.; Craciun, A.M.; Mangalagiu, I.I.; Zbancioc, G.; Danac, R. Design, Synthesis, Molecular Modelling and Anticancer Activities of New Fused Phenanthrolines. Molecules 2020, 25, 527. [Google Scholar] [CrossRef] [PubMed]
- Zbancioc, G.; Mangalagiu, I.I.; Moldoveanu, C. The Effective Synthesis of New Benzoquinoline Derivatives as Small Molecules with Anticancer Activity. Pharmaceuticals 2024, 17, 52. [Google Scholar] [CrossRef] [PubMed]
- Abraham, J.; Staffurth, J. Hormonal therapy for cancer. Medicine 2016, 44, 30–33. [Google Scholar] [CrossRef]
- Esfahani, K.; Roudaia, L.; Buhlaiga, N.; Del Rincon, S.V.; Papneja, N.; Miller, W.H., Jr. A review of cancer immunotherapy: From the past, to the present, to the future. Curr. Oncol. 2020, 27 (Suppl. S2), 87–97. [Google Scholar] [CrossRef] [PubMed]
- Czogała, W.; Czogała, M.; Kwiecinska, K.; Bik-Multanowski, M.; Tomasik, P.; Hałubiec, P.; Łazarczyk, A.; Miklusiak, K.; Skoczen, S. The Expression of Genes Related to Lipid Metabolism and Metabolic Disorders in Children before and after Hematopoietic StemCell Transplantation—A Prospective Observational Study. Cancers 2021, 13, 3614. [Google Scholar] [CrossRef]
- Kinch, M.S. An analysis of FDA–approved drugs for oncology. Drug Discov. Today 2014, 19, 1831–1835. [Google Scholar] [CrossRef] [PubMed]
- Magalhaes, L.G.; Ferreira, L.L.G.; Andricopulo, A.D. Recent Advances and Perspectives in Cancer Drug Design. An. Acad. Bras. Cienc. 2018, 90, 1233–1236. [Google Scholar] [CrossRef]
- Barreca, M.; Spanò, V.; Raimondi, M.V.; Tarantelli, C.; Spriano, F.; Bertoni, F.; Barraja, P.; Montalbano, A. Recurrence of the oxazolemotif in tubulin colchicine site inhibitors with anti-tumor activity. Eur. J Med. Chem. Rep. 2021, 1, 100004. [Google Scholar] [CrossRef]
- Barreca, M.; Spanò, V.; Rocca, R.; Bivacqua, R.; Abel, A.C.; Maruca, A.; Montalbano, A.; Raimondi, M.V.; Tarantelli, C.; Gaudio, E.; et al. Development of [1,2]oxazoloisoindoles tubulin polymerization inhibitors: Further chemical modifications and potential therapeutic effects against lymphomas. Eur. J. Med. Chem. 2022, 243, 114744. [Google Scholar] [CrossRef]
- Vieira, A.M.G.; Silvestre, O.F.; Silva, B.F.B.; Ferreira, C.J.O.; Lopes, I.; Gomes, A.C.; Espiña, B.; Sárria, M.P. pH-sensitive nanoliposomes for passive and CXCR-4-mediated marine yessotoxin delivery for cancer therapy. Nanomedicine 2022, 17, 717–739. [Google Scholar] [CrossRef] [PubMed]
- Shrivastava, N.; Naim, M.J.; Alam, M.J.; Nawaz, F.; Ahmed, S.; Alam, O. Benzimidazole Scaffold as Anticancer Agent: Synthetic Approaches and Structure-Activity Relationship. Arch. Pharm. 2017, 350, e201700040. [Google Scholar] [CrossRef]
- Amin, N.H.; El-Saadi, M.T.; Abdel-Fattah, M.M.; Mohammed, A.A.; Said, E.G. Development of certain aminoquinazoline scaffolds as potential multitarget anticancer agents with apoptotic and anti-proliferative effects: Design, synthesis and biological evaluation. Bioorg. Chem. 2023, 135, 106496. [Google Scholar] [CrossRef]
- Ilakiyalakshmi, M.; Arumugam Napoleon, A.A. Review on recent development of quinoline for anticancer activities. Arab. J. Chem. 2022, 15, 104168. [Google Scholar] [CrossRef]
- Park, G.Y.; Wilson, J.J.; Song, Y.; Lippard, S.J. Phenanthriplatin, a monofunctional DNA–binding platinum anticancer drugcandidate with unusual potency and cellular activity profile. Proc. Natl. Acad. Sci. USA 2012, 109, 11987–11992. [Google Scholar] [CrossRef]
- Zbancioc, G.; Ciobanu, C.-I.; Mangalagiu, I.I.; Moldoveanu, C. Ultrasound-Assisted Synthesis of Fluorescent Azatetracyclic Derivatives: An Energy-Efficient Approach. Molecules 2022, 27, 3180. [Google Scholar] [CrossRef] [PubMed]
- Shoemaker, H.R. The NCI60 human tumor cell line anticancer drug screen. Nat. Rev. Cancer 2006, 6, 813–823. [Google Scholar] [CrossRef]
- Skehan, P.; Storeng, R.; Scudiero, D.; Monks, A.; McMahon, J.; Vistica, D.; Warren, J.T.; Bokesch, H.; Kenney, S.; Boyd, M.R. Newcolorimetric cytotoxicity assay for anticancer–drug screening. J. Natl. Cancer Inst. 1990, 82, 1107–1112. [Google Scholar] [CrossRef] [PubMed]
- Boyd, R.B. The NCI In Vitro Anticancer Drug Discovery Screen. In Anticancer Drug Development Guide; Teicher, B.A., Ed.; Cancer Drug Discovery and Development; Humana Press: Totowa, NJ, USA, 1997; pp. 23–42. [Google Scholar]
- The COMPARE Program. Available online: https://dtp.cancer.gov/databases_tools/compare.htm (accessed on 12 January 2025).
- Development. Available online: https://next.cancer.gov/developmentResources/default.htm (accessed on 22 October 2024).
- Cell Lines in the In Vitro Screen. Available online: https://dtp.cancer.gov/discovery_development/nci-60/cell_list.htm (accessed on 14 November 2024).
- The Standard NCI/DTP Methodology of the In Vitro Cancer Screen. Available online: https://dtp.cancer.gov/discovery_development/nci-60/methodology.htm (accessed on 23 October 2024).
- Cruz, M.; Duarte-Rodrigues, J.; Campelo, M. Cardiotoxicity in anthracycline therapy: Prevention strategies. Rev. Port. Cardiol. 2016, 35, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Chandra Tjin, C.; Gao, X.; Xue, Y.; Jiao, H.; Zhang, R.; Wu, M.; He, Z.; Ellman, J.; Ha, Y. Pharmacological inhibition of PI5P4Kα/β disrupts cell energy metabolism and selectively kills p53-null tumor cells. Proc. Natl. Acad. Sci. USA 2021, 118, e2002486118. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Lagunin, A.A.; Dubovskaja, V.I.; Rudik, A.V.; Pogodin, P.V.; Druzhilovskiy, D.S.; Gloriozova, T.A.; Filimonov, D.A.; Sastry, N.G.; Poroikov, V.V. CLC-Pred: A freely available web-service for in silico prediction of human cell line cytotoxicity for drug-like compounds. PLoS ONE 2018, 12, e0191838. [Google Scholar] [CrossRef]
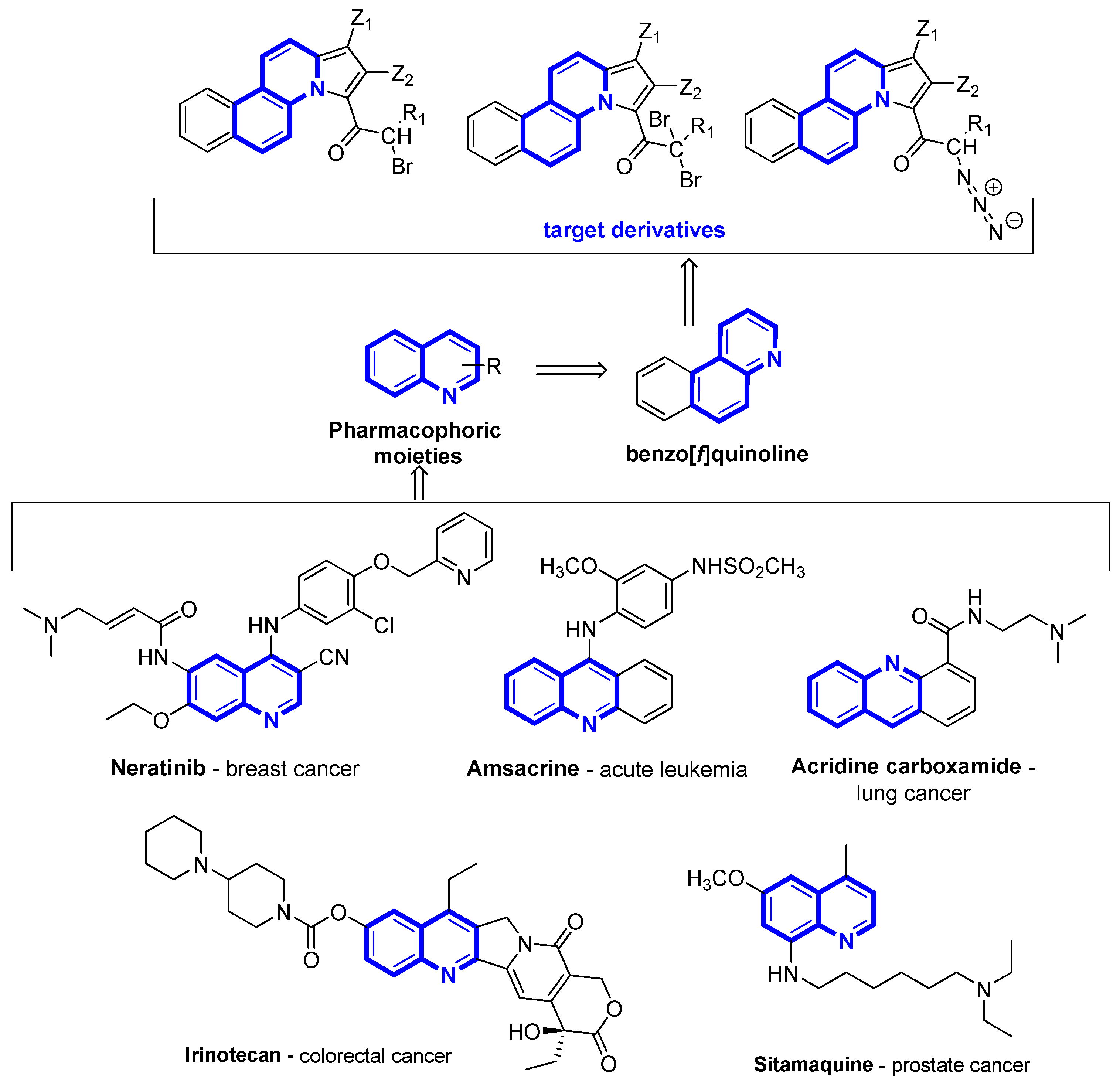
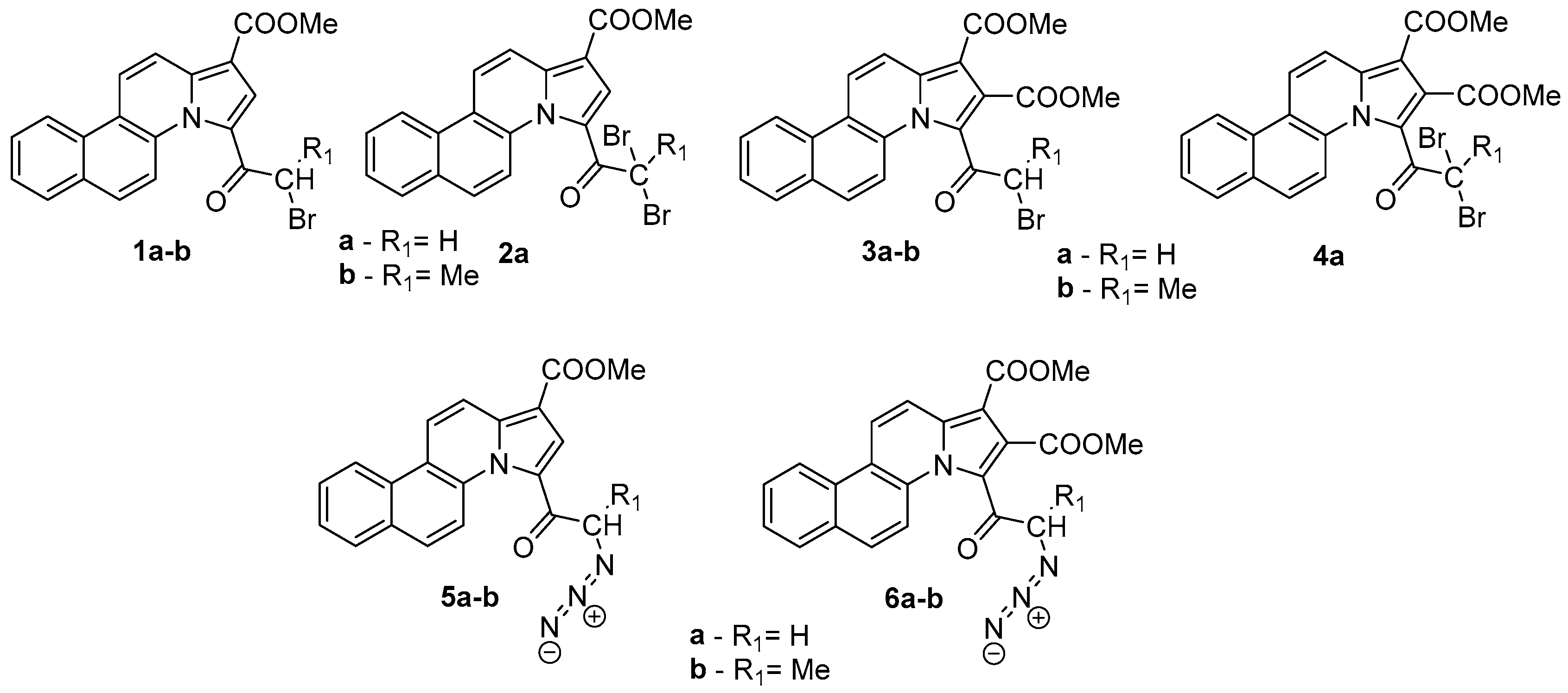
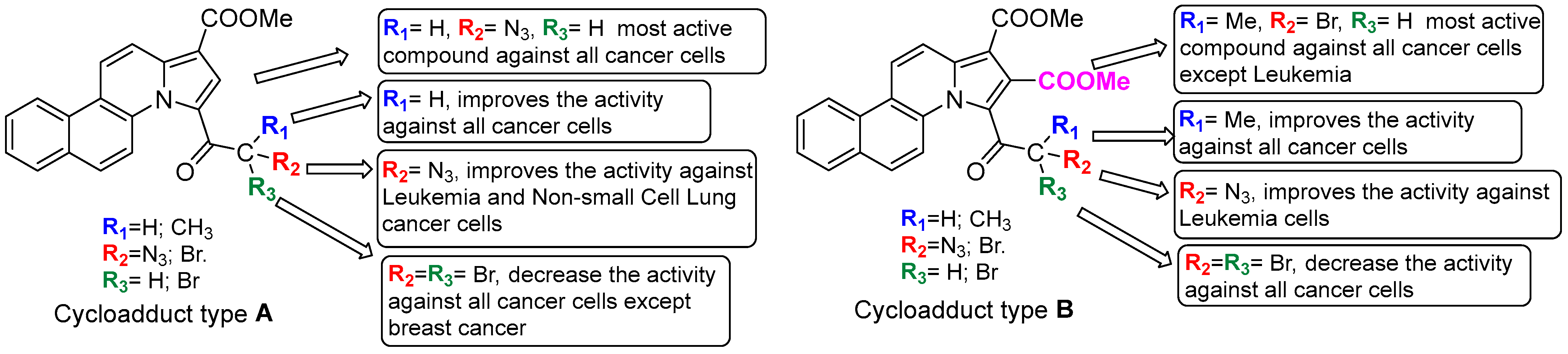


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Type | Compound/Percentage Growth Inhibition (PGI%) a | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1a | 1b | 2a | 3a | 3b | 4a | 5a | 5b | 6a | 6b | |
| Leukemia | ||||||||||
| CCRF–CEM | 87 | 65 | 65 | 28 | 100 (4) b | 42 | 100 (89) b | 59 | 68 | 100 (19) b |
| HL–60 (TB) | 51 | 18 | 24 | 7 | 61 | 10 | 100 (98) b | 100 (38) b | 33 | 66 |
| K–562 | 58 | 33 | 48 | 36 | 96 | 44 | 0 | 52 | 35 | 66 |
| MOLT–4 | 94 | 68 | 78 | 45 | 99 | 35 | 100 (99) b | 100 (96) b | 88 | 100 (93) b |
| RPMI–8226 | 62 | 29 | 46 | 31 | 97 | 29 | 100 (87) b | 84 | 55 | 100 (30) b |
| SR | 98 | 87 | 87 | 44 | 97 | 65 | - | 100 (59) b | 100 (6) b | 100 (97) b |
| Non–small Cell Lung Cancer | ||||||||||
| A549/ATCC | 69 | 46 | 60 | 8 | 50 | 0 | 100 (40) b | 51 | 6 | 0 |
| EKVX | 40 | 14 | 57 | 1 | 14 | 0 | 100 (44) b | 90 | 100 (1) b | 36 |
| HOP–62 | 52 | 25 | 44 | 13 | 33 | 0 | 100 (20) b | - | - | - |
| HOP–92 | 47 | 26 | 46 | 0 | 47 | 0 | 100 (6) b | 56 | 15 | 27 |
| NCI–H226 | 47 | 22 | 36 | 0 | 39 | 0 | 100 (2) b | 100 (5) b | 47 | 80 |
| NCI–H23 | 49 | 17 | 31 | 7 | 67 | 2 | 100 (2) b | 55 | 18 | 35 |
| NCI–H322M | 18 | 6 | 11 | 4 | 17 | 0 | 100 (15) b | 0 | 3 | 21 |
| NCI–460 | 61 | 60 | 82 | 0 | 83 | 0 | 100 (88) b | 77 | 17 | 49 |
| NCI–H522 | 41 | 28 | 40 | 27 | 100 (55) b | 10 | 93 | 46 | 41 | 100 (44) b |
| Colon Cancer | ||||||||||
| COLO 205 | 62 | 0 | 45 | 0 | 57 | 0 | 100 (44) b | 35 | 0 | 10 |
| HCC–2998 | 38 | 0 | 57 | 0 | 33 | 0 | 100 (9) b | 19 | 0 | 5 |
| HCT–116 | 65 | 23 | 51 | 5 | 86 | 9 | 77 | 47 | 19 | 49 |
| HCT–15 | 61 | 39 | 66 | 31 | 91 | 18 | 92 | 32 | 5 | 19 |
| HT29 | 56 | 16 | 37 | 14 | 77 | 5 | 67 | 20 | 13 | 24 |
| SW–620 | 45 | 34 | 55 | 6 | 91 | 29 | 64 | 61 | 0 | 34 |
| CNS Cancer | ||||||||||
| SF–268 | 26 | 4 | 32 | 0 | 31 | 1 | 77 | 76 | 16 | 47 |
| SF–295 | 63 | 22 | 47 | 0 | 22 | 0 | 100 (15) b | - | - | - |
| SF–539 | 57 | 27 | 46 | 2 | 83 | 3 | 100 (1) b | 45 | 10 | 36 |
| SNB–19 | 45 | 17 | 42 | 15 | 48 | 0 | 72 | 53 | 19 | 30 |
| SNB–75 | 10 | 2 | 34 | 0 | 6 | 0 | 100 (8) b | 72 | 50 | 49 |
| U251 | 61 | 50 | 56 | 30 | 77 | 13 | 75 | - | - | - |
| Melanoma | ||||||||||
| LOX IMVI | 69 | 58 | 59 | 56 | 100 (5) b | 28 | 71 | 52 | 36 | 64 |
| MDA–MB–435 | 26 | 0 | 21 | 17 | 90 | 4 | 39 | 0 | 0 | 10 |
| SK–MEL–2 | 0 | 0 | 0 | 10 | 74 | 0 | 61 | 20 | 24 | 87 |
| Ovarian Cancer | ||||||||||
| OVCAR–3 | 0 | 0 | 24 | 4 | 84 | 1 | 92 | 34 | 23 | 58 |
| OVCAR–8 | 69 | 44 | 62 | 20 | 85 | 16 | 74 | 65 | 26 | 44 |
| SK–OV–3 | 38 | 9 | 25 | 0 | 19 | 0 | 100 (61) b | 25 | 17 | 4 |
| Renal Cancer | ||||||||||
| 786–0 | 48 | 12 | 36 | 3 | 59 | 0 | 81 | 55 | 18 | 28 |
| A498 | 0 | 0 | 0 | 0 | 13 | 0 | 100 (47) b | 53 | 18 | 18 |
| ACHN | 83 | 31 | 64 | 0 | 57 | 0 | 100 (7) b | 65 | 31 | 31 |
| CAKI–1 | 47 | 30 | 42 | 3 | 29 | 4 | 77 | 52 | 23 | 53 |
| RXF 393 | 34 | 0 | 0 | 0 | 68 | 0 | 89 | - | - | - |
| SN12C | 49 | 33 | 47 | 4 | 65 | 0 | 93 | 70 | 24 | 22 |
| TK–10 | 16 | 12 | 35 | 0 | 42 | 0 | 99 | 61 | 33 | 43 |
| Prostate Cancer | ||||||||||
| DU–145 | 31 | 35 | 48 | 0 | 29 | 0 | 100 (2) b | 38 | 10 | 14 |
| Breast Cancer | ||||||||||
| MCF7 | 94 | 61 | 95 | 30 | 94 | 38 | 87 | 63 | 58 | 52 |
| HS 578T | 40 | 31 | 56 | 0 | 35 | 0 | 100 (19) b | 100 (12) b | 61 | 76 |
| T–47D | 43 | 24 | 99 | 40 | 64 | 23 | - | 99 | 48 | 42 |
| MDA–MB–468 | 52 | 0 | 100 (8) b | 15 | 50 | 0 | 100 (52) b | - | - | - |
| Cell Type | GI50 (μM) b | ||
|---|---|---|---|
| Compound 3b | Compound 5a | Phenstatin | |
| Leukemia | |||
| CCRF–CEM | 3.92 | 24.7 | 0.034 |
| HL–60 (TB) | 5.39 | 12.7 | 0.011 |
| K–562 | 6.48 | >42.2 | - |
| MOLT–4 | 3.30 | 6.58 | - |
| RPMI–8226 | 7.43 | 13.6 | - |
| SR | 5.87 | 8.32 | <0.010 |
| Non-small Cell Lung Cancer | |||
| A549/ATCC | >42.2 | 1.41 | - |
| EKVX | >42.2 | 8.77 | - |
| HOP–62 | 33.1 | >42.2 | 0.073 |
| HOP–92 | 15.2 | >42.2 | - |
| NCI–H226 | 25.6 | >42.2 | - |
| NCI–H23 | 12.9 | >42.2 | - |
| NCI–H322M | >42.2 | 12.8 | - |
| NCI–460 | 17.8 | 3.07 | 0.033 |
| NCI–H522 | 3.07 | >42.2 | 0.027 |
| Colon Cancer | |||
| COLO 205 | 38.8 | 4.18 | 4.86 |
| HCC–2998 | 39.9 | 6.73 | - |
| HCT–116 | 11.0 | >42.2 | 0.038 |
| HCT–15 | 6.17 | >42.2 | <0.010 |
| HT29 | 20.6 | >42.2 | 2.95 |
| KM12 | >42.2 | >42.2 | <0.010 |
| SW–620 | 7.26 | >42.2 | <0.010 |
| CNS Cancer | |||
| SF–268 | 34.4 | >42.2 | - |
| SF–295 | 28.3 | >42.2 | 0.367 |
| SF–539 | 10.9 | >42.2 | 0.011 |
| SNB–19 | >42.2 | >42.2 | - |
| SNB–75 | 24.7 | >42.2 | <0.010 |
| U251 | 18.3 | >42.2 | 0.043 |
| Melanoma | |||
| LOX IMVI | 4.35 | >42.2 | 0.013 |
| MALME–3M | 6.10 | >42.2 | - |
| M14 | 31.3 | >42.2 | <0.010 |
| MDA–MB–435 | 16.1 | >42.2 | <0.010 |
| SK–MEL–2 | 17.9 | >42.2 | 0.520 |
| SK–MEL–28 | 25.7 | >42.2 | 65.20 |
| SK–MEL–5 | 13.0 | >42.2 | 0.040 |
| UACC–257 | >42.2 | >42.2 | - |
| UACC–62 | 11.9 | >42.2 | 0.448 |
| Ovarian Cancer | |||
| IGROV1 | 33.2 | >42.2 | 0.18 |
| OVCAR–3 | 16.9 | >42.2 | 0.021 |
| OVCAR–4 | 25.6 | >42.2 | - |
| OVCAR–5 | 22.5 | >42.2 | - |
| OVCAR–8 | 14.9 | >42.2 | 0.042 |
| NCI/ADR–RES | 17.7 | >42.2 | 0.012 |
| SK–OV–3 | >42.2 | 11.9 | - |
| Renal Cancer | |||
| 786–0 | 21.1 | >42.2 | 0.905 |
| A498 | >42.2 | 5.71 | 2.28 |
| ACHN | 22.2 | >42.2 | 0.042 |
| CAKI–1 | 21.2 | 27.8 | - |
| RXF 393 | 2.79 | >42.2 | 0.016 |
| SN12C | 17.2 | 29.8 | - |
| TK–10 | >42.2 | >42.2 | - |
| UO–31 | 24.9 | >42.2 | 0.074 |
| Prostate cancer | |||
| PC–3 | 17.8 | >42.2 | 0.045 |
| DU–145 | >42.2 | 25.3 | 0.039 |
| Breast cancer | |||
| MCF7 | 4.58 | 24.5 | 0.033 |
| MDA–MB–231/ATCC | 4.72 | >42.2 | 0.029 |
| HS 578T | 18.8 | >42.2 | 0.031 |
| BT–549 | 6.18 | >42.2 | 0.034 |
| T–47D | 11.5 | 18.1 | 30.4 |
| MDA–MB–468 | 13.2 | 7.25 | 2.71 |
| ADME Parameter | 3b | 5a |
|---|---|---|
| Physicochemical Properties | ||
| Molecular weight | 468.30 g/mol | 358.35 g/mol |
| Log Po/w (MLOGP) | 3.24 | 1.94 |
| Number of H-bond acceptors | 5 | 6 |
| Number of H-bond donors | 0 | 0 |
| Number of rotatable bonds | 6 | 5 |
| TPSA | 74.08 Å2 | 97.53 Å2 |
| Pharmacokinetics | ||
| Gastrointestinal (GI) absorption | high | high |
| Blood–brain barrier (BBB) permeant | no | no |
| P-gp substrate | no | no |
| Drug-likeness | ||
| Log S (ESOL) | −6.55 | −6.02 |
| Water solubility class | poorly soluble | poorly soluble |
| Lipinski rule | no violation | no violation |
| Veber rule | no violation | no violation |
| Bioavailability | 0.55 | 0.55 |
| Medicinal Chemistry | ||
| PAINS alerts | 0 | 1 alert: azo |
| Brenk alerts | 3 alerts: alkyl_halide, more than 2 esters, polycyclic aromatic hydrocarbon | 4 alerts: azido_group, diazo_group, polycyclic aromatic hydrocarbon, quaternary nitrogen |
| Synthetic accessibility | 3.65 | 2.54 |
| 3b | |||
| Pa | Pi | Cell Line | Cell Type |
| 0.567 | 0.001 | MOLT-3 | T-lymphoblastic leukemia |
| 0.381 | 0.033 | CCRF-CEM | Childhood T acute lymphoblastic leukemia |
| 0.402 | 0.069 | NCI-H187 | Small cell lung carcinoma |
| 5a | |||
| Pa | Pi | Cell Line | Cell Type |
| 0.437 | 0.019 | SF-539 | Glioblastoma |
| 0.433 | 0.021 | UACC-62 | Melanoma |
| 0.419 | 0.028 | SN12C | Renal carcinoma |
| 0.419 | 0.028 | HOP-62 | Non-small cell lung carcinoma |
| 0.379 | 0.036 | OVCAR-3 | Ovarian adenocarcinoma |
| 0.318 | 0.060 | T47D | Breast carcinoma |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moldoveanu, C.; Mangalagiu, I.I.; Zbancioc, G.; Danac, R.; Tataringa, G.; Zbancioc, A.M. Anticancer Potential of Azatetracyclic Derivatives: In Vitro Screening and Selective Cytotoxicity of Azide and Monobrominated Compounds. Molecules 2025, 30, 702. https://doi.org/10.3390/molecules30030702
Moldoveanu C, Mangalagiu II, Zbancioc G, Danac R, Tataringa G, Zbancioc AM. Anticancer Potential of Azatetracyclic Derivatives: In Vitro Screening and Selective Cytotoxicity of Azide and Monobrominated Compounds. Molecules. 2025; 30(3):702. https://doi.org/10.3390/molecules30030702
Chicago/Turabian StyleMoldoveanu, Costel, Ionel I. Mangalagiu, Gheorghita Zbancioc, Ramona Danac, Gabriela Tataringa, and Ana Maria Zbancioc. 2025. "Anticancer Potential of Azatetracyclic Derivatives: In Vitro Screening and Selective Cytotoxicity of Azide and Monobrominated Compounds" Molecules 30, no. 3: 702. https://doi.org/10.3390/molecules30030702
APA StyleMoldoveanu, C., Mangalagiu, I. I., Zbancioc, G., Danac, R., Tataringa, G., & Zbancioc, A. M. (2025). Anticancer Potential of Azatetracyclic Derivatives: In Vitro Screening and Selective Cytotoxicity of Azide and Monobrominated Compounds. Molecules, 30(3), 702. https://doi.org/10.3390/molecules30030702