

Concise Synthesis of Naphthalene-Based 14-Aza-12-Oxasteroids


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
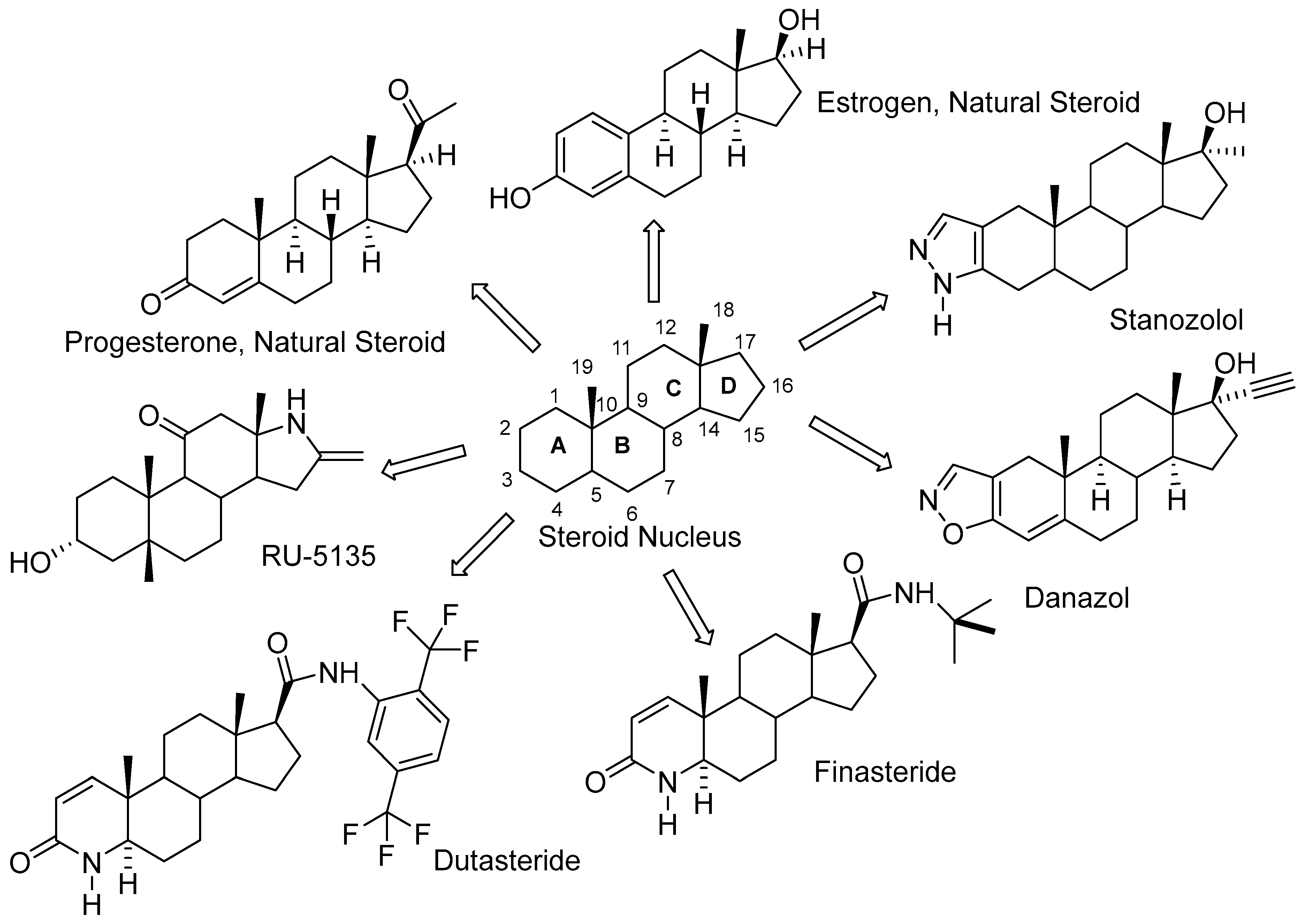
1. Introduction
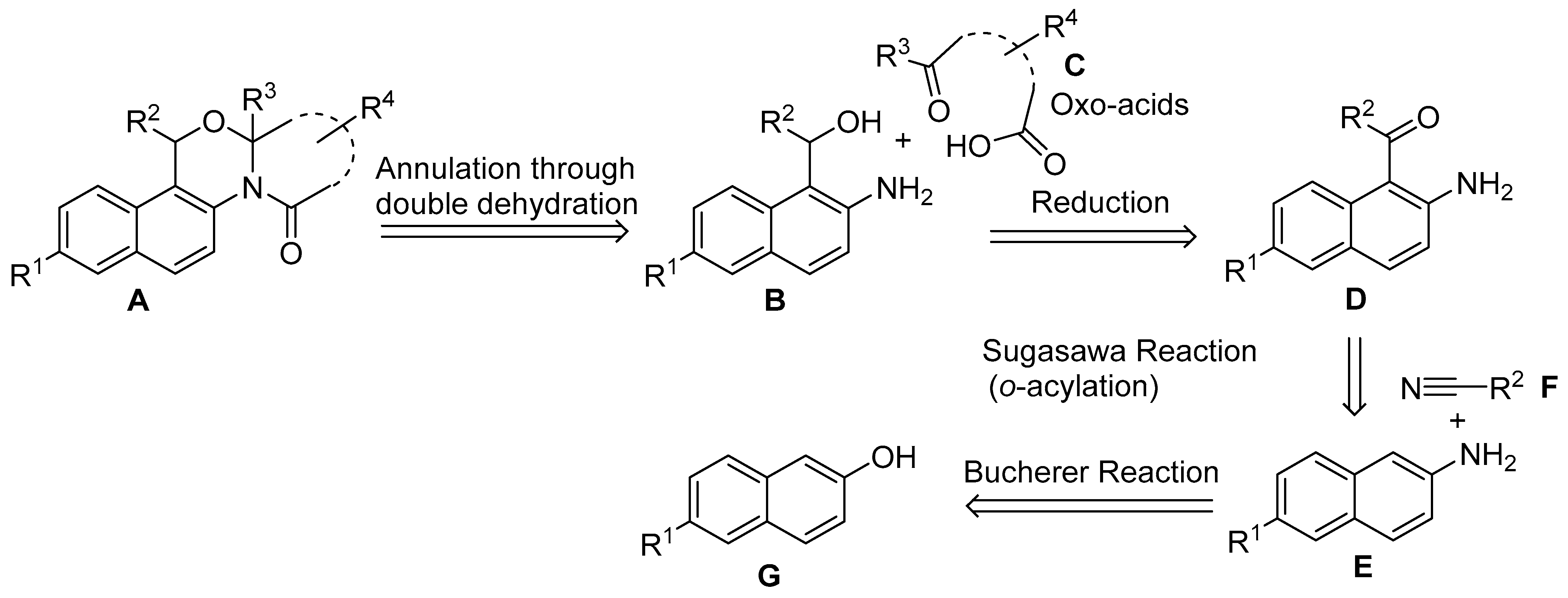
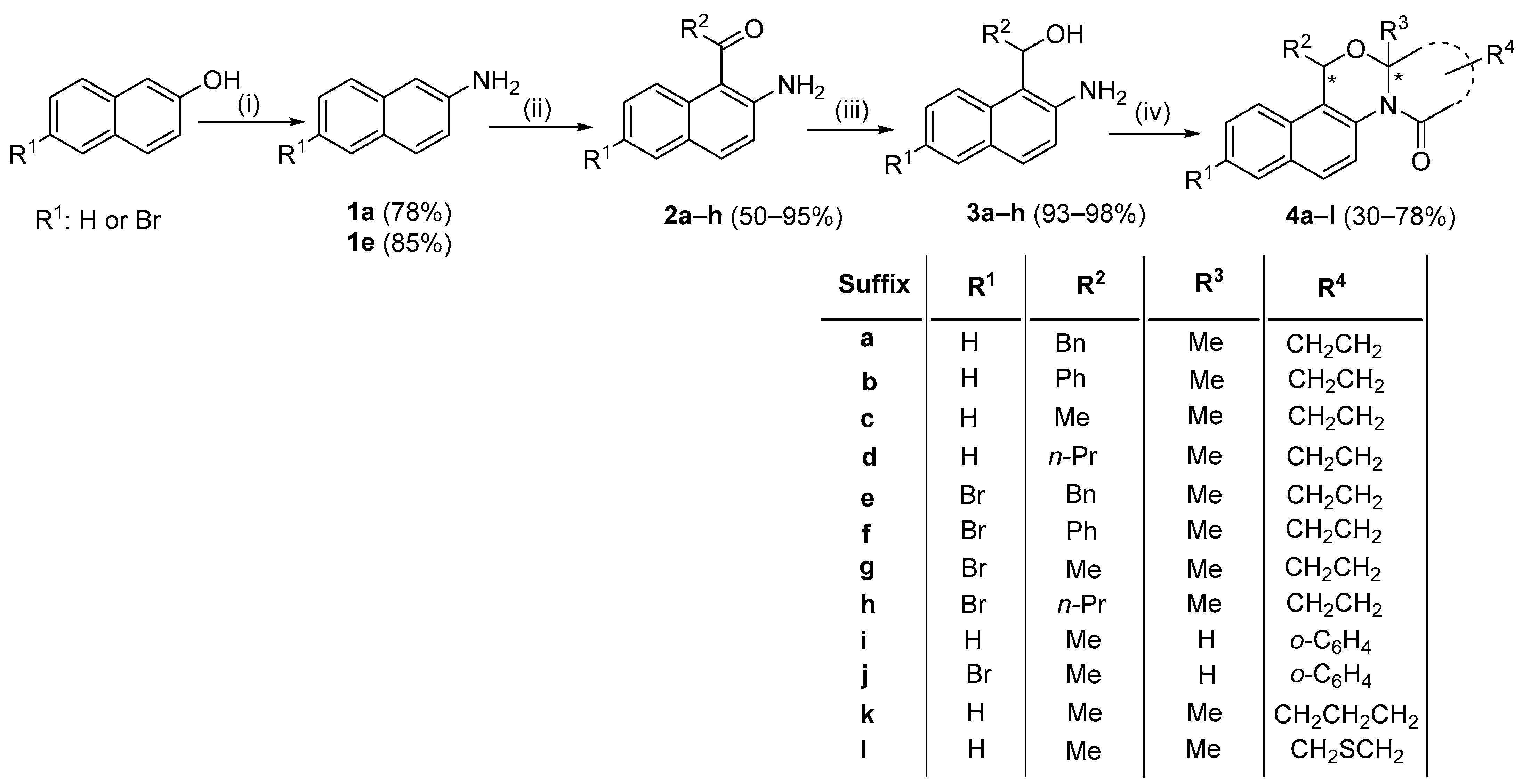
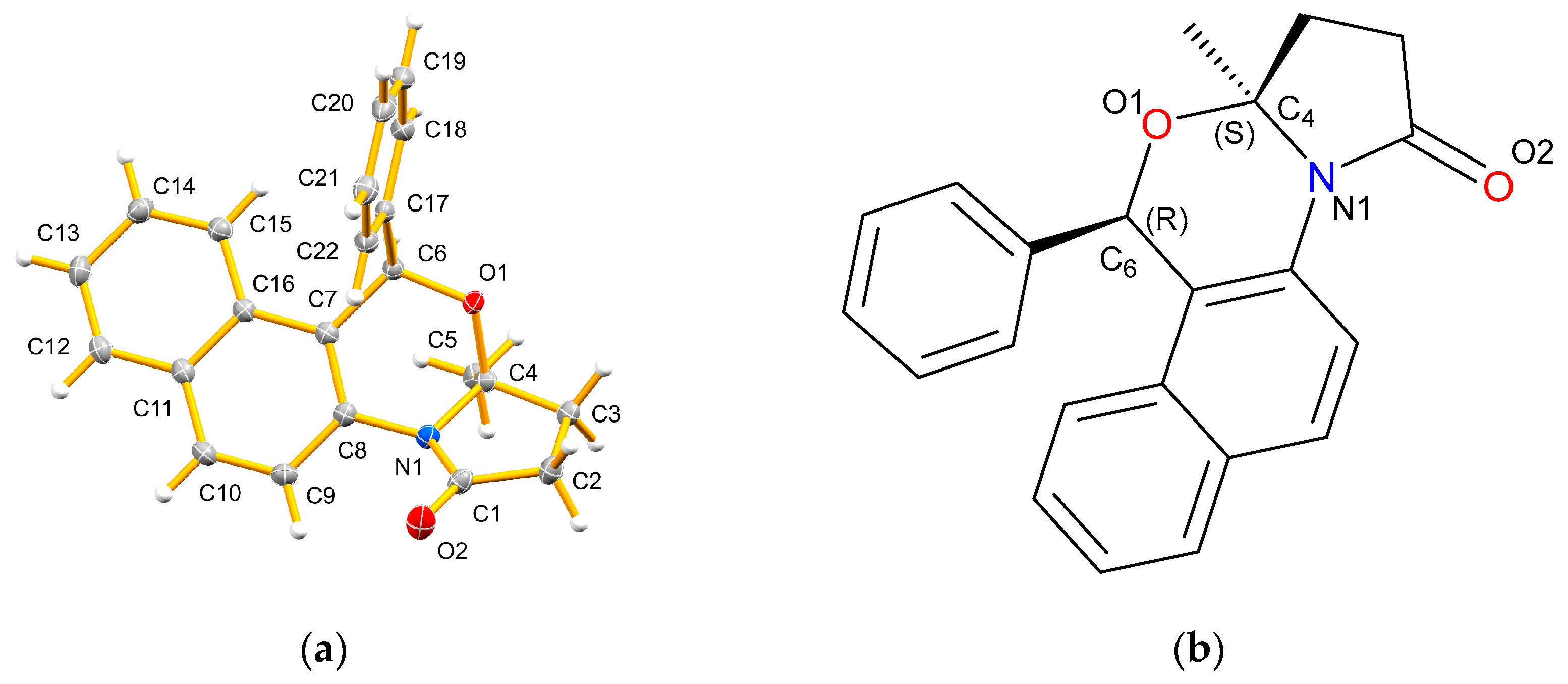
2. Results
3. Discussion
4. Materials and Methods
- 1-(2-Aminonaphthalen-1-yl)-2-phenylethanone (2a): Dark orange solid; 50% yield (1.81 g); m.p. 72–73 °C; 1H NMR (CDCl3, 300 MHz): δH 7.85 (d, 3JH-H = 9.0 Hz, 1H, ArH), 7.75 (d, 3JH-H = 9.0 Hz, 1H, ArH), 7.70 (d, 3JH-H = 9.0 Hz, 1H, ArH), 7.51 (t, 3JH-H = 7.5 Hz, 1H, ArH), 7.39–7.28 (m, 6H, ArH), 6.83 (d, 3JH-H = 9.0 Hz, 1H, ArH), 5.48 (brs, 2H, NH2), 4.36 (s, 2H, CH2Ph); 13C NMR (CDCl3, 75 MHz): δC 203.6 (CO), 146.0, 135.6, 133.4, 129.4, 128.7, 128.4, 127.4, 126.6, 124.1, 122.6, 122.6, 119.2, 115.4 (ArCs), 50.0 (CH2Ph); HRMS (ESI+) MeOH/CHCl3, m/z (RI): 262.1227 ([M + H]+, 100%). [M + H]+ calcd. for C18H15NO, 262.1226.
- (2-Aminonaphthalen-1-yl)(phenyl)methanone (2b): Yellow crystals; 60% yield (2.06 g); m.p. 166–167 °C; 1H NMR (DMSO-d6, 300 MHz): δH 7.78 (d, 3JH-H = 9.0 Hz, 1H, ArH), 7.71 (d, 3JH-H = 6.0 Hz, 1H, ArH), 7.66 (d, 3JH-H = 6.0 Hz, 2H, ArH), 7.59 (t, 3JH-H = 6.0 Hz, 1H, ArH), 7.46 (t, 3JH-H = 7.5 Hz, 2H, ArH), 7.17–7.06 (m, 4H, ArH), 5.93 (brs, 2H, NH2); 13C NMR (DMSO-d6, 75 MHz): δC 197.8 (CO), 145.7, 138.6, 132.8, 132.2, 131.4, 129.0, 128.5, 128.0, 126.3, 125.9, 123.3, 121.2, 118.9, 112.4 (ArCs); HRMS (ESI+) MeOH/CHCl3, m/z (RI): 248.1070 ([M + H]+, 100%). [M + H] calcd. for C17H13NO, 248.1070.
- 1-Acetyl-2-aminonaphthalene (2c): Orange solid; 63% yield (1.62 g); m.p. 107–108 °C; 1H NMR (CDCl3, 300 MHz): δH 7.82 (d, 3JH-H = 9.0 Hz, 1H, ArH), 7.68 (d, 3JH-H = 6.0 Hz, 1H, ArH), 7.65 (d, 3JH-H = 6.0 Hz, 1H, ArH), 7.46 (t, 3JH-H = 9.0 Hz, 1H, ArH), 7.27 (t, 3JH-H = 9.0 Hz, 1H, ArH), 6.85 (d, 3JH-H = 9.0 Hz, 1H, ArH), 5.80 (brs, 2H, NH2), 2.70 (s, 3H, CH3); 13C NMR (CDCl3, 75 MHz): δC 202.9 (CO), 146.4, 133.8, 132.6, 128.7, 127.7, 127.4, 124.4, 122.6, 119.5, 115.2 (ArCs), 32.3 (CH3); HRMS (ESI+) MeOH/CHCl3, m/z (RI): 186.2337 ([M + H]+, 100%). [M + H] calcd. for C12H11NO, 186.2335.
- 1-(2-Aminonaphalen-1-yl)butan-1-one (2d): Orange oil; 50% yield (1.33 g); 1H NMR (CDCl3, 300 MHz): δH 7.74–7.68 (m, 2H, ArH), 7.66 (d, 3JH-H = 9.0 Hz, 1H, ArH), 7.46 (t, 3JH-H = 9.0 Hz, 1H, ArH), 7.28 (t, 3JH-H = 7.5 Hz, 1H, ArH), 6.87 (d, 3JH-H = 9.0 Hz, 1H, ArH), 5.17 (brs, 2H, NH2), 2.99 (t, 3JH-H = 7.5 Hz, 2H, CH2CH2CH3), 1.86 (sx, 3JH-H = 7.8 Hz, 2H, CH2CH2CH3), 0.97 (t, 3JH-H = 6.0 Hz, 3H, CH2CH2CH3); 13C NMR (CDCl3, 75 MHz): δC 207.7 (CO), 144.8, 132.8, 131.2, 128.6, 127.6, 127.2, 124.0, 122.5, 119.6, 116.4 (ArCs), 46.2, 19.3, 13.8 (n-Pr); HRMS (ESI+) MeOH/CHCl3, m/z (RI): 214.1231 ([M + H]+, 100%). [M + H]+ calcd. for C14H15NO, 214.1226.
- 1-(2-Amino-6-bromonaphthalen-1-yl)-2-phenylethanone (2e): Dark purple solid; 75% yield (2.29 g); m.p. 101–102 °C; 1H NMR (CDCl3, 300 MHz): δH 7.84 (s, 1H, ArH), 7.65 (d, 3JH-H = 9.0 Hz, 1H, ArH), 7.55 (m, 2H, ArH), 7.34–7.26 (m, 5H, ArH), 6.83 (d, 3JH-H = 12.0 Hz, 1H, ArH), 5.46 (brs, 2H, NH2), 4.26 (s, 2H, CH2Ph); 13C NMR (CDCl3, 75 MHz): δC 203.2 (CO), 146.0, 135.3, 132.3, 130.6, 130.4, 129.3, 128.7, 128.5, 126.8, 125.7, 120.3, 115.9 (ArCs), 50.1 (CH2Ph); HRMS (ESI+) MeOH/CHCl3, m/z (RI): 340.0335 ([M + H]+, 98%). [M + H]+ calcd. for C18H14BrNO, 340.0332.
- (2-Amino-6-bromonaphthalen-1-yl)(phenyl)methanone (2f): Yellow crystals; 65% yield (1.90 g); m.p. 120–121 °C; 1H NMR (DMSO-d6, 300 MHz): δH 7.97 (d, 4JH-H = 1.5 Hz, 1H, ArH), 7.77 (d, 3JH-H = 12.0 Hz, 1H, ArH), 7.64 (d, 3JH-H = 6.0 Hz, 2H, ArH), 7.60 (d, 3JH-H = 6.0 Hz, 1H, ArH), 7.46 (t, 3JH-H = 7.5 Hz, 2H, ArH), 7.27 (d, 3JH-H = 6.0 Hz, 1H, ArH), 7.16 (d, 3JH-H = 9.0 Hz, 1H, ArH), 7.00 (d, 3JH-H = 9.0 Hz, 1H, ArH), 6.03 (s, 2H, NH2); 13C NMR (DMSO-d6, 75 MHz): δC 197.6 (CO), 146.4, 138.6, 133.3, 131.1, 130.9, 129.9, 129.3, 128.9, 127.4, 125.6, 120.4, 113.9, 112.3 (ArCs); HRMS (ESI+) MeOH/CHCl3, m/z (RI): 326.0184 ([M + H]+, 100%). [M + H]+ calcd. for C17H12BrNO, 326.0175.
- 1-Acetyl-2-amino-6-bromonaphthalene (2g): Orange solid; 95% yield (2.25 g); m.p. 135–137 °C. 1H NMR (CDCl3, 300 MHz): δH 7.80 (s, 1H, ArH), 7.68 (d, 3JH-H = 9.0 Hz, 1H, ArH), 7.55 (d, 3JH-H = 9.0 Hz, 2H, ArH), 6.84 (d, 3JH-H = 9.0 Hz, 1H, ArH), 5.94 (brs, 2H, NH2), 2.66 (s, 3H, CH3); 13C NMR (CDCl3, 75 MHz): δC 202.3 (CO), 147.0, 132.7, 131.2, 130.6, 130.4, 130.0, 128.8, 128.2, 125.9, 120.6 (ArCs), 32.2 (CH3); HRMS (ESI+) MeOH/CHCl3, m/z (RI): 265.1291 ([M + H]+, 100%). [M + H]+ calcd. for C12H11BrNO, 265.1295.
- 1-(2-Amino-6-bromonaphalen-1-yl)butan-1-one (2h): Brown solid; 60% yield (1.57 g); m.p. 42–43 °C; 1H NMR (CDCl3, 300 MHz): δH 7.81 (s, 1H, ArH), 7.59–7.47 (m, 3H, ArH), 6.86 (d, 3JH-H = 9.0 Hz, 1H, ArH), 5.02 (brs, 2H, NH2), 2.92 (t, 3JH-H = 7.5 Hz, 2H, CH2CH2CH3), 1.79 (dd, 3JH-H = 15.0 Hz, 9.0 Hz, 2H, CH2CH2CH3), 0.95 (t, 3JH-H = 7.5 Hz, 3H, CH2CH2CH3); 13C NMR (CDCl3, 75 MHz): δC 207.1 (CO), 145.1, 131.8, 130.7, 130.5, 130.2, 128.7, 125.6, 120.3, 116.1, 115.7 (ArCs), 46.2, 19.3, 13.8 (n-Pr); HRMS (ESI+) MeOH/CHCl3, m/z (RI): 292.0337 ([M + H]+, 100%). [M + H]+ calcd. for C14H14BrNO, 292.0332.
- 1-(2-Amimonaphthalen-1-yl)-2-phenylethanol (3a): Orange solid; 93% yield (467 mg); m.p. 129–131 °C; 1H NMR (CDCl3, 300 MHz): δH 7.82 (d, 3JH-H = 6.0 Hz, 1H, ArH), 7.73 (d, 3JH-H = 6.0 Hz, 1H, ArH), 7.61 (d, 3JH-H = 9.0 Hz, 1H, ArH), 7.44 (t, 3JH-H = 7.5 Hz, 1H, ArH), 7.39–7.25 (m, 6H, ArH), 6.89 (d, 3JH-H = 9.0, 1H, ArH), 5.85 (dd, 3JH-H = 9.0 Hz, 3.0 Hz, 1H, ArH), 3.81 (brs, 3H, NH2 and OH), 3.39 (dd, 2JH-H = 15.0 Hz, 3JH-H = 9.0 Hz, 1H, CH2Ph), 3.12 (dd, 2JH-H = 12.0 Hz, 3JH-H = 3.0 Hz, 1H, CH2Ph); 13C NMR (CDCl3, 75 MHz): δC 143.1, 138.6, 131.8, 129.5, 129.1, 128.8, 128.5, 128.1, 126.6, 126.6, 121.8, 120.6, 120.3, 115.9 (ArCs), 72.0 (CHOH), 40.7 (CH2Ph); HRMS (ESI+) MeOH/CHCl3, m/z (RI): 264.1383 ([M + H]+, 26%). [M + H]+ calcd. for C18H17NO, 264.1383.
- (2-Aminonaphthalen-1-yl)(phenyl)methanol (3b): Pale orange solid; 94% yield (474 mg); m.p. 131–133 °C; 1H NMR (DMSO-d6, 300 MHz): δH 7.97 (d, 3JH-H = 9.0 Hz, 1H, ArH), 7.67 (d, 3JH-H = 9.0 Hz, 1H, ArH), 7.59 (d, 3JH-H = 9.0 Hz, 1H, ArH), 7.38 (d, 3JH-H = 6.0 Hz, 2H, ArH), 7.31–7.23 (m, 3H, ArH), 7.19–7.09 (m, 2H, ArH), 7.02 (d, 3JH-H = 9.0 Hz, 1H, ArH), 6.62 (s, 1H, ArH), 6.16 (s, 1H, OH), 5.61 (brs, 2H, NH2); 13C NMR (DMSO-d6, 75 MHz): δC 144.7, 144.4, 132.8, 128.4, 128.3, 127.8, 127.0, 126.3, 126.0, 125.9, 122.1, 120.6, 119.9, 116.0 (ArCs), 68.5 (CHOH); HRMS (ESI+) MeOH/CHCl3, m/z (RI): 250.1214 ([M + H]+, 36%). [M + H]+ calcd. for C17H15NO, 250.1226.
- 1-(1-Hydroxyethyl)-2-aminonaphthalene (3c): Colorless solid; 98% yield (499 mg); m.p. 89.9–91.2 °C; 1H NMR (CDCl3, 300 MHz): δH 7.78 (d, 3JH-H = 9.0 Hz, 1H, ArH), 7.70 (d, 3JH-H = 9.0 Hz, 1H, ArH), 7.58 (d, 3JH-H = 9.0 Hz, 1H, ArH), 7.41 (t, 3JH-H = 6.0 Hz, 1H, ArH), 7.23 (t, 3JH-H = 6.0 Hz, 1H, ArH), 6.87 (d, 3JH-H = 9.0 Hz, 1H, ArH), 5.91 (q, 3JH-H = 7.5 Hz, 1H, ArH), 4.82 (brs, 1H, OH), 3.33 (brs, 2H, NH2), 1.65 (d, 3JH-H = 7.5 Hz, 3H, CH3); 13C NMR (CDCl3, 75 MHz): δC 142.8, 131.6, 128.8, 128.7, 127.9, 126.6, 121.8, 120.6, 120.2, 117.4 (ArCs), 67.0 (CHOH), 20.2 (CH3); HRMS (ESI+) MeOH/CHCl3, m/z (RI): 188.1068 ([M + H]+, 100%). [M + H]+ calcd. for C12H13NO, 188.1070.
- 1-(2-Aminonaphthalen-1-yl)butan-1-ol (3d): Orange solid; 95% yield (480 mg); m.p. 86–88 °C; 1H NMR (DMSO-d6, 300 MHz): δH 7.87 (d, 3JH-H = 6.0 Hz, 1H, ArH), 7.64 (d, 3JH-H = 9.0 Hz, 1H, ArH), 7.52 (d, 3JH-H = 9.0 Hz, 1H, ArH), 7.32 (t, 3JH-H = 7.5 Hz, 1H, ArH), 7.10 (t, 3JH-H = 7.5 Hz, 1H, ArH), 6.97 (d, 3JH-H = 9.0, 1H, ArH), 5.64 (brs, 2H, CHOH, OH), 5.53 (brs, 2H, NH2), 2.01–1.89 (m, 1H, CH2CH2CH3), 1.73–1.62 (m, 1H, CH2CH2CH3), 1.57–1.46 (m, 1H, CH2CH2CH3), 1.34–1.23 (m, 1H, CH2CH2CH3), 0.89 (t, 3JH-H = 7.5 Hz, 3H, CH2CH2CH3); 13C NMR (DMSO-d6, 75 MHz): δC 144.5, 132.2, 128.4, 127.7, 126.9, 121.2, 120.5, 120.0, 116.5, 68.1 (CHOH), 37.8, 19.1, 14.1 (CH2CH2CH3 and CH3); HRMS (ESI+) MeOH/CHCl3, m/z (RI): 216.1375 ([M + H]+, 100%). [M + H]+ calcd. for C14H17NO, 216.1383.
- 1-(2-Amimo-6-bromonaphthalen-1-yl)-2-phenylethanol (3e): Purple solid; 93% yield (468 mg); m.p. 144–146 °C; 1H NMR (DMSO-d6, 300 MHz): δH 7.86 (s, 1H, ArH), 7.54 (d, J = 9.0 Hz, 1H, ArH), 7.35 (d, 3JH-H = 9.0 Hz, 1H, ArH), 7.25–7.21 (m, 4H, ArH), 7.18–7.12 (m, 2H, ArH), 7.06 (d, 3JH-H = 9.0, 1H, ArH), 5.80 (brs, 2H, NH2), 5.61 (brs, 1H, OH), δ 5.69 (s, 1H, CHOH), 3.20 (dd, 2JH-H = 15.0 Hz, 3JH-H = 9.0 Hz, 1H, CH2Ph), 2.96 (dd, 3JH-H = 12.0 Hz, 3JH-H = 6.0 Hz, 1H, CH2Ph); 13C NMR (DMSO-d6, 75 MHz): δC 145.1, 140.5, 139.3, 130.7, 129.9, 129.5, 128.4, 128.2, 127.9, 127.3, 125.8, 121.1, 115.9, 113.0 (ArCs), 69.6 (CHOH), 40.4 (CH2Ph); HRMS (ESI+) MeOH/CHCl3, m/z (RI): 342.0495 ([M + H]+, 100%). [M + H]+ calcd. for C18H16BrNO, 342.0488.
- (2-Amino-6-bromonaphthalen-1-yl)(phenyl)methanol (3f): Very pale orange solid; 94% yield (474 mg); m.p. 171–172 °C; 1H NMR (DMSO-d6, 300 MHz): δH 7.94–7.89 (m, 2H, ArH), 7.58 (d, 3JH-H = 9.0 Hz, 1H, ArH), 7.34 (d, 3JH-H = 9.0 Hz, 3H, ArH), 7.25 (t, 3JH-H = 7.5 Hz, 2H, ArH), 7.16 (t, 3JH-H = 6.0 Hz, 1H, ArH), 7.06 (d, 3JH-H = 9.0 Hz, 1H, ArH), 6.54 (s, 1H, ArH), 6.17 (s, 1H), 5.72 (brs, 2H, NH2); 13C NMR (DMSO-d6, 75 MHz): δC 145.2, 144.2, 131.4, 129.8, 128.4, 127.8, 127.7, 126.3, 125.9, 124.9, 120.9, 116.1, 113.1 (ArCs), 68.2 (CHOH); HRMS (ESI+) MeOH/CHCl3, m/z (RI): 328.0319 ([M + H]+, 76%). [M + H]+ calcd. for C17H14BrNO, 328.0332.
- 1-(1-Hydroxyethyl)-2-amino-6-bromonaphthalene (3g): Colorless solid; 98% yield (495 mg); m.p. 120–123 °C; 1H NMR (DMSO-d6, 300 MHz): δH 7.90 (s, 1H, ArH), 7.87 (d, 3JH-H = 3.0 Hz, 1H, ArH), 7.50 (d, 3JH-H = 9.0 Hz, 1H, ArH), 7.39 (d, 3JH-H = 3.0 Hz, 1H, ArH), 6.99 (d, 3JH-H = 9.0 Hz, 1H, ArH), 5.74 (brs, 2H, NH2), 5.59 (q, 3JH-H = 6.0 Hz, 1H, CHOH), 5.52 (brs, 1H, OH), 1.41 (d, 3JH-H = 6.0 Hz, 3H, CH3); 13C NMR (DMSO-d6, 75 MHz): δC 144.6, 130.3, 129.9, 128.4, 128.2, 126.9, 123.7, 121.0, 117.1, 112.9 (ArCs), 64.4 (CHOH), 21.1 (CH3); HRMS (ESI+) MeOH/CHCl3, m/z (RI): 266.0173 ([M + H]+, 100%). [M + H]+ calcd. for C12H12BrNO, 266.0175.
- 1-(2-Amino-6-bromonaphthalen-1-yl)butan-1-ol (3h): Brown solid; 98% yield (493 mg); m.p. 138–140 °C; 1H NMR (DMSO-d6, 300 MHz): δH 7.87 (d, 4JH-H = 1.5 Hz, 2H, ArH), 7.52 (d, 3JH-H = 9.0 Hz, 1H, ArH), 7.40 (d, 3JH-H = 9.0 Hz, 1H, ArH), 7.01 (d, 3JH-H = 9.0, 1H, ArH), 5.80 (brs, 2H, NH2), 5.61 (brs, 1HOH), 5.42 (t, 3JH-H = 6.0 Hz, 1H, CHOH), 1.96–1.84 (m, 1H, CH2CH2CH3), 1.69–1.56 (m, 1H, CH2CH2CH3), 1.54–1.42 (m, 1H, CH2CH2CH3), 1.30–1.18 (m, 1H, CH2CH2CH3), 0.87 (t, 3JH-H = 7.5 Hz, 3H, CH2CH2CH3); 13C NMR (DMSO-d6, 75 MHz): δC 144.7, 130.8, 129.9, 128.4, 127.0, 121.1, 116.9, 113.0 (ArCs), 68.0 (CHOH), 36.9, 19.1, 14.1 (CH2CH2CH3); HRMS (ESI+) MeOH/CHCl3, m/z (RI) (RI): 294.0490 ([M + H]+, 3%), 276.0390 ([M + H-H2O]+, 100%). [M + H]+ calcd. for C14H16BrNO, 294.0488.
- rac-(11R,12aS)-11-benzyl-12a-methyl-11,12a-dihydro-1H-naphtho[2,1-d]pyrrolo[2,1-b][1,3]oxazin-3(2H)-one (4a): Orange solid; 40% yield (104 mg); Rf 0.55 (2% MeOH in DCM); m.p. 124–126 °C; 1H NMR (CDCl3, 300 MHz): δH 8.16 (d, 3JH-H = 9.0 Hz, 1H, ArH), 7.93 (t, 3JH-H = 7.5 Hz, 2H, ArH), 7.84 (d, 3JH-H = 9.0 Hz, 1H, ArH), 7.62 (t, 3JH-H = 7.5 Hz, 1H, ArH), 7.52 (t, 3JH-H = 7.5 Hz, 1H, ArH), 7.27–7.22 (m, 3H, ArH), 7.07 (d, 4JH-H = 3.0, 2H, ArH), 5.85 (d, 3JH-H = 3.0 Hz, 1H, PhCH2CH), 3.43 (d, 2JH-H = 12.0 Hz, 1H, PhCH2CH), 3.06 (dd, 2JH-H = 15.0 Hz, 3JH-H = 6.0 Hz, 1H, PhCH2CH), 2.58 (t, 3JH-H = 7.5 Hz, 2H, CH2CH2CO), 2.37–2.20 (m, 2H, CH2CH2CO), 1.42 (s, 3H, CH3); 13C NMR (DMSO, 75 MHz): δC 172.1 (CO), 137.7, 131.4, 129.5, 129.1, 128.4, 127.8, 126.5, 126.5, 124.9, 122.6, 121.8, 120.7 (ArCs), 89.3 (C12), 72.1 (C11), 42.8 (PhCH2), 32.6, 29.8, 22.4 (CH2CH2 and CH3); HRMS (ESI+) MeOH/CHCl3, m/z (RI): 344.1644 ([M + H]+, 100%). [M + H]+ calcd. for C23H21NO2, 344.1645.
- rac-(11R,12aS)-12a-methyl-11-phenyl-11,12a-dihydro-1H-naphtho[2,1-d]pyrrolo[2,1-b][1,3]oxazin-3(2H)-one (4b): Yellow-orange powder; 45% yield (119 mg); Rf 0.46 (2% MeOH in DCM); m.p. 189–191 °C; 1H NMR (CDCl3, 300 MHz): δH 8.46 (d, 3JH-H = 9.0 Hz, 1H, ArH), 7.90 (d, 3JH-H = 9.0 Hz, 1H, ArH), 7.83 (d, 3JH-H = 9.0 Hz, 1H, ArH), 7.48 (d, 3JH-H = 9.0 Hz, 1H, ArH), 7.36 (t, 3JH-H = 6.0 Hz, 1H, ArH), 7.32–7.26 (m, 6H, ArH), 6.45 (s, 1H, PhCH), 2.71–2.66 (m, 2H, CH2CH2CO), 2.29 (t, 3JH-H = 9.0 Hz, 2H, CH2CH2CO), 1.64 (s, 3H, CH3); 13C NMR (DMSO-d6, 75 MHz): δC 172.6 (CO), 141.6, 132.3, 131.5, 129.7, 129.5, 129.2, 129.0, 129.0, 128.8, 126.6, 125.2, 124.6, 120.7, 120.6 (ArCs), 90.6 (C12), 75.8 (C11), 33.1, 30.6, 22.0 (CH2CH2 and CH3); HRMS (ESI+) MeOH/CHCl3, m/z (RI): 330.1489 ([M + H]+, 100%). [M + H]+ calcd. for C22H19NO2, 330.1489.
- rac-(11R,12aS)-11,12a-dimethyl-11,12a-dihydro-1H-naphtho[2,1-d]pyrrolo[2,1-b][1,3]oxazin-3(2H)-one (4c): Viscous orange liquid; 34% yield (96 mg); Rf 0.39 (2% MeOH in DCM); 1H NMR (CDCl3, 300 MHz): δH 8.35 (d, 4JH-H = 1.5 Hz, 1H, ArH), 7.87–7.78 (m, 3H, ArH), 7.53–7.43 (m, 2H, ArH), 5.66 (q, 3JH-H = 6.0 Hz, 1H, CH3CH), 2.67 (t, 3JH-H = 9.0 Hz, 2H, CH2CH2CO), 2.30 (t, 3JH-H = 9.0 Hz, 2H, CH2CH2CO), 1.65 (d, 3JH-H = 6.0 Hz, 3H, CH3), 1.47 (s, 3H, CH3); 13C NMR (CDCl3, 75 MHz): δC 172.1 (CO), 131.2, 130.2, 129.2, 128.9, 128.2, 126.2, 124.8, 123.3, 123.1, 120.4 (ArCs), 89.3 (C12), 68.0 (C11), 32.8, 30.1, 23.1, 21.5 (CH2CH2 and CH3); HRMS (ESI+) MeOH/CHCl3, m/z (RI): 268.1332 ([M + H]+, 100%). [M + H]+ calcd. for C17H17NO2, 268.1331.
- rac-(11R,12aS)-12a-methyl-11-propyl-11,12a-dihydro-1H-naphtho[2,1-d]pyrrolo[2,1-b][1,3]oxazin-3(2H)-one (4d): Orange solid; yield 45% (123 mg); Rf 0.40 (2% MeOH in DCM); m.p. 123–125 °C; 1H NMR (CDCl3, 300 MHz): δH 8.27 (d, 3JH-H = 9.0 Hz, 1H, ArH), 7.87–7.76 (m, 3H, ArH), 7.52 (t, 3JH-H = 9.0 Hz, 1H, ArH), 7.45 (t, 3JH-H = 9.0 Hz, 1H, ArH), 5.58 (dd, 3JH-H = 6.0 Hz, 4JH-H = 1.5 Hz, 1H, CH3CH), 2.67 (t, 3JH-H = 7.5 Hz, 2H, CH2CH2CO), 2.30 (t, 3JH-H = 7.5 Hz, 2H, CH2CH2CO), 2.12–2.01 (m, 1H, CH2CH2CH3), 1.87–1.75 (m, 1H, CH2CH2CH3), 1.55–1.44 (m, 1H, CH2CH2CH3), 1.44 (s, 3H, CH3), 1.32–1.21 (m, 1H, CH2CH2CH3), 0.87 (t, 3JH-H = 6.0 Hz, 3H, CH2CH2CH3); 13C NMR (DMSO-d6, 75 MHz): δC 172.3 (CO), 131.2, 131.0, 129.2, 128.9, 128.1, 126.2, 124.8, 122.9, 122.6, 120.5 (ArCs), 89.3 (C12), 71.3 (C11), 38.8, 32.8, 30.1, 21.9, 18.0, 13.9 (CH2CH2, CH3, n-Pr); HRMS (ESI+) MeOH/CHCl3, m/z (RI): 296.1631 ([M + H]+, 100%). [M + H]+ calcd. for C19H21NO2, 296.1645.
- rac-(11R,12aS)-11-benzyl-8-bromo-12a-methyl-11,12a-dihydro-1H-naphtho[2,1-d]pyrrolo[2,1-b][1,3]oxazin-3(2H)-one (4e): Purple solid; 33% yield (82 mg); Rf 0.49 (2% MeOH in DCM); m.p. 106–108 °C; 1H NMR (CDCl3, 300 MHz): δH 8.16 (d, 3JH-H = 9.0 Hz, 1H, ArH), 8.05 (d, 4JH-H = 1.5 Hz, 1H), 7.79 (d, 3JH-H = 9.0 Hz, 1H, ArH), 7.72 (d, 3JH-H = 9.0 Hz, 1H, ArH), 7.66 (d, 3JH-H = 9.0 Hz, 1H, ArH), 7.23–7.19 (m, 3H, ArH), 7.00–6.99 (m, 2H, ArH), 5.79 (dd, 3JH-H = 6.0 Hz, 4JH-H = 3.0 Hz, 1H, PhCH2CH), 3.36 (dd, 2JH-H = 15.0 Hz, 3JH-H = 3.0 Hz, 1H, PhCH2CH), 3.04 (dd, 2JH-H = 15.0 Hz, 3JH-H = 6.0 Hz, 1H, PhCH2CH), 2.56 (t, 3JH-H = 7.5 Hz, 2H, CH2CH2CO), 2.18–2.34 (m, 2H, CH2CH2CO), 1.40 (s, 3H, CH3); 13C NMR (DMSO-d6, 75 MHz): δC 172.0 (CO), 137.2, 132.3, 131.7, 131.0, 129.8, 129.4, 127.8, 127.5, 126.6, 124.4, 122.0, 121.8, 118.8 (ArCs), 71.9 (C12), 89.3 (C11), 42.8 (PhCH2), 32.6, 29.8, 22.3 (CH2CH2, CH3); HRMS (ESI+) MeOH/CHCl3, m/z (RI): 421.0740 ([M + H]+, 100%). [M + H]+ calcd. for C23H20BrNO2, 421.0677.
- rac-(11R,12aS)-8-bromo-12a-methyl-11-phenyl-11,12a-dihydro-1H-naphtho[2,1-d]pyrrolo[2,1-b][1,3]oxazin-3(2H)-one (4f): Purple-brown solid; 30% yield (75 mg); Rf 0.46 (2% MeOH in DCM); m.p. 203–205 °C; 1H NMR (CDCl3, 300 MHz): δH 8.49 (d, 3JH-H = 9.0 Hz, 1H, ArH), 7.97 (s, 1H, ArH), 7.79 (d, 3JH-H = 9.0 Hz, 1H, ArH), 7.31–7.27 (m, 5H, ArH), 7.23–7.21 (m, 2H, ArH), 6.39 (s, 1H, PhCH), 2.70–2.65 (m, 2H, CH2CH2CO), 2.29 (t, 3JH-H = 6.0 Hz, 2H, CH2CH2CO), 1.64 (s, 3H, CH3); 13C NMR (DMSO-d6, 75 MHz): δC 172.2 (CO), 140.9, 132.3, 132.2, 130.6, 129.5, 128.9, 128.5, 128.1, 127.8, 125.9, 121.4, 120.3, 118.8 (ArCs), 90.2 (C12), 75.2 (C11), 32.7, 30.1, 21.6 (CH2CH2, CH3); HRMS (ESI+) MeOH/CHCl3, m/z (RI): 408.0584 ([M + H]+, 100%). [M + H]+ calcd. for C22H18BrNO2, 408.0594.
- rac-(11R,12aS)-8-bromo-11,12a-dimethyl-11,12a-dihydro-1H-naphtho[2,1-d]pyrrolo[2,1-b][1,3]-oxazin-3(2H)-one (4g): Viscous orange liquid; 35% yield (92 mg); Rf 0.41 (2% MeOH in DCM); 1H NMR (CDCl3, 300 MHz): δH 8.35 (d, 4JH-H = 1.5 Hz, 1H, ArH), 7.98 (s, 1H, ArH), 7.69–7.39 (m, 3H, ArH), 5.58 (q, 3JH-H = 6.0 Hz, 1H, CH3CH), 2.65 (t, 3JH-H = 7.5 Hz, 2H, CH2CH2CO), 2.27 (t, 3JH-H = 7.5 Hz, 2H, CH2CH2CO), 1.59 (d, 3JH-H = 6.0 Hz, 3H, CH3), 1.43 (s, 3H, CH3); 13C NMR (CDCl3, 75 MHz): δC 172.2 (CO), 132.3, 130.7, 129.4, 127.7, 127.2, 124.8, 123.5, 121.4, 119.0, 118.7 (ArCs), 89.3 (C12), 67.7 (C11), 32.7, 30.0, 23.1, 21.4 (CH2CH2, CH3); HRMS (ESI+) MeOH/CHCl3, m/z (RI): 346.0433 ([M + H]+, 100%). [M + H]+ calcd. for C17H16BrNO2, 346.0437.
- rac-(11R,12aS)-8-Bromo-12a-methyl-11-propyl-11,12a-dihydro-1H-naphtho[2,1-d]pyrrolo[2,1-b][1,3]-oxazin-3(2H)-one (4h): Purple oil; 40% yield (102 mg); Rf 0.62 (2% MeOH in DCM); 1H NMR (CDCl3, 300 MHz): δH 8.29 (d, 3JH-H = 9.0 Hz, 1H, ArH), 7.99 (d, 4JH-H = 1.5 Hz, 1H, ArH), 7.70–7.62 (m, 2H, ArH), 7.56 (d, 3JH-H = 9.0 Hz, 1H, ArH), 5.53 (dd, 3JH-H = 6.0 Hz, 3.0 Hz, 1H, CH3CH), 2.66 (t, 3JH-H = 7.5 Hz, 2H, CH2CH2CO), 2.29 (t, 3JH-H = 7.5 Hz, 2H, CH2CH2CO), 2.06–1.95 (m, 1H, CH2CH2CH3), 1.82–1.70 (m, 1H, CH2CH2CH3), 1.51–1.39 (m, 1H, CH2CH2CH3), 1.43 (s, 3H, CH3), 1.23–1.15 (m, 1H, CH2CH2CH3), 0.85 (t, 3JH-H = 7.5 Hz, 3H, CH2CH2CH3); 13C NMR (DMSO-d6, 75 MHz): δC 172.3 (CO), 132.4, 131.3, 130.8, 129.4, 127.6, 127.2, 124.6, 122.7, 121.5, 118.7 (ArCs), 89.3 (C12), 71.1 (C11), 38.8, 32.8, 30.0, 21.8, 17.9, 13.8 (CH2CH2, CH3, n-Pr); HRMS (ESI+) MeOH/CHCl3, m/z (RI): 374.0739 ([M + H]+, 100%). [M + H]+ calcd. for C19H20BrNO2, 374.0750.
- 7-Methyl-7H-naphtho[2′,1′:4,5][1,3]oxazino[2,3-a]isoindol-13(8aH)-one (4i): Viscous orange liquid; 78% yield (249 mg). Rf 0.79 (2% MeOH in DCM); mixture of two diastereomers (6:4). Data for the major diastereomer are presented here; 1H NMR (CDCl3, 300 MHz): δH 8.56 (d, 4JH-H = 1.5 Hz, 1H, ArH), 7.94–7.43 (m, 9H, ArH), 5.95 (q, 3JH-H = 9.0 Hz, 1H, CH3CH), 5.79 (s, 1H, NCHO), 1.72 (d, 3JH-H = 9.0 Hz, 3H, CH3); 13C NMR (CDCl3, 75 MHz): δC 164.8 (CO), 140.2, 132.7, 130.2, 128.9, 128.5, 126.6, 126.4, 124.7, 124.6, 124.0, 123.5, 123.2, 123.1, 122.4, 120.8, 119.0 (ArCs), 83.1, 72.4, 23.3 (CH3); HRMS (ESI+) MeOH/CHCl3, m/z (RI): 302.1173 ([M + H]+, 100%). [M + H]+ calcd. for C20H15NO2, 302.1176.
- 4-Bromo-7-methyl-7H-naphtho[2′,1′:4,5][1,3]oxazino[2,3-a]isoindol-13(8aH)-one (4j): Colorless solid; 40% yield (112 mg); Rf 0.85 (2% MeOH in DCM); m.p. 230–233 °C. Mixture of two diastereomers (8:2). Data for the major diastereomer are presented here; 1H NMR (CDCl3, 300 MHz): δH 8.58 (d, 3JH-H = 9.0 Hz, 1H, ArH), 8.03 (s, 1H, ArH), 7.95 (d, 3JH-H = 9.0 Hz, 1H, ArH), 8.03–7.59 (m, 6H, ArH), 5.94 (q, 3JH-H = 6.0 Hz, 1H, CH3CH), 5.87 (s, 1H, NCHO), 1.70 (d, 3JH-H = 6.0 Hz, 3H, CH3); 13C NMR (CDCl3, 75 MHz): δC 164.9 (CO), 140.2, 132.9, 132.2, 131.0, 130.5, 130.0, 129.7, 128.2, 127.7, 124.9, 124.2, 123.6, 122.5, 120.3, 118.6 (ArCs), 83.2 (C-12), 72.3 (C-11), 23.4 (CH3); HRMS (ESI+) MeOH/CHCl3, m/z (RI): 380.0284 ([M + H]+, 96%). [M + H]+ calcd. for C20H14BrNO2, 380.0281.
- rac-(12R,13aS)-12,13a-dimethyl-1,2,3,13a-tetrahydronaphtho[2,1-d]pyrido[2,1-b][1,3]oxazin-4(12H)-one (4k): Viscous orange liquid; 30% yield (91 mg); Rf 0.61 (2% MeOH in DCM); 1H NMR (CDCl3, 300 MHz): δH 7.87–7.70 (m, 4H, ArH), 7.55–7.46 (m, 2H, ArH), 5.65 (q, 3JH-H = 6.0 Hz, 1H, CH3CH), 2.63 (t, 3JH-H = 9.0 Hz, 2H, CH2CH2CH2), 2.13 (t, 3JH-H = 9.0 Hz, 2H, CH2CH2CH2), 1.63 (d, 3JH-H = 6.0 Hz, 3H, CH3), 1.38 (s, 3H, CH3), 0.83 (t, 3JH-H = 9.0 Hz, 2H, CH2CH2CH2); 13C NMR (CDCl3, 75 MHz): δC 170.1 (CO), 132.8, 131.6, 128.9, 128.8, 127.6, 127.1, 126.2, 125.2, 122.5 (ArCs), 86.1 (C12), 67.3 (C11), 37.0, 34.1, 25.0, 23.5, 17.0 (CH2CH2CH2, CH3); HRMS (ESI+) MeOH/CHCl3, m/z (RI): 282.3619 ([M + H]+, 100%). [M + H]+ calcd. for C18H19NO2: 282.3625.
- rac-(12R,13aS)-12,13a-dimethyl-12,13a-dihydro-1H-naphtho[2,1-d][1,4]thiazino[3,4-b][1,3]oxazin-4(3H)-one (4l): Viscous orange liquid; 30% yield (92 mg); Rf 0.62 (2% MeOH in DCM); 1H NMR (CDCl3, 300 MHz): δH 7.87–7.66 (m, 4H, ArH), 7.56–7.47 (m, 2H, ArH), 5.65 (q, 3JH-H = 7.5 Hz, 1H, CH3CH), 3.60 (m, 2H, CH2SCH2), 3.06 (s, 2H, CH2SCH2), 1.68 (d, 3JH-H = 7.5 Hz, 3H, CH3), 1.54 (s, 3H, CH3); 13C NMR (CDCl3, 75 MHz): δC 165.6 (CO), 132.1, 131.6, 128.8, 128.7, 127.2, 126.3, 125.5, 124.6, 122.5 (ArCs), 87.7 (C12), 67.5 (C11), 39.0, 33.8, 23.4, 23.3 (CH2SCH2), 2xCH3); HRMS (ESI+) MeOH/CHCl3, m/z (RI): 300.1047 ([M + H]+, 100%). [M + H]+ calcd. for C17H17NO2S: 300.1053.
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ibrahim-Ouali, M. Recent advances in oxasteroids chemistry. Steroids 2007, 72, 475–508. [Google Scholar] [CrossRef] [PubMed]
- Cole, T.J.; Short, K.L.; Hopper, S.B. The science of Steroid. Semin. Fetal Neonatal Med. 2019, 24, 170–175. [Google Scholar] [CrossRef] [PubMed]
- White, R.; Parker, M.G. Molecular mechanisms of steroid hormone action. Endocr.-Relat. Cancer 1998, 5, 1–14. [Google Scholar] [CrossRef]
- Frye, C.A. Steroids, reproductive endocrine function, and affect: A review. Minerva Ginecol. 2009, 61, 541–562. [Google Scholar] [PubMed]
- Wall, E.H.; Hewitt, S.C.; Case, L.K.; Lin, C.-Y.; Korach, K.S.; Teuscher, C. The role of genetics in estrogen responses: A critical piece of an intricate puzzle. FASEB J. 2014, 12, 5042–5054. [Google Scholar] [CrossRef]
- Chen, L.; Xu, T.; Lou, J.; Zhang, T.; Wu, S.; Xie, R.; Xu, J. The beneficial roles and mechanism of estrogens in immune health and infection disease. Steroids 2024, 207, 109426. [Google Scholar] [CrossRef]
- Taraborrelli, S. Physiology, production and action of progesterone. Acta Obstet. Gynecol. Scand. 2015, 94, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Bansal, R.; Acharya, P.C. Man-made cytotoxic steroids: Exemplary agents for cancer therapy. Chem. Rev. 2014, 114, 6986–7005. [Google Scholar] [CrossRef] [PubMed]
- Guon, Z. The modification of natural products for medical use. Acta Pharm. Sin. B 2017, 7, 119–136. [Google Scholar] [CrossRef]
- Kerru, N.; Gummidi, L.; Maddila, S.; Gangu, K.K.; Jonnalagadda, S.B. A review on recent advances in nirogen containing molecules and their biological applications. Molecules 2020, 25, 1909. [Google Scholar] [CrossRef]
- Singh, H.; Jindal, D.P.; Yadav, M.R.; Kumar, M. Heterosteroids and drug research. In Progress in Medicinal Chemistry; Ellis, G.P., West, G.B., Eds.; Elsevier: Amsterdam, The Netherlands, 1991; pp. 233–300. [Google Scholar] [CrossRef]
- Ibrahim-Ouali, M.; Rocheblave, L. Recent advances in azasteroids chemistry. Steroids 2008, 73, 375–407. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim-Ouali, M.; Santelli, M. Recent advances in thiasteroids chemistry. Steroids 2006, 71, 1025–1044. [Google Scholar] [CrossRef] [PubMed]
- Burbiel, J.; Bracher, F. Azasteroids as antifungals. Steroids 2003, 68, 587–594. [Google Scholar] [CrossRef]
- Dembitsky, V.M. Steroidsbearing heteroatom as potential drugs for medicine. Biomediciens 2023, 11, 2698–2785. [Google Scholar] [CrossRef]
- Helfman, T.; Falanga, D. Miami and Florida, Stanozolol as a novel therapeutic agent in dermatology. J. Am. Acad. Dermatol. 1995, 33, 254–258. [Google Scholar] [CrossRef] [PubMed]
- Cottreau, C.M.; Ness, R.B.; Modugno, F.; Allen, G.O.; Goodman, M.T. Endometriosis and its treatment with danazol or lupron in relation to ovarian cancer. Clin. Cancer Res. 2003, 9, 5142–5144. [Google Scholar] [PubMed]
- Curtis, D.R.; Malik, R. Glycine antagonism by RU 5135. Eur. J. Pharmacol. 1985, 110, 383–384. [Google Scholar] [CrossRef] [PubMed]
- Edwards, J.; Moore, R.A. Finasteride in the treatment of clinical benign prostatic hyperplasia: A systematic review of randomised trials. BMC Urol. 2002, 2, 14–30. [Google Scholar] [CrossRef] [PubMed]
- Pohlman, G.D.; Pohlman, E.A.; Crawford, E.D. Dutasteride: A review of its use in the management of prostate disorders. Clin. Med. Insights Ther. 2011, 3, 172–177. [Google Scholar] [CrossRef]
- Oumzil, K.; Ibrahim-Ouali, M.; Santelli, M. First total synthesis of (±)-3-aza-11-oxa-1,3,5(10)-trieno steroids. Steroids 2006, 71, 886–894. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Panda, G. L-Proline derived nitrogenous steroidal systems: An asymmetric approach to 14-azasteroids. RSC Adv. 2013, 3, 19533–19544. [Google Scholar] [CrossRef]
- Bernath, G.; Fueloep, F.; Argay, G.; Kalman, A.; Sohar, P. Stereochemical studies. Saturated heterocycles. A simple stereospecific synthesis of oxazasteroids. Tetrahedron Lett. 1981, 22, 3797–3800. [Google Scholar] [CrossRef]
- Abdelkhalik, A.M.; Paul, N.K.; Jha, A. Concise synthesis of 12a-methul-11-aryl-1, 2-dihydrobenzo[f]pyrrolo [1,2-a]quinolin-3(12aH)-ones as racemic 14-azaestrongen analogues. Steroids 2015, 98, 107–113. [Google Scholar] [CrossRef]
- Jha, A.; Chou, T.-Y.; Jaroudi, Z.A.L.; Ellis, B.D.; Cameron, T.S. Aza-Diels–Alder reaction between N-aryl-1-oxo-1H-isoindolium ions and tert-enamides: Steric effects on reaction outcome. Beilstein J. Org. Chem. 2014, 10, 848–857. [Google Scholar] [CrossRef] [PubMed]
- Jha, A.; Naidu, A.B.; Abdelkhalik, A.M. Transition metal-free one-pot cascade synthesis of 7-oxa-2-azatricyclo [7.4.0.02,6] trideca-1(9), 10, 12-trien-3-ones from biomass-derived levulinic acid under mild conditions. Org. Biomol. Chem. 2013, 11, 7559–7565. [Google Scholar] [CrossRef]
- Sugasawa, T.; Toyoda, T.; Adachi, M.; Sasakura, K. Aminohaloborane in organic synthesis. 1. Specific ortho substitution reaction of anilines. J. Am. Chem. Soc. 1978, 100, 4842–4852. [Google Scholar] [CrossRef]
- Canete, A.; Melendrez, M.X.; Saitz, C.; Zanocco, A.L. Synthesis of amino naphthalene derivatives using the Bucherer reaction under microwave irradiation. Synth. Commun. 2001, 31, 2143–2148. [Google Scholar] [CrossRef]
- Sartori, G.; Maggi, R. Use of solid catalysts in Friedel−Crafts acylation reactions. Chem. Rev. 2006, 106, 1077–1104. [Google Scholar] [CrossRef] [PubMed]
- González-Morales, A.; Díaz-Coutiño, D.; Fernánez-Zertuche, M.; García-Barradas, O.; Ordóñez, M. Preparation of dimethyl (R)- and (S)-2-(2-aminophenyl)-2-hydroxyethylphosphonate from anthranilic acid. Tetrahedron Asymmetry 2004, 15, 457–463. [Google Scholar] [CrossRef]
- Zhao, Y.; Huang, B.; Yang, C.; Chen, Q.; Xia, W. Sunlight-driven forging of amide/ester bonds from three independent components: An approach to carbamates. Org. Lett. 2016, 18, 5572–5575. [Google Scholar] [CrossRef] [PubMed]
- Orloff, H.D. The stereoisomerism of cyclohexane derivatives. Chem. Rev. 1954, 54, 347–447. [Google Scholar] [CrossRef]
- Imoto, H.; Fujii, R.; Naka, K. Solid-state emissive diaminomaleimide dimers and a polymer—Syntheses, structures and optical properties. Eur. J. Org. Chem. 2019, 2019, 3086–3092. [Google Scholar] [CrossRef]
- Allen, F.H.; Kennard, O.; Watson, D.G.; Brammer, L.; Orpen, A.G.; Taylor, R. Tables of bond lengths determined by X-Ray and neutron diffraction. Part I. Bond lengths in organic compounds. J. Chem. Soc. Perkin Trans. 1987, 2, S1–S19. [Google Scholar] [CrossRef]
- Brink, M. “Long range”—Kopplungen in einigen acetonylderivaten. Tetrahedron Lett. 1971, 29, 2753–2756. [Google Scholar] [CrossRef]
- APEX 3; Bruker AXS Inc.: Madison, WI, USA, 2018.
- SAINT; Bruker AXS Inc.: Madison, WI, USA, 2016.
- SADABS; Bruker AXS Inc.: Madison, WI, USA, 2016.
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal structure determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar] [CrossRef]
- Spek, A.L. Structure validation in chemical crystallography. Acta Cryst. 2009, D65, 148–155. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Srivastava, S.; Luo, J.; Whalen, D.; Robertson, K.N.; Jha, A. Concise Synthesis of Naphthalene-Based 14-Aza-12-Oxasteroids. Molecules 2025, 30, 415. https://doi.org/10.3390/molecules30020415
Srivastava S, Luo J, Whalen D, Robertson KN, Jha A. Concise Synthesis of Naphthalene-Based 14-Aza-12-Oxasteroids. Molecules. 2025; 30(2):415. https://doi.org/10.3390/molecules30020415
Chicago/Turabian StyleSrivastava, Smriti, Jun Luo, Daniel Whalen, Katherine N. Robertson, and Amitabh Jha. 2025. "Concise Synthesis of Naphthalene-Based 14-Aza-12-Oxasteroids" Molecules 30, no. 2: 415. https://doi.org/10.3390/molecules30020415
APA StyleSrivastava, S., Luo, J., Whalen, D., Robertson, K. N., & Jha, A. (2025). Concise Synthesis of Naphthalene-Based 14-Aza-12-Oxasteroids. Molecules, 30(2), 415. https://doi.org/10.3390/molecules30020415