2.1. Enthalpies of Formation in the Liquid Phase
The standard state correction, known as Washburn correction, Δ
UΣ, is determined as recommended in the literature for organic compounds [
8]. The internal energy for the isothermal bomb process, Δ
U(IBP), is calculated using the Equation (1), where
εcal is the energy equivalent of the calorimeter,
εf is the energy equivalent of the content in the final state, ∆
Tad is the corrected temperature rise obtained in each experiment, and ∆
U(ign) is the electrical energy of ignition. The remaining terms have been previously described [
8].
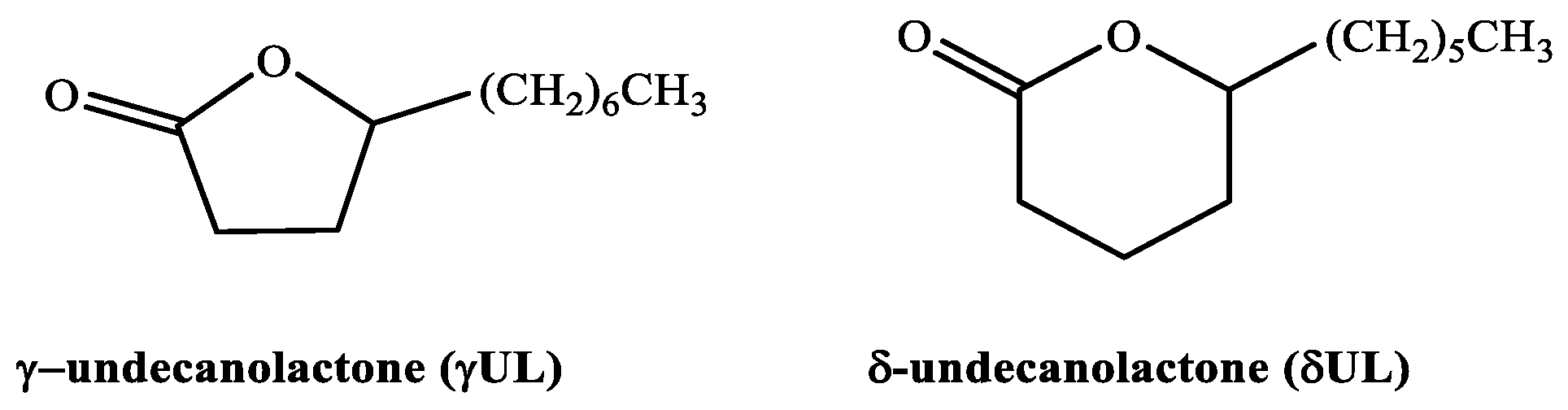
γUL and δUL being structural isomers, have the same molecular formula. Thus, the respective standard massic energy of combustion is referred to in the combustion reaction described by the chemical Equation (2).
The individual values of standard massic energies of combustion, Δ
cu°, of each compound studied, as well as the mean values with the corresponding standard deviation of the mean, are collected in the
Table 2.
The results of standard molar internal energies of combustion,
, enthalpy of combustion,
, and standard molar enthalpy of formation,
, in the liquid phase, at
T = 298.15 K, are reported in the
Table 3. The uncertainties associated with the standard molar internal energy and enthalpy of combustion are twice the overall standard deviation of the mean and include the uncertainties in the calibration with benzoic acid, and in the auxiliary materials used in the combustion experiments [
9,
10]. To calculate
from
, the standard molar enthalpies of formation, at
T = 298.15 K, were used for H
2O(l), −(285.830 ± 0.040) kJ·mol
–1 [
11], and CO
2(g), −(393.51 ± 0.13) kJ·mol
–1 [
11].
2.2. Enthalpies of Vaporization
For each compound studied, six experiments were carried out in the Calvet microcalorimeter at
T~365 K, and the final results are shown in
Table 4.
The standard molar enthalpy of vaporization,
, of each lactone was calculated from Equation (3), where the term,
, corresponds to the observed enthalpy of vaporization at the experimental temperature (
T~365 K), and the term
corresponds to the correction of the experimental temperature to
T = 298.15 K. The last parameter (Equation (4)) is obtained using the gas phase molar heat capacities,
, derived from statistical thermodynamics and the vibrational frequencies obtained at the B3LYP/6-31G(d) level [
12], scaled by a factor of 0.9613 [
13].
The values of
are well fitted to third-order polynomials, which are represented in the Equations (5) and (6), for γ-undecanolactone (γUL) and δ-undecanolactone (δUL), respectively. More details about each value of standard molar heat capacity of the lactones studied are given in the
Supplementary Information (Table S3).
2.3. Experimental Enthalpies of Formation in the Gas Phase
The standard (
p° = 0.1 MPa) molar enthalpies of formation, at
T = 298.15 K, in the gas phase,
, of the lactones were calculated by combining the standard molar enthalpies of formation in the liquid phase and the standard molar enthalpies of vaporization; the results are summarized in the
Table 5.
The enthalpic increment for the presence of a methylene group was calculated for the system γ-pentanolactone/γ-hexanolactone [
14], −24.4 kJ·mol
–1. Thus, using the
value of γ-nonanolactone and adding the energetic contribution of two methylene groups, it is possible to estimate the
of the γ-undecanolactone, −538.9 kJ·mol
–1, which is in agreement with the experimental results obtained in this work. The
value of δ-undecanolactone was also estimated, −535.4 kJ·mol
–1, summing the energetic contribution of two methylene groups (−24.4 kJ·mol
–1) with the
value of δ-nonanolactone [
15]. These results give us confidence concerning the reliability of the experimental results obtained.
In parallel to the experimental study, the standard molar enthalpy of formation in the gas phase was calculated using quantum chemical calculations.
2.4. Enthalpies of Formation in the Gas Phase Derived from Theoretical Calculations
The investigation of the two lactones started with the identification of the lowest energy conformations. First, the optimum gas-phase geometries of the two lactone rings were optimized using the M06-2X/6-311++G(d,p) approach (
Figure 2). Then, the geometry of the molecule with the CH
3 group instead of the side chain was optimized. The rotational barrier of the attached CH
3 group was computed as well. The found value of ca. 15 kJ·mol
–1 is in accordance with the published barrier of the CH
3 group in the methoxy group of anisole [
16,
17]. For the CH
2–CH
3 side chain, we also performed a 2D scan when both, the CH
2 and CH
3 fragments, were rotated, to find the lowest energy conformation. For each additional CH
2 fragment, the rotational barrier remains ca. 15 kJ·mol
–1. For species involved in working reactions, corresponding conformations were retained. Thus, the calculated reaction enthalpies and enthalpies of formation are not biased with the effect of conformational changes. From M06-2X/6-311++G(d,p), G4 or G4(MP2) total enthalpies, the reaction enthalpies of individual working reactions were found (
Table 6 and
Table 7). Using the available enthalpies of formation (
Table S4) for the compounds in the working reactions, the enthalpies of formation for the lactone were obtained. From five values, the average, and its uncertainty,
u95%, for the 95% confidence interval were found. In comparison to the experimentally determined enthalpies of formation, it can be concluded that the gas-phased M06-2X/6-311++G(d,p) enthalpies of formation of the two lactones are less negative than experimental ones. However, considering the uncertainties of theoretical and experimental values, M06-2X functional offers results in fair agreement with experiment. Although the composite ab initio G4 method gives results in excellent agreement with the experiment, the computational costs of the G4 calculations are ca. six times higher than M06-2X/6-311++G(d,p). Therefore, the G4(MP2) approach, which uses an approximation based on second-order perturbation theory to lower computational costs [
18], may represent a rational choice for thermochemical calculations. With costs analogous to the M06-2X/6-311++G(d,p) method, the discovered results can be considered practically identical to the G4 ones. Although higher errors were reported [
19] when the G4(MP2) method was applied to larger organic molecules, for the studied lactones, it provides reliable enthalpies of formation.
For the estimation of the enthalpy of formation of γUL and δUL in the gas phase, a set of five working reactions, (7) to (11) and (12) to (16), respectively, were selected considering the experimental data available in the literature. The computational results at the M06-2X, G4 or G4(MP2) levels are summarized in
Table 6 and
Table 7. In general, the M06-2X method gives less negative values in comparison to the experimentally determined enthalpies of formation with deviations of 8–12 kJ·mol
–1. In the case of γUL, Reaction (8) is the one with a result closest to the experimental value, with small deviations of 1.9 kJ·mol
–1 and –1.4 kJ·mol
–1, for the G4 and G4(MP2) methods, respectively. The other reactions give results less negative than the discovered experimental value. For δUL, the best agreement between experimental and computed enthalpy of formation shows Reaction (15) with deviations of 2.6 kJ·mol
–1 for G4 method and 2.9 kJ·mol
–1 for G4(MP2). The worst agreement shows Reaction (14), with roughly 10 kJ·mol
–1 deviations for both methods. The results in
Table 6 and
Table 7 also indicate that the M06-2X functional may represent a rational choice for calculations of larger organic molecules.
2.5. Estimates of Formation and Vaporization Enthalpies of γ- and δ-Lactones
In this work, the estimation of enthalpies of formation in liquid and gaseous phases, as well as the enthalpies of vaporization of γ- and δ-undecanolactones, was carried out. For this purpose, empirical correlation models were obtained by fitting the experimental literature values available for each set of lactones [
14,
15], as well as the experimental results obtained in this work.
Figure 3 presented the structures of lactones used in these correlations and the data are collected in
Table 8 and
Table 9 for γ- and δ-lactones, respectively.
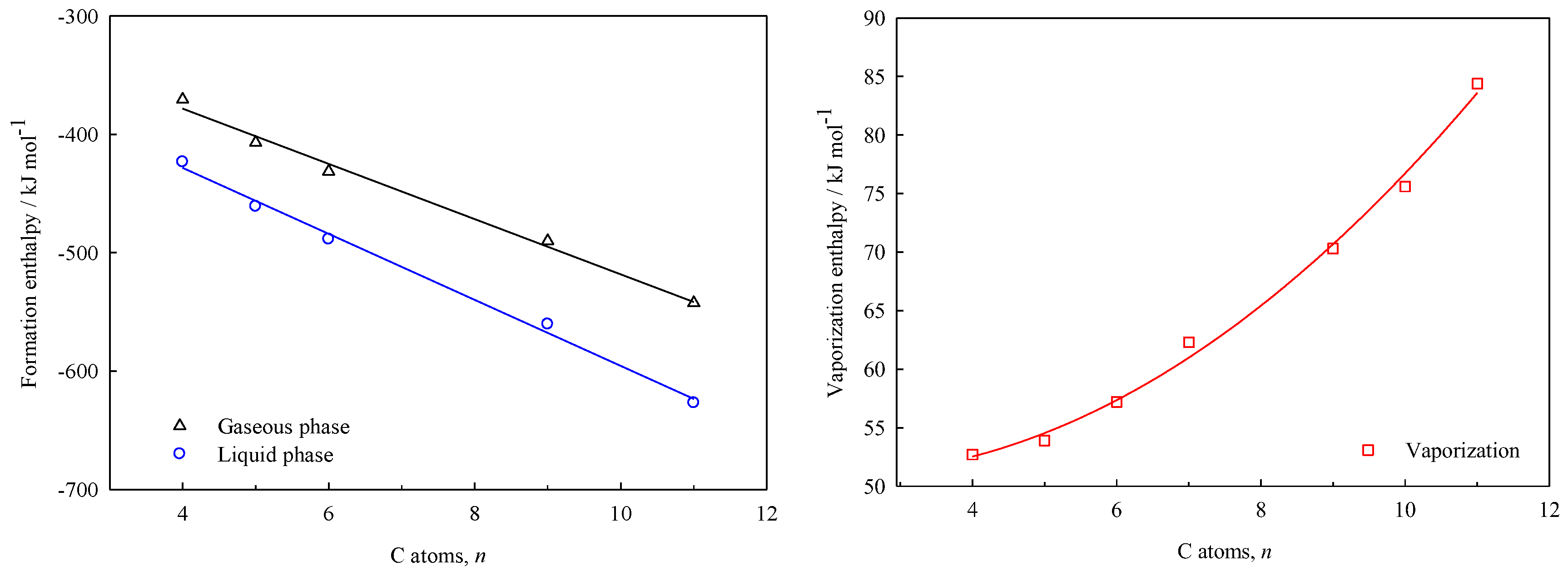
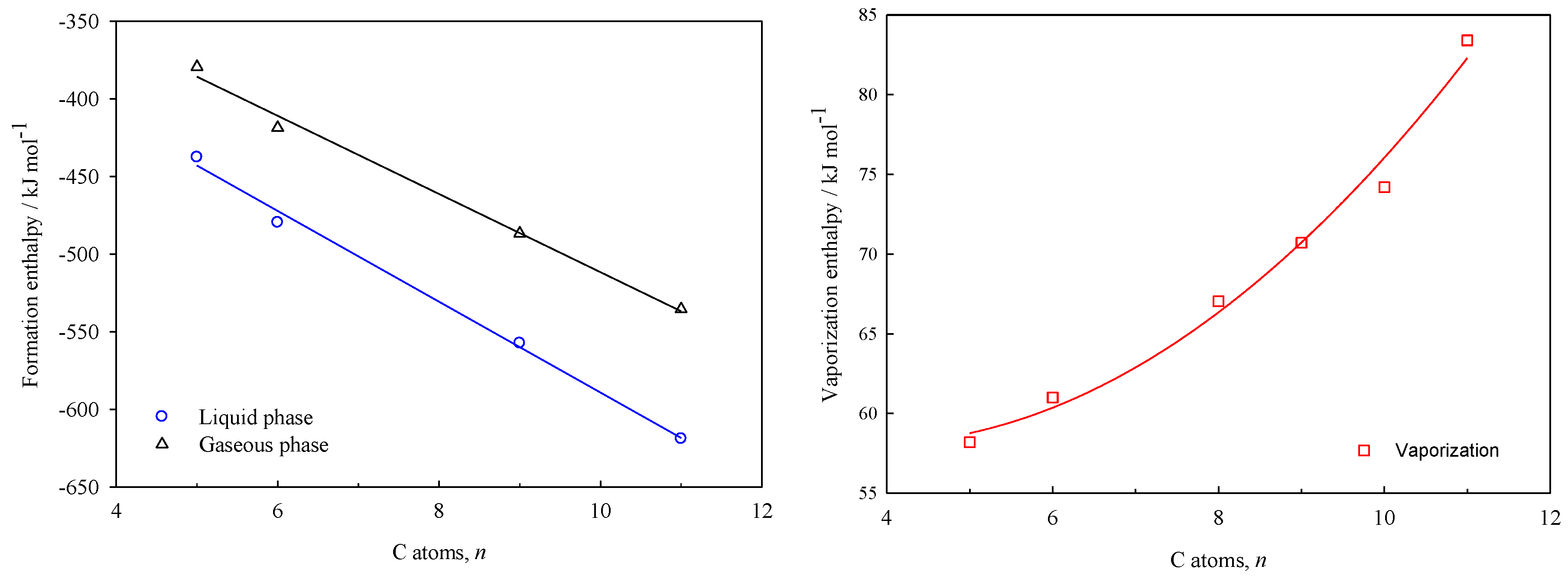
The plots of the enthalpies of formation in liquid and gaseous phases, as well as the enthalpies of vaporization vs. the number of carbon atoms (
n) of each molecule, are shown in
Figure 4 and
Figure 5, for γ-lactones and δ-lactones, respectively.
The equations derived from the linear and non-linear fits depicted above, are shown in the Equations (17) to (19) for the γ-lactones, and the Equations (20) to (22) for the δ-lactones. This enables the estimation of the energetic properties (, , ) for others hydrocarbon-chain lactones that are not experimentally feasible to study, with a standard error of estimates maximum of 8 kJ·mol–1.
Finally, in
Table 10, the estimated values of the standard molar enthalpies of formation in the liquid and gas phase of lactones with seven, eight and ten carbon atoms in their molecular structure are reported. Additionally, it includes the estimated enthalpy of vaporization values for the eight-carbon γ-lactone and the seven-carbon δ-lactone.
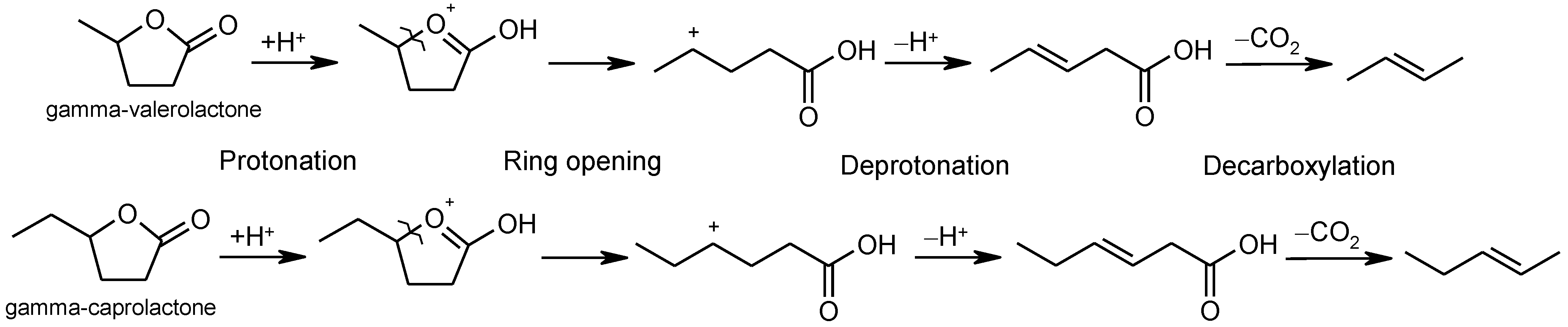
2.6. Ring-Opening Hydrogenation of Biomass-Derived Cyclic Oxygenates
Two five-membered ring lactones, i.e., γ-valerolactone and γ-caprolactone containing a methyl and ethyl group instead of a long alkyl side chain, were selected for the investigation of lactone ring opening and successive decarboxylation thermochemistry. These calculations were performed using the (SMD) M06-2X/6-311++G(d,p) and (SMD) G4 methods for aqueous solution. In general, the overall reaction can be formally divided into several steps: (1) protonation of carbonyl group O-atom; (2) ring opening, i.e., C–O bond cleavage; (3) deprotonation of the formed cation; and (4) decarboxylation (
Figure 6). The reaction Gibbs free energies of individual steps are summarized in
Table 11. We have also calculated the Gibbs free energies of ring O-atom protonation. However, the found results indicate that this process is considerably endergonic and cannot represent the thermodynamically preferred pathway. For the two lactones, the M06-2X method gives a Gibbs free energy of ring O-atom protonation ca. 80 kJ·mol
–1, and G4 provided ca. 50 kJ·mol
–1. For the overall reaction, the M06-2X reaction Gibbs free energy predicts a slightly endergonic character, while the G4 data indicate practically zero reaction Gibbs free energy. Data in
Table 11 indicate that the length of side chain plays a negligible role in the thermochemistry of the studied reaction. For γ-valerolactone, the experimentally determined standard reaction enthalpy (44.7 ± 4.3 kJ·mol
–1) and the reaction entropy (71.9 ± 9.1 J·mol
–1·K
–1) of lactone ring opening and the formation of pentenoic acid [
24] give reaction Gibbs free energy of 23 kJ·mol
–1 at 298.15 K. With respect to the uncertainty of experimental standard reaction enthalpy, this estimate is in fair agreement with the calculated reaction G4 and M06-2X Gibbs free energies of 29 kJ·mol
–1 and 31 kJ·mol
–1 (representing the sum of reaction Gibbs free energies of first three steps), respectively.
We have also performed calculations to identify the transition states and to find the plausible reaction pathway leading to the formation of final products, i.e., alkene and CO
2. Due to the geometry optimizations in the small basis set, composite ab initio methods are not appropriate for the calculations of potential energy surfaces and transition states. Therefore, M06-2X calculations were performed. The discovered reaction pathway is identical for both lactones. In
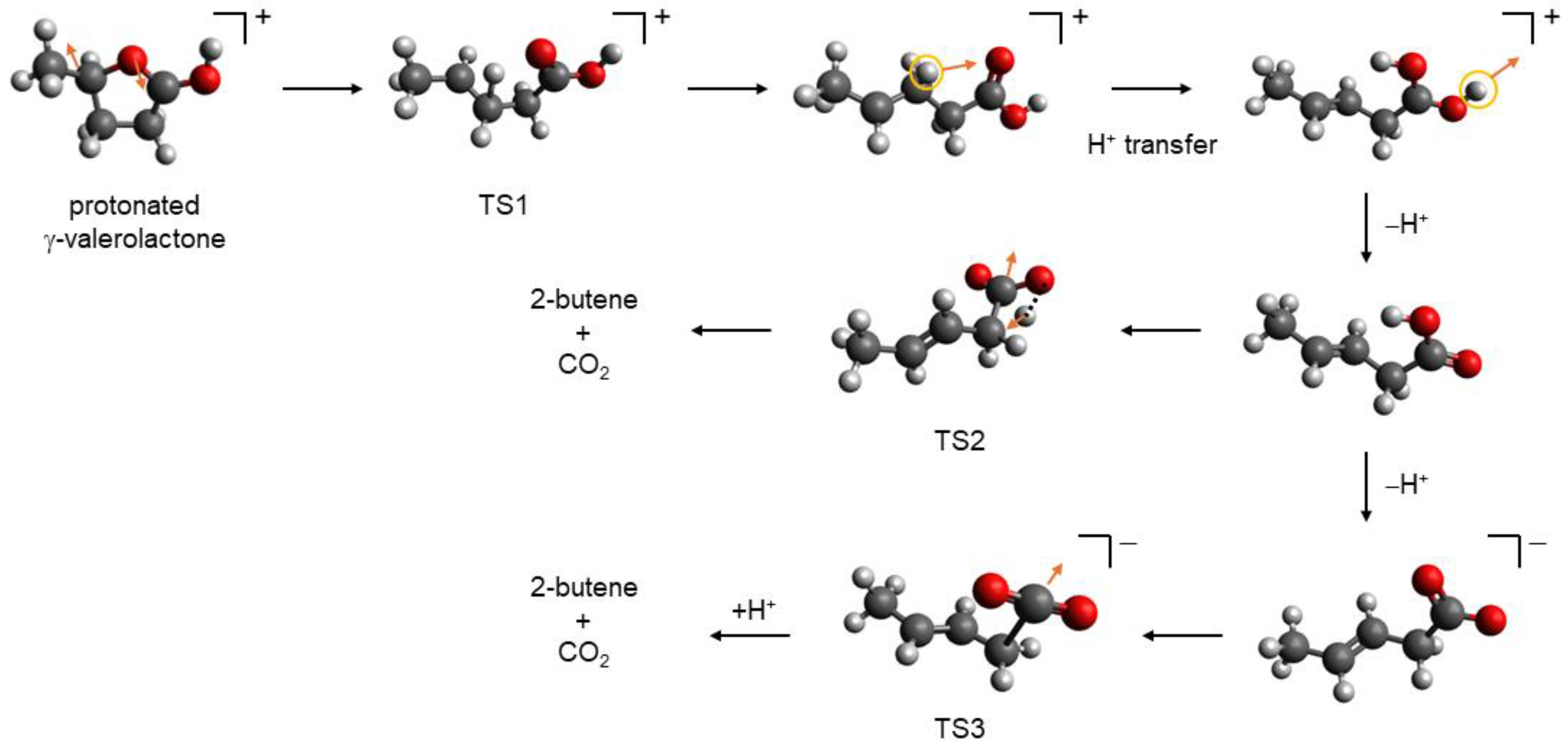
Figure 7, the individual steps of the plausible mechanism are shown for γ-valerolactone.
After the protonation of carbonyl group O-atom, ring opening, i.e., C–O bond cleavage occurs. The ring O-atom moves in the opposite direction to the C-atom bearing the methyl group. This movement is caused by the change in dihedral angle defined by four ring C-atoms. For the ring-opening process, the transition state (TS1 in
Figure 7), with the barrier of ∆
G≠ = 93 kJ·mol
–1, was identified. For γ-caprolactone, this barrier is lower by ca. 2 kJ·mol
–1. The ring-opening process is endergonic with the M06-2X reaction Gibbs free energies of 83 and 85 kJ·mol
–1 (
Table 11) for γ-valerolactone and γ-caprolactone, respectively. After the ring opening, the formed cation undergoes H
+ transfer to O-atom (their distance is ca. 2.5 Å). This step is exergonic (–40 kJ·mol
–1) and results in the structure depicted in the right upper corner in
Figure 7. For both lactones, this H
+ transfer requires relatively low activation Gibbs energy of 23 kJ·mol
–1. Then, exergonic (–34 kJ·mol
–1) deprotonation from the second O-atom occurs. Successive decarboxylation involves H atom transfer and CO
2 loss, see the arrows in TS2 in
Figure 7. Corresponding reaction Gibbs free energy reached the value of –24 kJ·mol
–1. The activation Gibbs free energy of 279 kJ·mol
–1 obtained for the formation of TS2 transition state is identical for the two lactones. The high activation Gibbs free energy explains why the decarboxylation after the lactone ring opening proceeds as a catalyzed reaction at elevated temperatures [
24,
25,
26]. The remaining results obtained for γ-caprolactone can be considered analogous to those for γ-valerolactone. Proton transfer from the C-atom to the neighboring O-atom and consecutive deprotonation are both exergonic with reaction Gibbs free energies of –43 kJ·mol
–1 and –30 kJ·mol
–1, respectively. The last step, i.e., the H atom transfer and CO
2 abstraction, is slightly more exergonic with ∆
rGo = –27 kJ·mol
–1 in comparison to γ-valerolactone.
Because carboxylic acids in aqueous solutions can be deprotonated, we have also considered the abstraction of the CO
2 molecule from the corresponding carboxylate anions with successive protonation of formed carbanions. The activation Gibbs free energy value related to corresponding transition state, TS3 in
Figure 7, is significantly lower than the value found for TS2. In the case of γ-valerolactone, its value reached 217 kJ·mol
–1. Although the formation of the carboxylate anion (77 kJ·mol
–1) and the abstraction of CO
2 molecule (200 kJ·mol
–1) are endergonic, the large negative reaction Gibbs free energy of formed carbanion protonation (–301 kJ·mol
–1) means that the overall process is exergonic with the reaction Gibbs free energy of –24 kJ·mol
–1. Almost identical results were obtained for γ-caprolactone: the activation Gibbs free energy of TS3 formation reached the value of 215 kJ·mol
–1; the reaction Gibbs free energies of carboxylate anion formation, CO
2 abstraction, and carbanion protonation resulting in 2-pentene formation are 75 kJ·mol
–1, 196 kJ·mol
–1, and –298 kJ·mol
–1, respectively.
We should also note that protonation of C3 carbon atom, instead of the C1 atom of carbanion, may lead to the formation of 1-butene from γ-valerolactone or 1-pentene from γ-caprolactone. This pathway is not thermodynamically favored, however; these protonations are only less exergonic by 12 kJ·mol–1.
In general, the computational data for the two studied model lactones clearly indicate that the energetics of the investigated processes can be considered practically identical and both reaction and activation. The Gibbs free energies may not depend on the length of the side chain. In general, the mechanism in
Figure 7 is compatible with the one proposed in Bond et al. [
24,
26].


 ,
, 





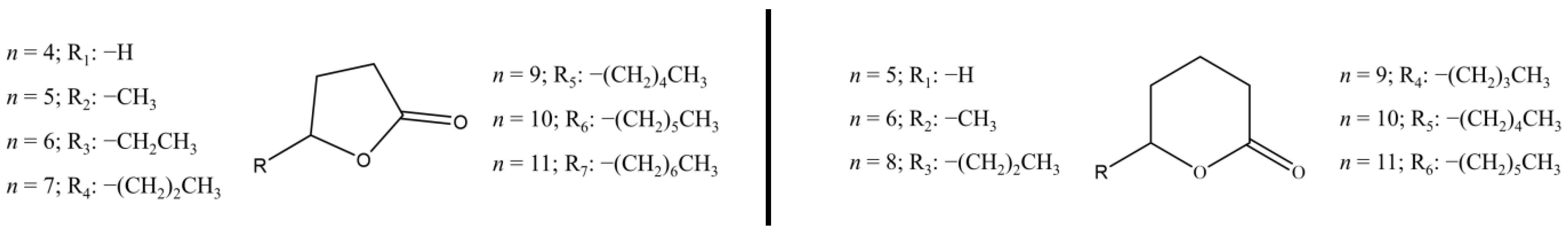

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}