
Application of Protein Structure Encodings and Sequence Embeddings for Transporter Substrate Prediction



Abstract
1. Introduction
1.1. Motivation and Background
1.2. Membrane Transporter Substrate Prediction with Machine Learning
1.3. Applications of Deep Learning Models in Bioinformatics
1.4. Predictive Methods Based on Protein Structures
1.5. Outline
2. Results
2.1. Classifying Substrates of Solute Carriers in A. thaliana
2.1.1. Dataset Analysis
2.1.2. Feature Comparison
2.1.3. Automatic Outlier Removal
2.1.4. Deep Neural Network Model for Membrane Transporter Substrate Prediction
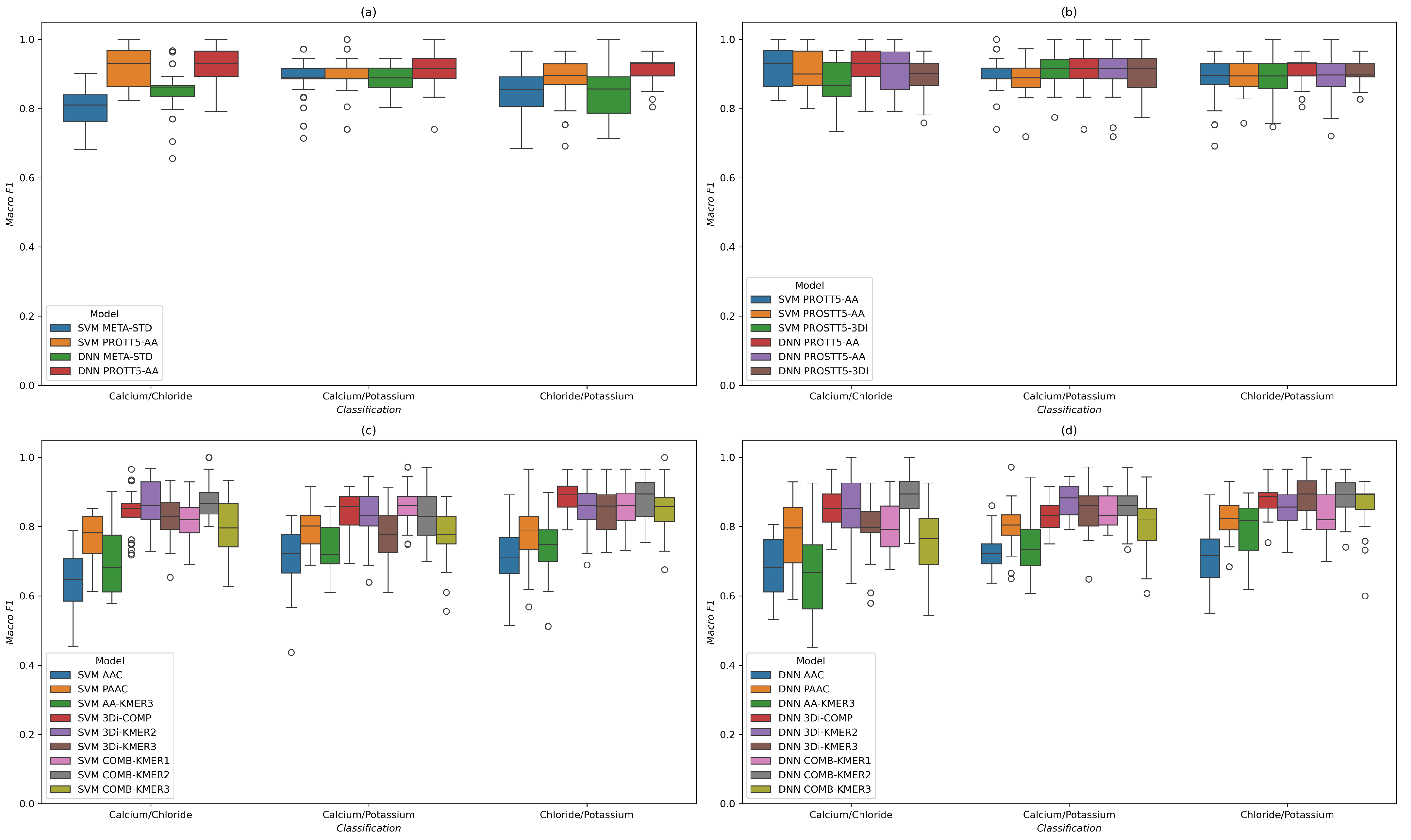
2.2. Categorization of Human Ion Channels
2.2.1. Dataset Analysis
2.2.2. Results for SVM and DNN Models
3. Methods
3.1. Data Availability
3.2. Protein Dataset Creation
3.3. Gene Ontology Graph and GO Annotations
3.4. Blast Databases and PSSM Calculation
3.5. Predicted 3D Structures and 3Di Sequences
3.6. Machine Learning Features Derived from Amino Acid Sequence
3.7. Machine Learning Features Derived from 3Di Sequence
3.8. Embeddings of Protein Sequence and Structure
3.9. Dummy Feature
3.10. Support Vector Machine (SVM) Models
3.11. Deep Neural Network (DNN) Model
3.12. Automatic Outlier Detection
3.13. Evaluation Methods
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Feature | Balanced Accuracy | F1 Amino Acid | F1 Sugar |
|---|---|---|---|
| DUMMY | 0.463 ± 0.122 | 0.393 ± 0.171 | 0.509 ± 0.118 |
| AAC | 0.711 ± 0.159 | 0.705 ± 0.152 | 0.700 ± 0.202 |
| PAAC | 0.892 ± 0.087 | 0.893 ± 0.086 | 0.881 ± 0.099 |
| AA-KMER3 | 0.883 ± 0.082 | 0.883 ± 0.080 | 0.870 ± 0.099 |
| MULTI-PSSM | 0.816 ± 0.122 | 0.815 ± 0.118 | 0.799 ± 0.153 |
| META | 0.818 ± 0.134 | 0.813 ± 0.139 | 0.808 ± 0.153 |
| META-STD | 0.869 ± 0.125 | 0.866 ± 0.129 | 0.864 ± 0.128 |
| COMB-KMER1 | 0.897 ± 0.082 | 0.894 ± 0.084 | 0.894 ± 0.088 |
| COMB-KMER2 | 0.907 ± 0.079 | 0.908 ± 0.076 | 0.898 ± 0.092 |
| COMB-KMER3 | 0.947 ± 0.072 | 0.947 ± 0.072 | 0.942 ± 0.078 |
| PROTT5-AA | 0.910 ± 0.058 | 0.900 ± 0.070 | 0.917 ± 0.049 |
| PROSTT5-AA | 0.913 ± 0.079 | 0.902 ± 0.102 | 0.921 ± 0.064 |
| PROSTT5-3Di | 0.909 ± 0.074 | 0.899 ± 0.087 | 0.917 ± 0.067 |
| 3Di-COMP | 0.929 ± 0.106 | 0.917 ± 0.127 | 0.936 ± 0.091 |
| 3Di-KMER2 | 0.971 ± 0.044 | 0.969 ± 0.046 | 0.970 ± 0.045 |
| 3Di-KMER3 | 0.920 ± 0.054 | 0.916 ± 0.058 | 0.920 ± 0.055 |
Appendix B
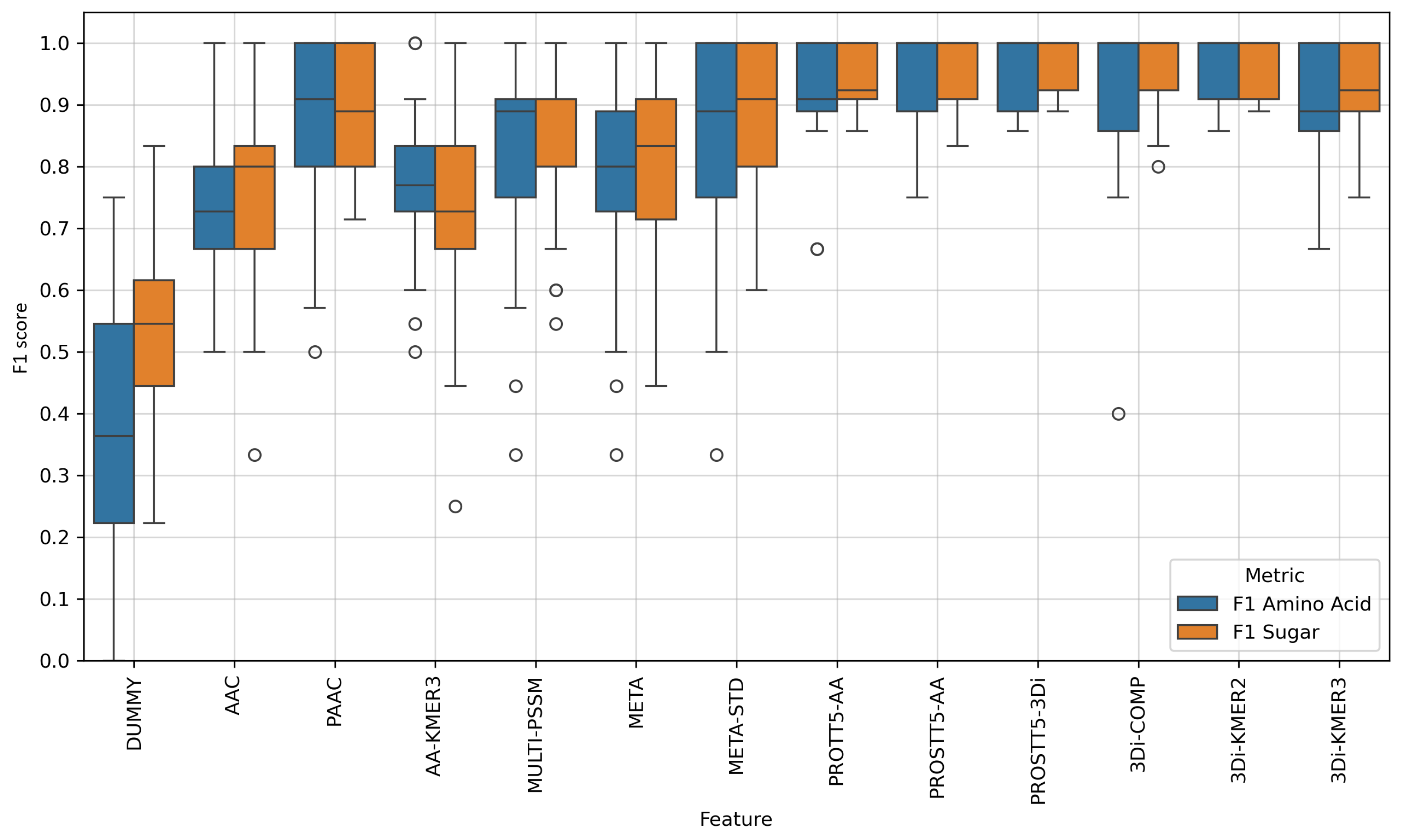
Appendix C
| Feature | Balanced Accuracy | F1 Amino Acid | F1 Sugar |
|---|---|---|---|
| DUMMY | 0.480 ± 0.136 | 0.354 ± 0.232 | 0.534 ± 0.156 |
| AAC | 0.766 ± 0.116 | 0.731 ± 0.139 | 0.770 ± 0.147 |
| PAAC | 0.881 ± 0.105 | 0.864 ± 0.134 | 0.879 ± 0.095 |
| AA-KMER3 | 0.770 ± 0.133 | 0.774 ± 0.126 | 0.727 ± 0.171 |
| MULTI-PSSM | 0.842 ± 0.138 | 0.812 ± 0.173 | 0.847 ± 0.133 |
| META | 0.810 ± 0.154 | 0.783 ± 0.181 | 0.813 ± 0.153 |
| META-STD | 0.869 ± 0.135 | 0.844 ± 0.176 | 0.876 ± 0.126 |
| COMB-KMER1 | 0.951 ± 0.080 | 0.945 ± 0.091 | 0.951 ± 0.086 |
| COMB-KMER2 | 0.938 ± 0.083 | 0.934 ± 0.086 | 0.933 ± 0.092 |
| COMB-KMER3 | 0.914 ± 0.088 | 0.906 ± 0.097 | 0.917 ± 0.086 |
| PROTT5-AA | 0.921 ± 0.073 | 0.910 ± 0.092 | 0.938 ± 0.051 |
| PROSTT5-AA | 0.948 ± 0.060 | 0.942 ± 0.069 | 0.954 ± 0.052 |
| PROSTT5-3Di | 0.949 ± 0.055 | 0.942 ± 0.062 | 0.959 ± 0.044 |
| 3Di-COMP | 0.934 ± 0.098 | 0.915 ± 0.140 | 0.954 ± 0.065 |
| 3Di-KMER2 | 0.967 ± 0.046 | 0.961 ± 0.054 | 0.968 ± 0.044 |
| 3Di-KMER3 | 0.899 ± 0.078 | 0.891 ± 0.088 | 0.912 ± 0.073 |
Appendix D
| Feature Name | Data Source | Type | Dimension |
|---|---|---|---|
| DUMMY | numpy.random | Unif. random values | 1024 |
| AAC | Amino acid sequence | 1-mer frequencies | 20 |
| PAAC | Amino acid sequence | 2-mer frequencies | 400 |
| AA-KMER3 | Amino acid sequence | 3-mer frequencies | 8000 |
| PSSM-50-1 | PSIBLAST (Uniref50, 1 iteration) | Evol. information | 400 |
| PSSM-50-3 | PSIBLAST (Uniref50, 3 iterations) | Evol. information | 400 |
| PSSM-90-1 | PSIBLAST (Uniref90, 1 iteration) | Evol. information | 400 |
| PSSM-90-3 | PSIBLAST (Uniref90, 3 iterations) | Evol. information | 400 |
| MULTI-PSSM | PSIBLAST (Four PSSMs) | Evol. information | 1600 |
| META | AAC + PAAC + MULTI-PSSM | Combined | 2020 |
| META-STD | META, standardized samples | Fingerprint | 2020 |
| 3Di-COMP | 3Di sequence | 1-mer frequencies | 20 |
| 3Di-KMER2 | 3Di sequence | 2-mer frequencies | 400 |
| 3Di-KMER3 | 3Di sequence | 3-mer frequencies | 8000 |
| COMB-KMER1 | AAC + 3Di-COMP | 1-mer frequencies | 40 |
| COMB-KMER2 | PAAC + 3Di-KMER2 | 2-mer frequencies | 800 |
| COMB-KMER3 | AA-KMER3 + 3Di-KMER3 | 3-mer frequencies | 1600 |
| PROTT5-AA | Amino acid sequence | pLM embedding | 1024 |
| PROSTT5-AA | Amino acid sequence | pLM embedding | 1024 |
| PROSTT5-3Di | 3Di sequence | pLM embedding | 1024 |
References
- Ferrada, E.; Superti-Furga, G. A structure and evolutionary-based classification of solute carriers. iScience 2022, 25, 105096. [Google Scholar] [CrossRef]
- Drew, D.; Boudker, O. Shared molecular mechanisms of membrane transporters. Annu. Rev. Biochem. 2016, 85, 543–572. [Google Scholar] [CrossRef]
- Forrest, L.R.; Krämer, R.; Ziegler, C. The structural basis of secondary active transport mechanisms. Biochim. Biophys. Acta (BBA)-Bioenerg. 2011, 1807, 167–188. [Google Scholar] [CrossRef]
- Mueckler, M.; Thorens, B. The SLC2 (GLUT) family of membrane transporters. Mol. Asp. Med. 2013, 34, 121–138. [Google Scholar] [CrossRef]
- Drew, D.; North, R.A.; Nagarathinam, K.; Tanabe, M. Structures and general transport mechanisms by the major facilitator superfamily (MFS). Chem. Rev. 2021, 121, 5289–5335. [Google Scholar] [CrossRef]
- Vastermark, A.; Wollwage, S.; Houle, M.E.; Rio, R.; Saier, M.H., Jr. Expansion of the APC superfamily of secondary carriers. Proteins Struct. Funct. Bioinform. 2014, 82, 2797–2811. [Google Scholar] [CrossRef]
- Chen, L.Q.; Hou, B.H.; Lalonde, S.; Takanaga, H.; Hartung, M.L.; Qu, X.Q.; Guo, W.J.; Kim, J.G.; Underwood, W.; Chaudhuri, B.; et al. Sugar transporters for intercellular exchange and nutrition of pathogens. Nature 2010, 468, 527–532. [Google Scholar] [CrossRef]
- Ji, J.; Yang, L.; Fang, Z.; Zhang, Y.; Zhuang, M.; Lv, H.; Wang, Y. Plant SWEET family of sugar transporters: Structure, evolution and biological functions. Biomolecules 2022, 12, 205. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.; Tampé, R. Structural and mechanistic principles of ABC transporters. Annu. Rev. Biochem. 2020, 89, 605–636. [Google Scholar] [CrossRef] [PubMed]
- Schaadt, N.S.; Christoph, J.; Helms, V. Classifying substrate specificities of membrane transporters from Arabidopsis thaliana. J. Chem. Inf. Model. 2010, 50, 1899–1905. [Google Scholar] [CrossRef] [PubMed]
- Schaadt, N.; Helms, V. Functional classification of membrane transporters and channels based on filtered TM/non-TM amino acid composition. Biopolymers 2012, 97, 558–567. [Google Scholar] [CrossRef]
- Chen, S.A.; Ou, Y.Y.; Lee, T.Y.; Gromiha, M.M. Prediction of transporter targets using efficient RBF networks with PSSM profiles and biochemical properties. Bioinformatics 2011, 27, 2062–2067. [Google Scholar] [CrossRef]
- Mishra, N.K.; Chang, J.; Zhao, P.X. Prediction of membrane transport proteins and their substrate specificities using primary sequence information. PLoS ONE 2014, 9, e100278. [Google Scholar] [CrossRef]
- Alballa, M.; Aplop, F.; Butler, G. TranCEP: Predicting the substrate class of transmembrane transport proteins using compositional, evolutionary, and positional information. PLoS ONE 2020, 15, e0227683. [Google Scholar] [CrossRef]
- Kroll, A.; Niebuhr, N.; Butler, G.; Lercher, M.J. SPOT: A machine learning model that predicts specific substrates for transport proteins. PLoS Biol. 2024, 22, e3002807. [Google Scholar] [CrossRef] [PubMed]
- Malik, M.S.; Le, V.T.; Ou, Y.Y. NA_mCNN: Classification of Sodium Transporters in Membrane Proteins by Integrating Multi-Window Deep Learning and ProtTrans for Their Therapeutic Potential. J. Proteome Res. 2025, 24, 2324–2335. [Google Scholar] [CrossRef]
- Denger, A.; Helms, V. Optimized Data Set and Feature Construction for Substrate Prediction of Membrane Transporters. J. Chem. Inf. Model. 2022, 62, 6242–6257. [Google Scholar] [CrossRef]
- Denger, A.; Helms, V. Identifying optimal substrate classes of membrane transporters. PLoS ONE 2024, 19, e0315330. [Google Scholar] [CrossRef] [PubMed]
- Vaswani, A.; Shazeer, N.; Parmar, N.; Uszkoreit, J.; Jones, L.; Gomez, A.N.; Kaiser, L.; Polosukhin, I. Attention is all you need. Adv. Neural Inf. Process. Syst. 2017, 30, 5998. [Google Scholar]
- LeCun, Y.; Bottou, L.; Bengio, Y.; Haffner, P. Gradient-based learning applied to document recognition. Proc. IEEE 2002, 86, 2278–2324. [Google Scholar] [CrossRef]
- Scarselli, F.; Gori, M.; Tsoi, A.C.; Hagenbuchner, M.; Monfardini, G. The graph neural network model. IEEE Trans. Neural Netw. 2008, 20, 61–80. [Google Scholar] [CrossRef]
- Kingma, D.P.; Welling, M. Auto-encoding variational bayes. arXiv 2013, arXiv:1312.6114. [Google Scholar]
- Elnaggar, A.; Heinzinger, M.; Dallago, C.; Rehawi, G.; Wang, Y.; Jones, L.; Gibbs, T.; Feher, T.; Angerer, C.; Steinegger, M.; et al. Prottrans: Toward understanding the language of life through self-supervised learning. IEEE Trans. Pattern Anal. Mach. Intell. 2021, 44, 7112–7127. [Google Scholar] [CrossRef] [PubMed]
- Rives, A.; Meier, J.; Sercu, T.; Goyal, S.; Lin, Z.; Liu, J.; Guo, D.; Ott, M.; Zitnick, C.L.; Ma, J.; et al. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences. Proc. Natl. Acad. Sci. USA 2021, 118, e2016239118. [Google Scholar] [CrossRef] [PubMed]
- Madani, A.; Krause, B.; Greene, E.R.; Subramanian, S.; Mohr, B.P.; Holton, J.M.; Olmos, J.L., Jr.; Xiong, C.; Sun, Z.Z.; Socher, R.; et al. Large language models generate functional protein sequences across diverse families. Nat. Biotechnol. 2023, 41, 1099–1106. [Google Scholar] [CrossRef]
- Ferruz, N.; Schmidt, S.; Höcker, B. ProtGPT2 is a deep unsupervised language model for protein design. Nat. Commun. 2022, 13, 4348. [Google Scholar] [CrossRef]
- Verkuil, R.; Kabeli, O.; Du, Y.; Wicky, B.I.; Milles, L.F.; Dauparas, J.; Baker, D.; Ovchinnikov, S.; Sercu, T.; Rives, A. Language models generalize beyond natural proteins. bioRxiv 2022. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Lin, Z.; Akin, H.; Rao, R.; Hie, B.; Zhu, Z.; Lu, W.; Smetanin, N.; Verkuil, R.; Kabeli, O.; Shmueli, Y.; et al. Evolutionary-scale prediction of atomic-level protein structure with a language model. Science 2023, 379, 1123–1130. [Google Scholar] [CrossRef]
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 373, 871–876. [Google Scholar] [CrossRef]
- Heinzinger, M.; Littmann, M.; Sillitoe, I.; Bordin, N.; Orengo, C.; Rost, B. Contrastive learning on protein embeddings enlightens midnight zone. NAR Genom. Bioinform. 2022, 4, lqac043. [Google Scholar] [CrossRef]
- Bileschi, M.L.; Belanger, D.; Bryant, D.H.; Sanderson, T.; Carter, B.; Sculley, D.; Bateman, A.; DePristo, M.A.; Colwell, L.J. Using deep learning to annotate the protein universe. Nat. Biotechnol. 2022, 40, 932–937. [Google Scholar] [CrossRef]
- Kulmanov, M.; Hoehndorf, R. DeepGOPlus: Improved protein function prediction from sequence. Bioinformatics 2020, 36, 422–429. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Song, H.; Zeng, M.; Li, Y.; Kurgan, L.; Li, M. DeepFunc: A deep learning framework for accurate prediction of protein functions from protein sequences and interactions. Proteomics 2019, 19, 1900019. [Google Scholar] [CrossRef] [PubMed]
- Littmann, M.; Heinzinger, M.; Dallago, C.; Olenyi, T.; Rost, B. Embeddings from deep learning transfer GO annotations beyond homology. Sci. Rep. 2021, 11, 1160. [Google Scholar] [CrossRef]
- Cao, Y.; Shen, Y. TALE: Transformer-based protein function Annotation with joint sequence–Label Embedding. Bioinformatics 2021, 37, 2825–2833. [Google Scholar] [CrossRef]
- Chen, J.Y.; Wang, J.F.; Hu, Y.; Li, X.H.; Qian, Y.R.; Song, C.L. Evaluating the advancements in protein language models for encoding strategies in protein function prediction: A comprehensive review. Front. Bioeng. Biotechnol. 2025, 13, 1506508. [Google Scholar] [CrossRef] [PubMed]
- Hashemifar, S.; Neyshabur, B.; Khan, A.A.; Xu, J. Predicting protein–protein interactions through sequence-based deep learning. Bioinformatics 2018, 34, i802–i810. [Google Scholar] [CrossRef]
- Jha, K.; Saha, S.; Singh, H. Prediction of protein–protein interaction using graph neural networks. Sci. Rep. 2022, 12, 8360. [Google Scholar] [CrossRef]
- Brandes, N.; Goldman, G.; Wang, C.H.; Ye, C.J.; Ntranos, V. Genome-wide prediction of disease variant effects with a deep protein language model. Nat. Genet. 2023, 55, 1512–1522. [Google Scholar] [CrossRef]
- Pejaver, V.; Urresti, J.; Lugo-Martinez, J.; Pagel, K.A.; Lin, G.N.; Nam, H.J.; Mort, M.; Cooper, D.N.; Sebat, J.; Iakoucheva, L.M.; et al. Inferring the molecular and phenotypic impact of amino acid variants with MutPred2. Nat. Commun. 2020, 11, 5918. [Google Scholar] [CrossRef]
- Stärk, H.; Dallago, C.; Heinzinger, M.; Rost, B. Light attention predicts protein location from the language of life. Bioinform. Adv. 2021, 1, vbab035. [Google Scholar] [CrossRef]
- Thumuluri, V.; Almagro Armenteros, J.J.; Johansen, A.R.; Nielsen, H.; Winther, O. DeepLoc 2.0: Multi-label subcellular localization prediction using protein language models. Nucleic Acids Res. 2022, 50, W228–W234. [Google Scholar] [CrossRef]
- Xiao, Y.; Zhao, W.; Zhang, J.; Jin, Y.; Zhang, H.; Ren, Z.; Sun, R.; Wang, H.; Wan, G.; Lu, P.; et al. Protein large language models: A comprehensive survey. arXiv 2025, arXiv:2502.17504. [Google Scholar]
- Bepler, T.; Berger, B. Learning the protein language: Evolution, structure, and function. Cell Syst. 2021, 12, 654–669. [Google Scholar] [CrossRef]
- Kulmanov, M.; Khan, M.A.; Hoehndorf, R. DeepGO: Predicting protein functions from sequence and interactions using a deep ontology-aware classifier. Bioinformatics 2018, 34, 660–668. [Google Scholar] [CrossRef]
- You, R.; Zhang, Z.; Xiong, Y.; Sun, F.; Mamitsuka, H.; Zhu, S. GOLabeler: Improving sequence-based large-scale protein function prediction by learning to rank. Bioinformatics 2018, 34, 2465–2473. [Google Scholar] [CrossRef]
- Varadi, M.; Bertoni, D.; Magana, P.; Paramval, U.; Pidruchna, I.; Radhakrishnan, M.; Tsenkov, M.; Nair, S.; Mirdita, M.; Yeo, J.; et al. AlphaFold Protein Structure Database in 2024: Providing structure coverage for over 214 million protein sequences. Nucleic Acids Res. 2024, 52, D368–D375. [Google Scholar] [CrossRef]
- Zhang, L.; Jiang, Y.; Yang, Y. Gnngo3d: Protein function prediction based on 3d structure and functional hierarchy learning. IEEE Trans. Knowl. Data Eng. 2023, 36, 3867–3878. [Google Scholar] [CrossRef]
- Gligorijević, V.; Renfrew, P.D.; Kosciolek, T.; Leman, J.K.; Berenberg, D.; Vatanen, T.; Chandler, C.; Taylor, B.C.; Fisk, I.M.; Vlamakis, H.; et al. Structure-based protein function prediction using graph convolutional networks. Nat. Commun. 2021, 12, 3168. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Liu, T.; Wang, Z. PANDA-3D: Protein function prediction based on AlphaFold models. NAR Genom. Bioinform. 2024, 6, lqae094. [Google Scholar] [CrossRef] [PubMed]
- Lai, B.; Xu, J. Accurate protein function prediction via graph attention networks with predicted structure information. Briefings Bioinform. 2022, 23, bbab502. [Google Scholar] [CrossRef]
- Julian, A.T.; Mascarenhas dos Santos, A.C.; Pombert, J.F. 3DFI: A pipeline to infer protein function using structural homology. Bioinform. Adv. 2021, 1, vbab030. [Google Scholar] [CrossRef]
- Van Kempen, M.; Kim, S.S.; Tumescheit, C.; Mirdita, M.; Lee, J.; Gilchrist, C.L.; Söding, J.; Steinegger, M. Fast and accurate protein structure search with Foldseek. Nat. Biotechnol. 2024, 42, 243–246. [Google Scholar] [CrossRef]
- Sledzieski, S.; Devkota, K.; Singh, R.; Cowen, L.; Berger, B. TT3D: Leveraging precomputed protein 3D sequence models to predict protein–protein interactions. Bioinformatics 2023, 39, btad663. [Google Scholar] [CrossRef]
- Heinzinger, M.; Weissenow, K.; Sanchez, J.G.; Henkel, A.; Mirdita, M.; Steinegger, M.; Rost, B. Bilingual language model for protein sequence and structure. NAR Genom. Bioinform. 2024, 6, lqae150. [Google Scholar] [CrossRef]
- Raffel, C.; Shazeer, N.; Roberts, A.; Lee, K.; Narang, S.; Matena, M.; Zhou, Y.; Li, W.; Liu, P.J. Exploring the limits of transfer learning with a unified text-to-text transformer. J. Mach. Learn. Res. 2020, 21, 1–67. [Google Scholar]
- Blum, M.; Andreeva, A.; Florentino, L.C.; Chuguransky, S.R.; Grego, T.; Hobbs, E.; Pinto, B.L.; Orr, A.; Paysan-Lafosse, T.; Ponamareva, I.; et al. InterPro: The protein sequence classification resource in 2025. Nucleic Acids Res. 2025, 53, D444–D456. [Google Scholar] [CrossRef]
- Roux, B. Ion channels and ion selectivity. Essays Biochem. 2017, 61, 201–209. [Google Scholar] [CrossRef]
- Parekh, A.B.; Putney, J.W., Jr. Store-operated calcium channels. Physiol. Rev. 2005, 85, 757–810. [Google Scholar] [CrossRef] [PubMed]
- Bateman, A.; Martin, M.J.; Orchard, S.; Magrane, M.; Adesina, A.; Ahmad, S.; Bowler-Barnett, E.H.; Bye-A-Jee, H.; Carpentier, D.; Denny, P.; et al. UniProt: The universal protein knowledgebase in 2025. Nucleic Acids Res. 2024, 52, D609–D617. [Google Scholar] [CrossRef]
- Saier, M.H., Jr.; Reddy, V.S.; Moreno-Hagelsieb, G.; Hendargo, K.J.; Zhang, Y.; Iddamsetty, V.; Lam, K.J.K.; Tian, N.; Russum, S.; Wang, J.; et al. The transporter classification database (TCDB): 2021 update. Nucleic Acids Res. 2021, 49, D461–D467. [Google Scholar] [CrossRef]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef]
- Consortium, G. The gene ontology resource: 20 years and still GOing strong. Nucleic Acids Res. 2019, 47, D330–D338. [Google Scholar]
- Huntley, R.P.; Sawford, T.; Mutowo-Meullenet, P.; Shypitsyna, A.; Bonilla, C.; Martin, M.J.; O’Donovan, C. The GOA database: Gene ontology annotation updates for 2015. Nucleic Acids Res. 2015, 43, D1057–D1063. [Google Scholar] [CrossRef]
- Smith, B.; Ashburner, M.; Rosse, C.; Bard, J.; Bug, W.; Ceusters, W.; Goldberg, L.J.; Eilbeck, K.; Ireland, A.; Mungall, C.J.; et al. The OBO Foundry: Coordinated evolution of ontologies to support biomedical data integration. Nat. Biotechnol. 2007, 25, 1251–1255. [Google Scholar] [CrossRef]
- Hagberg, A.; Swart, P.J.; Schult, D.A. Exploring Network Structure, Dynamics, and Function Using NetworkX; Technical report; Los Alamos National Laboratory (LANL): Los Alamos, NM, USA, 2008. [Google Scholar]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Suzek, B.E.; Wang, Y.; Huang, H.; McGarvey, P.B.; Wu, C.H.; Consortium, U. UniRef clusters: A comprehensive and scalable alternative for improving sequence similarity searches. Bioinformatics 2015, 31, 926–932. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinf. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Wolf, T.; Debut, L.; Sanh, V.; Chaumond, J.; Delangue, C.; Moi, A.; Cistac, P.; Rault, T.; Louf, R.; Funtowicz, M.; et al. Huggingface’s transformers: State-of-the-art natural language processing. arXiv 2019, arXiv:1910.03771. [Google Scholar]
- Harris, C.R.; Millman, K.J.; Van Der Walt, S.J.; Gommers, R.; Virtanen, P.; Cournapeau, D.; Wieser, E.; Taylor, J.; Berg, S.; Smith, N.J.; et al. Array programming with NumPy. Nature 2020, 585, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Cortes, C.; Vapnik, V. Support-vector networks. Mach. Learn. 1995, 20, 273–297. [Google Scholar] [CrossRef]
- Abadi, M.; Barham, P.; Chen, J.; Chen, Z.; Davis, A.; Dean, J.; Devin, M.; Ghemawat, S.; Irving, G.; Isard, M.; et al. TensorFlow: A system for Large-Scale machine learning. In Proceedings of the 12th USENIX symposium on operating systems design and implementation (OSDI 16), Savannah, GA, USA, 2–4 November 2016; pp. 265–283. [Google Scholar]
- Chollet, F. Keras. 2015. Available online: https://keras.io (accessed on 6 June 2025).
- Kingma, D.P. Adam: A method for stochastic optimization. arXiv 2014, arXiv:1412.6980. [Google Scholar]
- Liu, F.T.; Ting, K.M.; Zhou, Z.H. Isolation-based anomaly detection. ACM Trans. Knowl. Discov. Data (TKDD) 2012, 6, 1–39. [Google Scholar] [CrossRef]
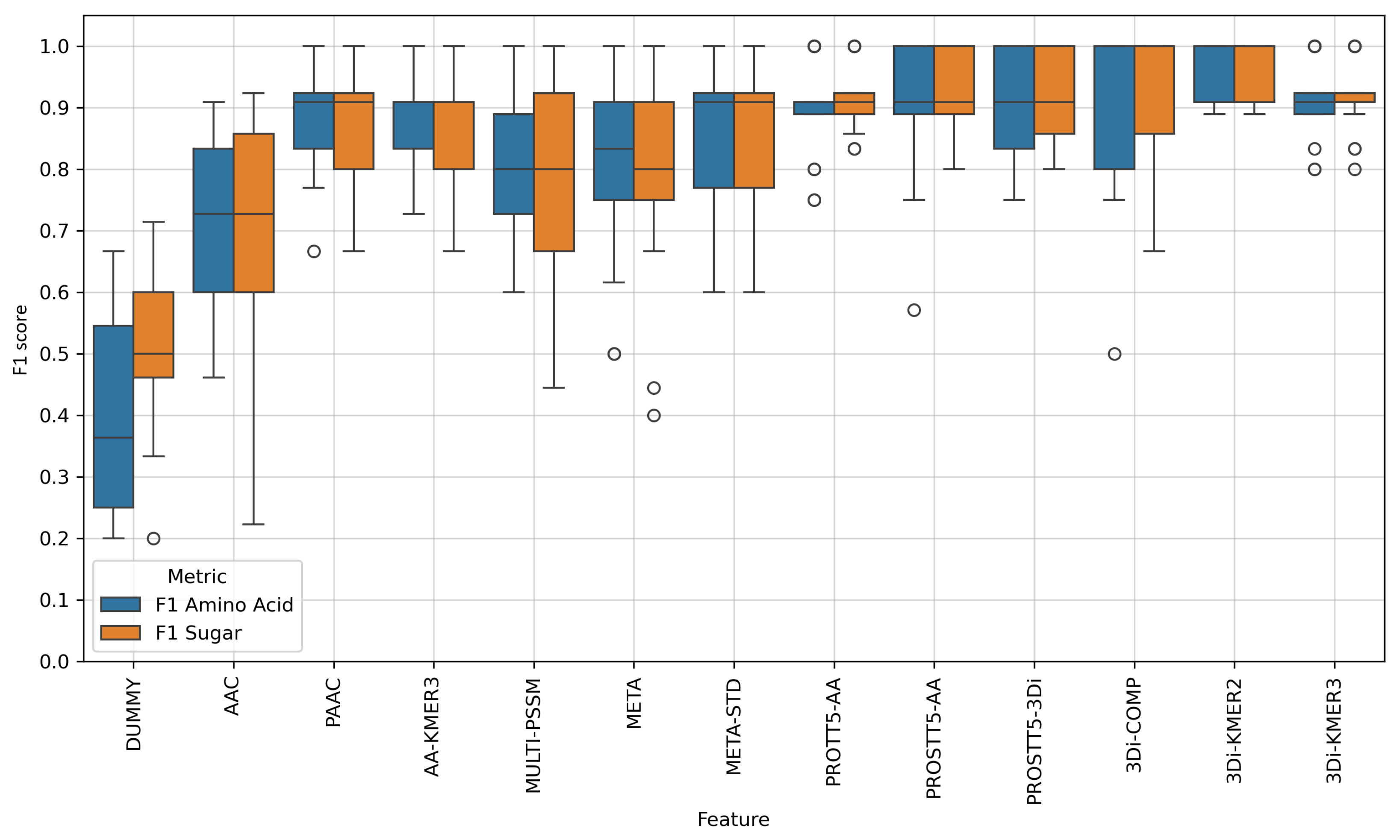

| Feature | Balanced Accuracy | F1 Amino Acid | F1 Sugar |
|---|---|---|---|
| DUMMY | 0.475 ± 0.120 | 0.400 ± 0.216 | 0.477 ± 0.207 |
| AAC | 0.754 ± 0.155 | 0.742 ± 0.156 | 0.754 ± 0.171 |
| PAAC | 0.925 ± 0.061 | 0.926 ± 0.059 | 0.915 ± 0.073 |
| AA-KMER3 | 0.915 ± 0.080 | 0.919 ± 0.075 | 0.899 ± 0.103 |
| MULTI-PSSM | 0.815 ± 0.122 | 0.807 ± 0.121 | 0.813 ± 0.141 |
| META | 0.859 ± 0.099 | 0.855 ± 0.093 | 0.851 ± 0.123 |
| META-STD | 0.889 ± 0.091 | 0.883 ± 0.097 | 0.890 ± 0.096 |
| COMB-KMER1 | 0.917 ± 0.084 | 0.909 ± 0.100 | 0.915 ± 0.085 |
| COMB-KMER2 | 0.945 ± 0.057 | 0.945 ± 0.058 | 0.942 ± 0.064 |
| COMB-KMER3 | 0.945 ± 0.052 | 0.947 ± 0.051 | 0.940 ± 0.058 |
| PROTT5-AA | 0.918 ± 0.063 | 0.910 ± 0.071 | 0.923 ± 0.058 |
| PROSTT5-AA | 0.944 ± 0.047 | 0.941 ± 0.050 | 0.946 ± 0.046 |
| PROSTT5-3DI | 0.937 ± 0.051 | 0.933 ± 0.053 | 0.938 ± 0.053 |
| 3Di-COMP | 0.959 ± 0.056 | 0.953 ± 0.065 | 0.963 ± 0.049 |
| 3Di-KMER2 | 0.959 ± 0.047 | 0.958 ± 0.049 | 0.959 ± 0.048 |
| 3Di-KMER3 | 0.959 ± 0.067 | 0.955 ± 0.074 | 0.963 ± 0.061 |
| Model | DNN | SVM | ||||
|---|---|---|---|---|---|---|
| Substrate | Ca/Cl | Ca/K | Cl/K | Ca/Cl | Ca/K | Cl/K |
| Feature | ||||||
| DUMMY | 0.522 ± 0.088 | 0.477 ± 0.062 | 0.498 ± 0.088 | 0.459 ± 0.084 | 0.505 ± 0.080 | 0.471 ± 0.078 |
| AAC | 0.684 ± 0.087 | 0.725 ± 0.053 | 0.712 ± 0.088 | 0.645 ± 0.084 | 0.703 ± 0.086 | 0.705 ± 0.081 |
| PAAC | 0.775 ± 0.094 | 0.805 ± 0.075 | 0.828 ± 0.063 | 0.767 ± 0.067 | 0.795 ± 0.059 | 0.787 ± 0.083 |
| AA-KMER3 | 0.663 ± 0.122 | 0.752 ± 0.080 | 0.798 ± 0.078 | 0.700 ± 0.097 | 0.743 ± 0.076 | 0.741 ± 0.077 |
| 3Di-COMP | 0.844 ± 0.060 | 0.831 ± 0.044 | 0.876 ± 0.050 | 0.839 ± 0.068 | 0.832 ± 0.055 | 0.885 ± 0.043 |
| 3Di-KMER2 | 0.854 ± 0.082 | 0.877 ± 0.049 | 0.845 ± 0.058 | 0.860 ± 0.068 | 0.832 ± 0.075 | 0.854 ± 0.065 |
| 3Di-KMER3 | 0.795 ± 0.078 | 0.855 ± 0.076 | 0.894 ± 0.060 | 0.825 ± 0.066 | 0.777 ± 0.076 | 0.845 ± 0.061 |
| COMB-KMER1 | 0.798 ± 0.077 | 0.848 ± 0.049 | 0.837 ± 0.071 | 0.817 ± 0.063 | 0.857 ± 0.063 | 0.864 ± 0.061 |
| COMB-KMER2 | 0.889 ± 0.063 | 0.861 ± 0.054 | 0.882 ± 0.059 | 0.880 ± 0.056 | 0.837 ± 0.066 | 0.880 ± 0.060 |
| COMB-KMER3 | 0.755 ± 0.095 | 0.801 ± 0.080 | 0.861 ± 0.076 | 0.798 ± 0.082 | 0.773 ± 0.079 | 0.847 ± 0.058 |
| PSSM-50-1 | 0.769 ± 0.084 | 0.834 ± 0.067 | 0.791 ± 0.071 | 0.755 ± 0.061 | 0.808 ± 0.063 | 0.783 ± 0.071 |
| PSSM-50-3 | 0.796 ± 0.052 | 0.863 ± 0.061 | 0.811 ± 0.075 | 0.803 ± 0.072 | 0.839 ± 0.056 | 0.806 ± 0.069 |
| PSSM-90-1 | 0.769 ± 0.105 | 0.776 ± 0.050 | 0.778 ± 0.083 | 0.739 ± 0.070 | 0.742 ± 0.045 | 0.759 ± 0.067 |
| PSSM-90-3 | 0.784 ± 0.084 | 0.773 ± 0.062 | 0.781 ± 0.087 | 0.739 ± 0.070 | 0.742 ± 0.045 | 0.759 ± 0.067 |
| MULTI-PSSM | 0.780 ± 0.077 | 0.833 ± 0.072 | 0.785 ± 0.082 | 0.783 ± 0.057 | 0.797 ± 0.052 | 0.770 ± 0.077 |
| META | 0.782 ± 0.072 | 0.803 ± 0.084 | 0.812 ± 0.065 | 0.826 ± 0.062 | 0.812 ± 0.061 | 0.784 ± 0.082 |
| META-STD | 0.851 ± 0.069 | 0.891 ± 0.038 | 0.850 ± 0.080 | 0.807 ± 0.055 | 0.884 ± 0.061 | 0.839 ± 0.072 |
| PROTT5-AA | 0.921 ± 0.059 | 0.915 ± 0.056 | 0.918 ± 0.043 | 0.922 ± 0.058 | 0.899 ± 0.054 | 0.889 ± 0.056 |
| PROSTT5-AA | 0.909 ± 0.065 | 0.903 ± 0.065 | 0.898 ± 0.061 | 0.911 ± 0.062 | 0.893 ± 0.054 | 0.898 ± 0.045 |
| PROSTT5-3DI | 0.894 ± 0.057 | 0.908 ± 0.053 | 0.908 ± 0.038 | 0.878 ± 0.068 | 0.911 ± 0.050 | 0.890 ± 0.061 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Denger, A.; Helms, V. Application of Protein Structure Encodings and Sequence Embeddings for Transporter Substrate Prediction. Molecules 2025, 30, 3226. https://doi.org/10.3390/molecules30153226
Denger A, Helms V. Application of Protein Structure Encodings and Sequence Embeddings for Transporter Substrate Prediction. Molecules. 2025; 30(15):3226. https://doi.org/10.3390/molecules30153226
Chicago/Turabian StyleDenger, Andreas, and Volkhard Helms. 2025. "Application of Protein Structure Encodings and Sequence Embeddings for Transporter Substrate Prediction" Molecules 30, no. 15: 3226. https://doi.org/10.3390/molecules30153226
APA StyleDenger, A., & Helms, V. (2025). Application of Protein Structure Encodings and Sequence Embeddings for Transporter Substrate Prediction. Molecules, 30(15), 3226. https://doi.org/10.3390/molecules30153226