
An Ab Initio Study of Aqueous Copper(I) Speciation in the Presence of Chloride



Abstract
1. Introduction
2. Results
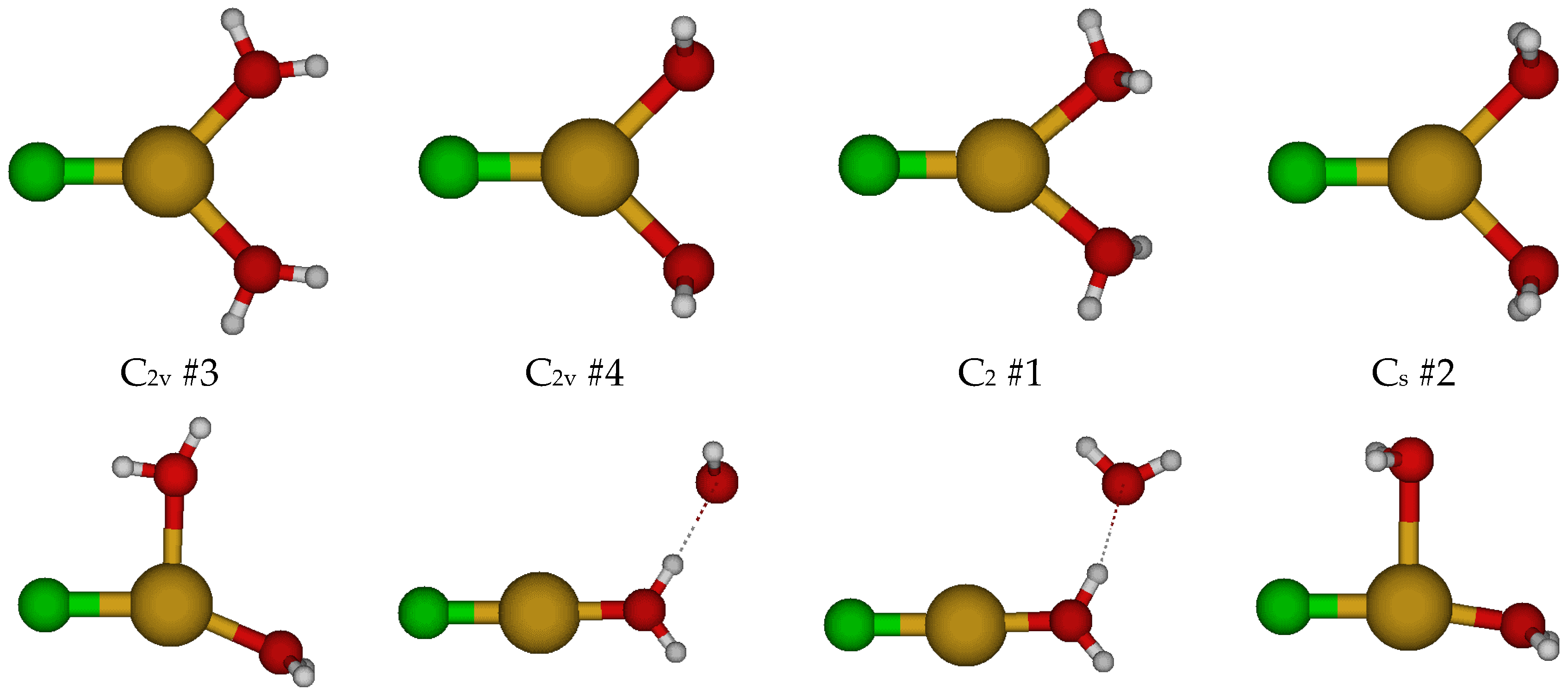
2.1. Structures of Monochloroaquacopper(I) Complexes, CuCl(H2O)n, n = 1–5, 7
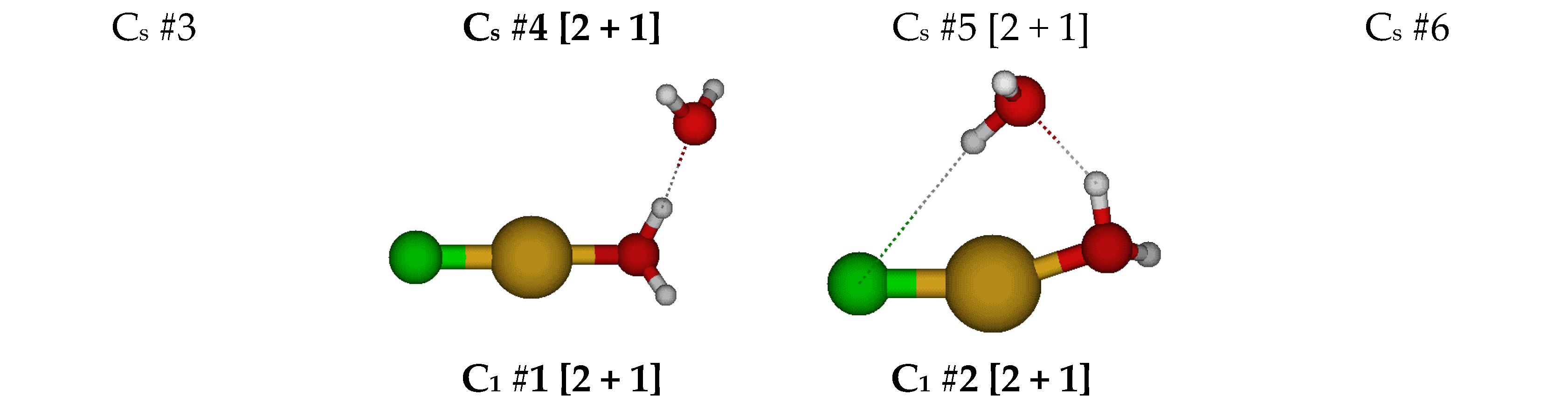
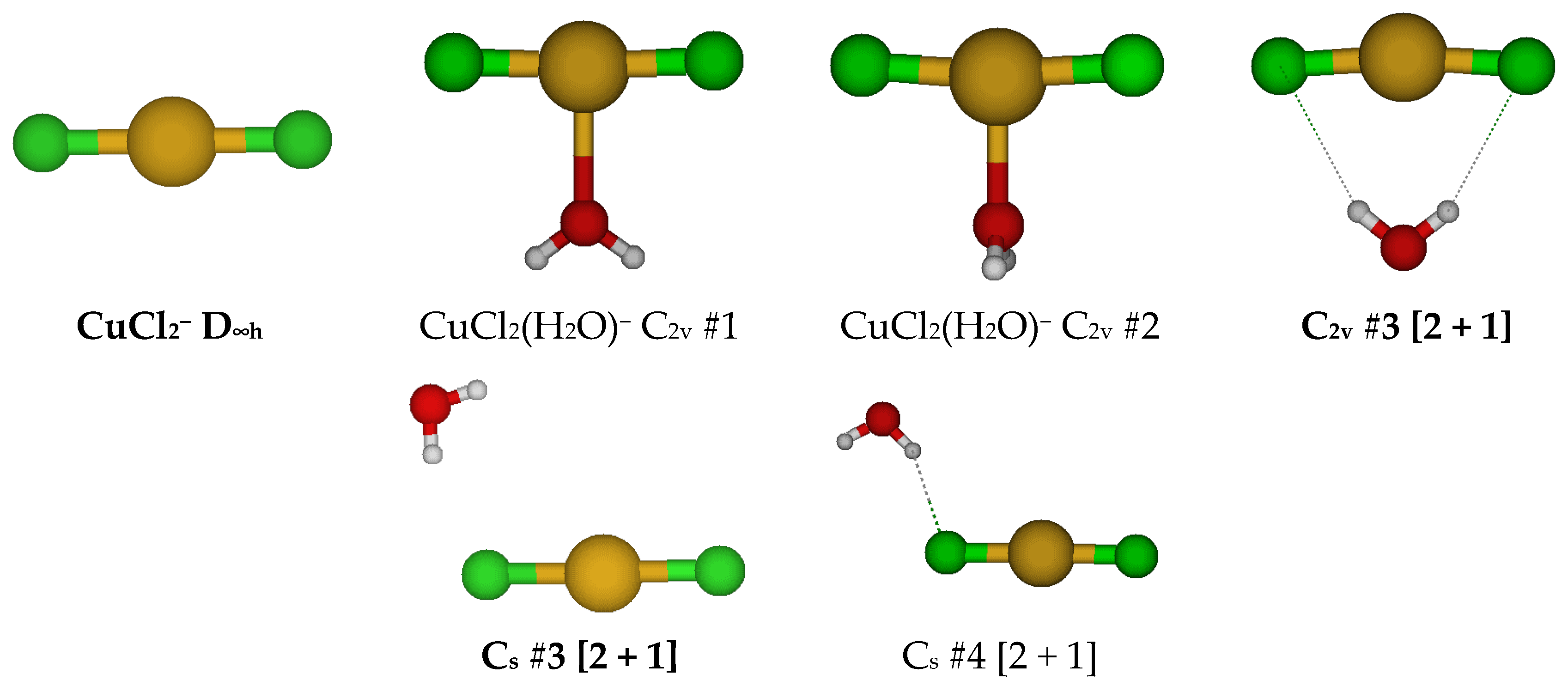
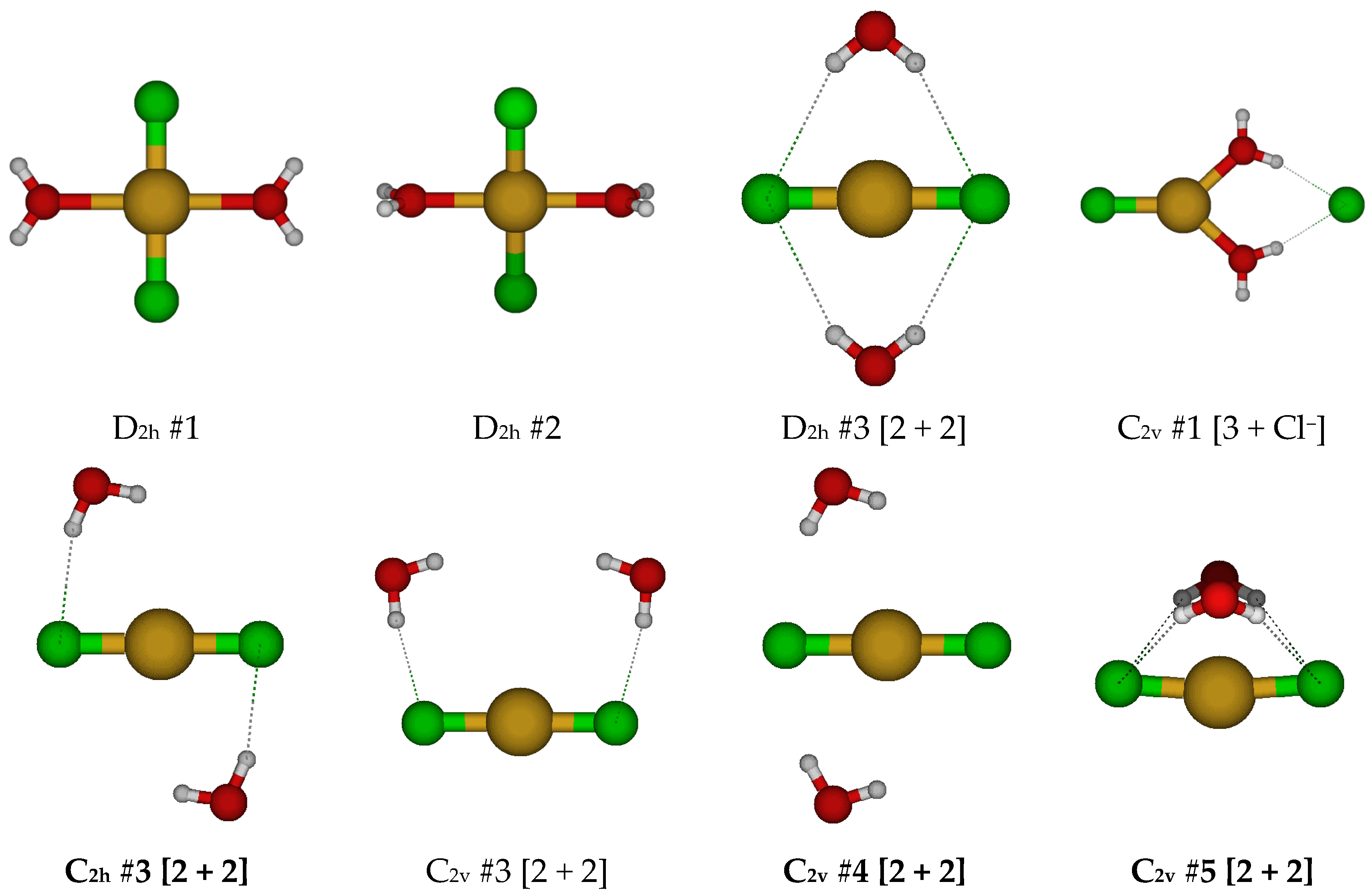
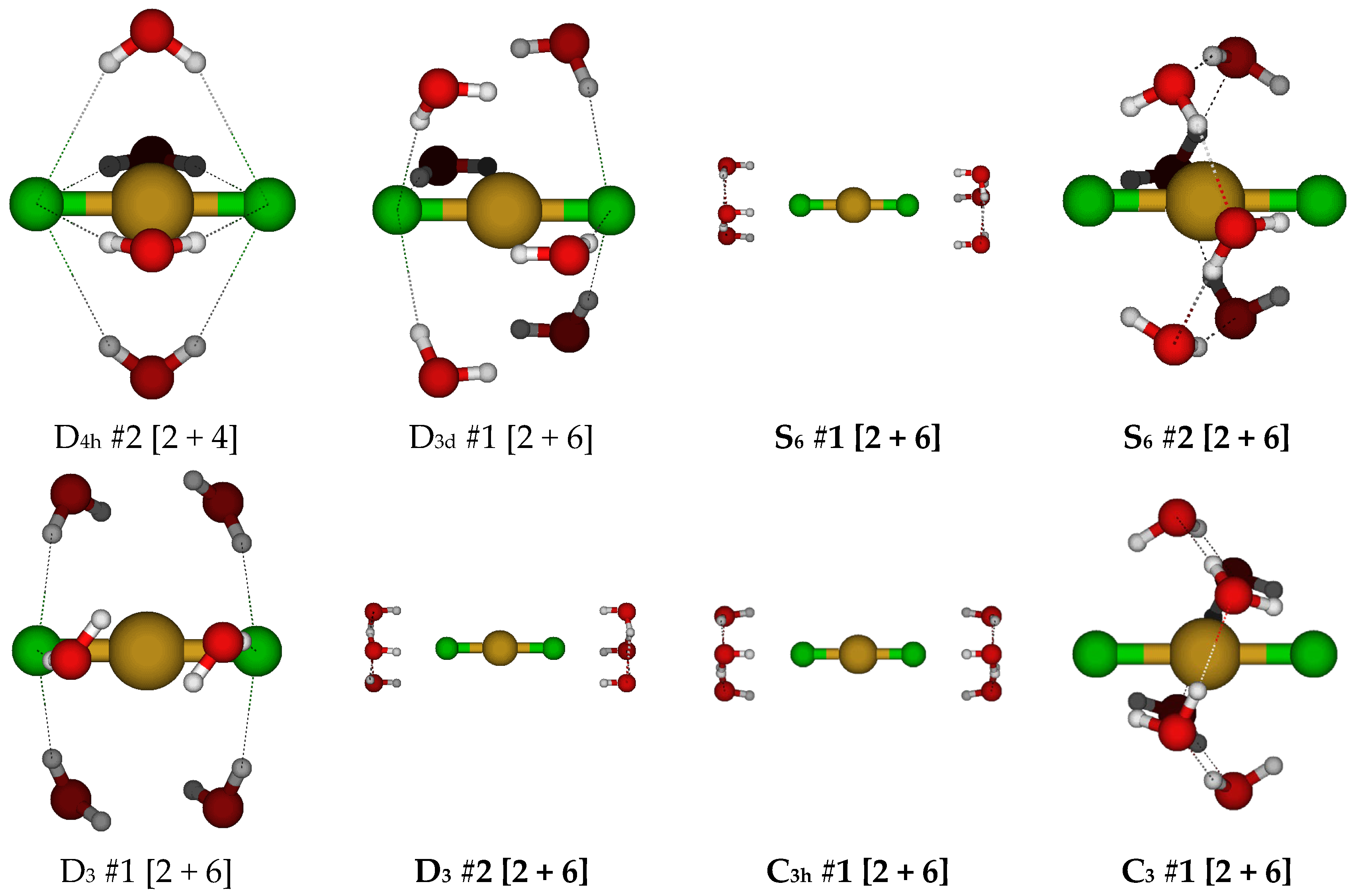
2.2. Structures of Dichloroaquacopper(I) Complexes, CuCl2(H2O)n−, n = 1–4, 6
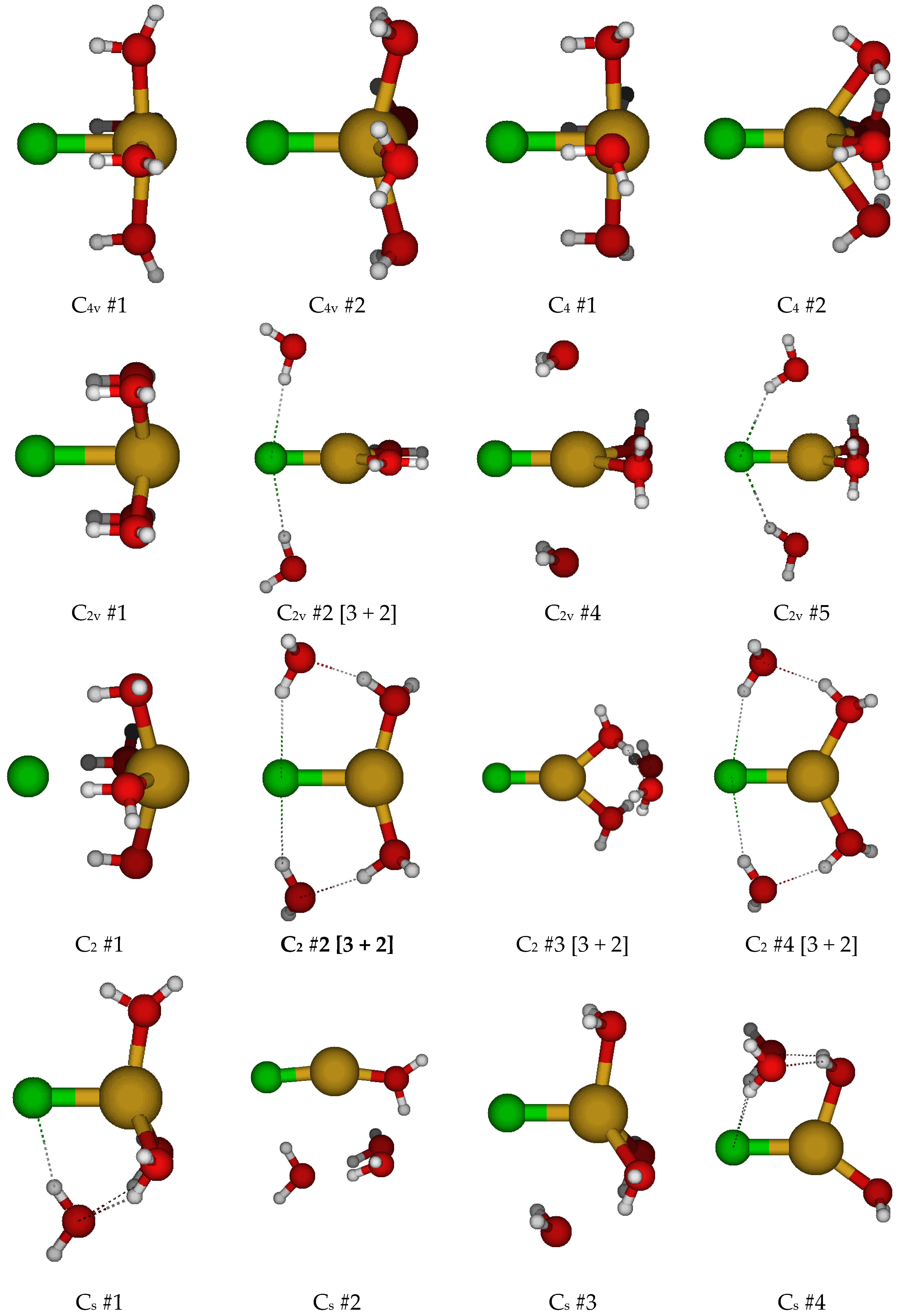
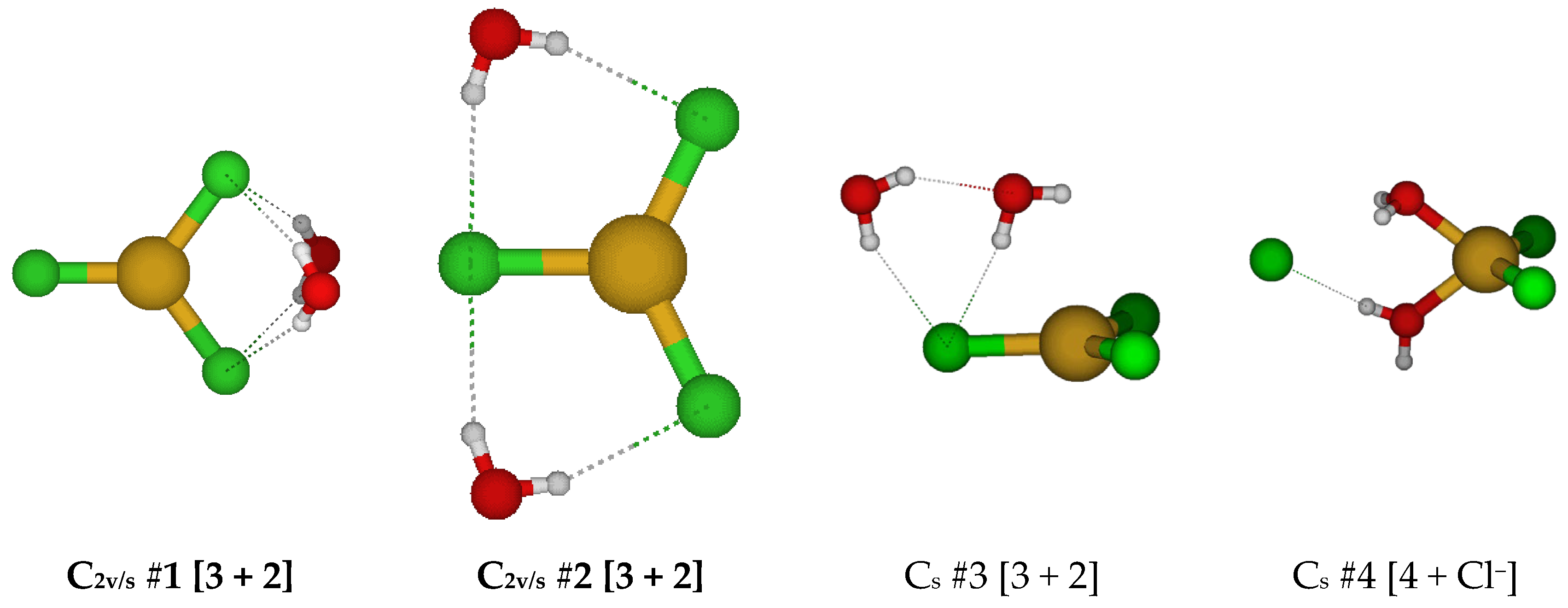
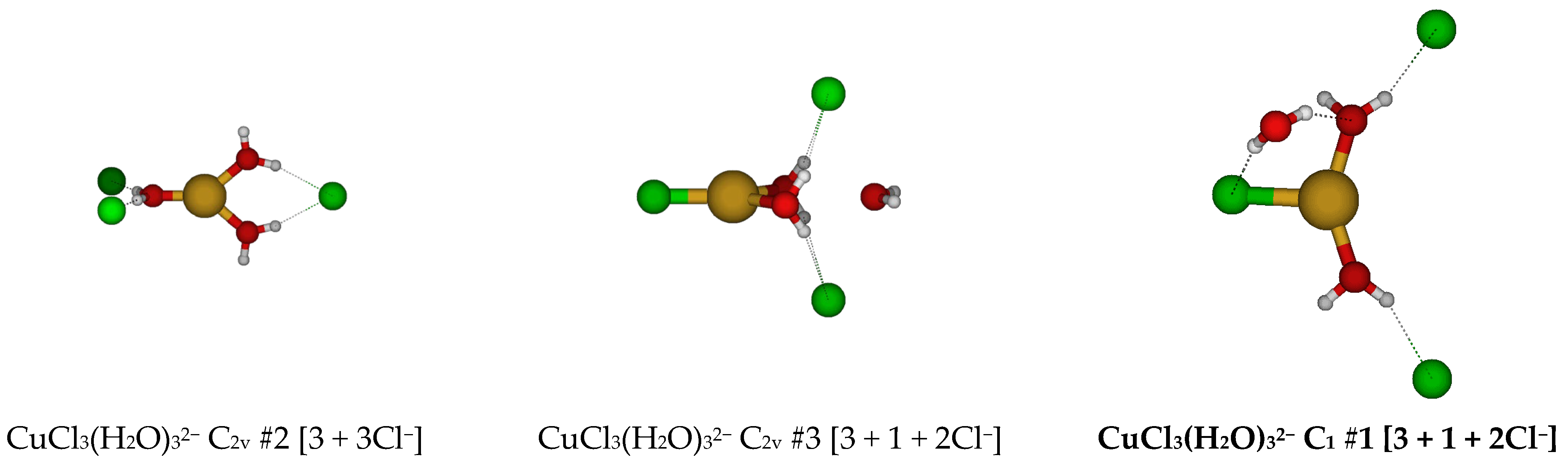
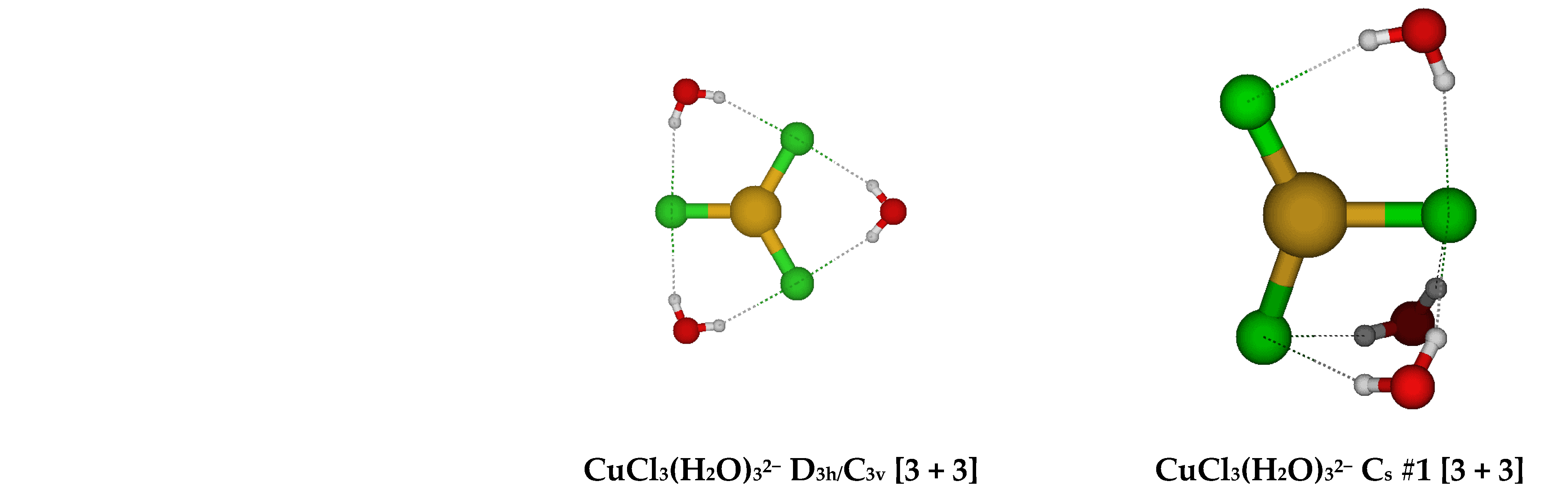
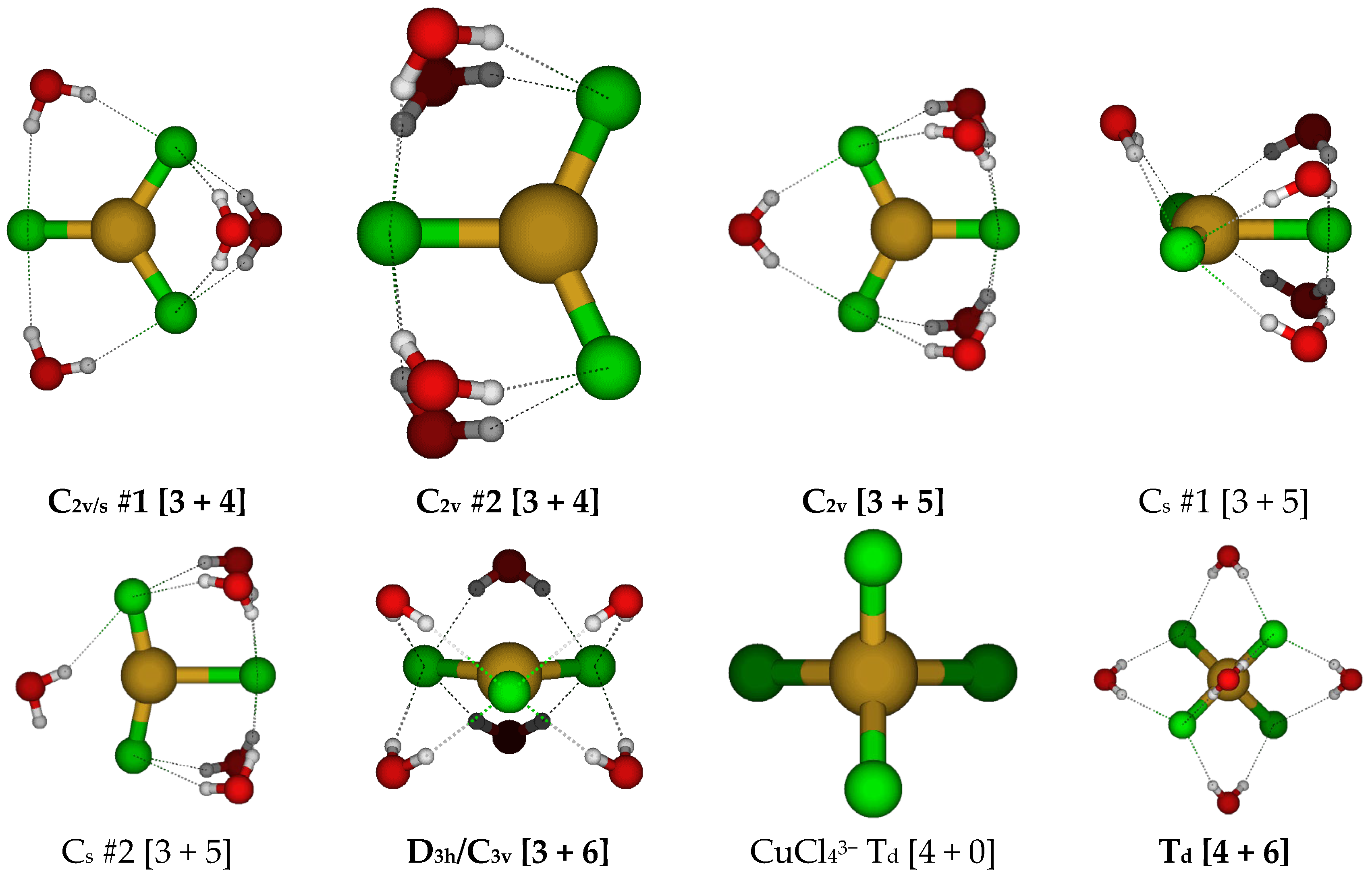
2.3. Structures of Trichloroaquacopper(I) Complexes, CuCl3(H2O)n2−, n = 1–4, 6
2.4. Structures of Tetrachloroaquacopper(I) Complexes, CuCl4(H2O)n3−, n = 0, 6
3. Discussion
4. Materials and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| HF | Hartree–Fock |
| MP2 | Second-order Moller–Plesset Perturbation Theory |
| B3LYP | Becke three-parameter exchange with Lee–Yang–Parr correlation functional |
References
- Klewe, B.; Pedersen, B. The Crystal Structure of Sodium Chloride Dihydrate. Acta Cryst. B 1974, 30, 2363–2371. [Google Scholar] [CrossRef]
- Journaux, B.; Pakhomova, A.; Collings, I.E.; Petitgirard, S.; Ballaran, T.B.; Brown, J.M.; Vance, S.D.; Chariton, S.; Prakapenka, V.B.; Huang, D.; et al. On the identification of hyperhydrated sodium chloride hydrates, stable at icy moon conditions. Proc. Nat. Acad. Sci. USA 2023, 120, e2217125120. [Google Scholar] [CrossRef]
- Pye, C.C.; Gunasekara, C.M. An ab Initio Investigation of the Hydration of Antimony(III). Liquids 2024, 4, 322–331. [Google Scholar] [CrossRef]
- Applegarth, L.M.S.G.A.; Corbeil, C.R.; Mercer, D.J.W.; Pye, C.C.; Tremaine, P.R. Raman and ab Initio Investigation of Aqueous Cu(I) Chloride Complexes from 25 to 80 °C. J. Phys. Chem. B 2014, 118, 204–214. [Google Scholar] [CrossRef]
- Pye, C.C.; Whynot, D.C.M.; Corbeil, C.R.; Mercer, D.J.W. Desymmetrization on geometry optimization: Application to an ab initio study of copper(I) hydration. Pure Appl. Chem. 2020, 92, 1643–1654. [Google Scholar] [CrossRef]
- Wyckoff, R.W.G.; Posnjak, E. The Crystal Structures of the Cuprous Halides. J. Am. Chem. Soc. 1922, 44, 30–36. [Google Scholar] [CrossRef]
- Wyckoff, R.W.G. Crystal Structures, 2nd ed.; Interscience: New York, NY, USA, 1963; Volume 1, pp. 85–237. [Google Scholar]
- Hull, S.; Keen, D.A. High-pressure polymorphism of the copper(I) halides: A neutron diffraction study to ~10 GPa. Phys. Rev. B 1994, 50, 5868–5885. [Google Scholar] [CrossRef]
- Rapoport, E.; Pistorius, C.W.F.T. Phase Diagrams of the Cuprous Halides to High Pressures. Phys. Rev. 1968, 172, 838–847. [Google Scholar] [CrossRef]
- Merrill, L. Behavior of the AB-Type Compounds at High Pressures and High Temperatures. J. Phys. Chem. Ref. Data 1977, 6, 1205–1252. [Google Scholar] [CrossRef]
- Brewer, L.; Lofgren, N.L. The Thermodynamics of Gaseous Cuprous Chloride, Monomer and Trimer. J. Am. Chem. Soc. 1950, 72, 3038–3045. [Google Scholar] [CrossRef]
- Klemperer, W.; Rice, S.A.; Berry, R.S. The Infrared Spectrum of Cuprous Chloride Vapor. J. Am. Chem. Soc. 1957, 79, 1810–1811. [Google Scholar] [CrossRef]
- Herzberg, G. Molecular Spectra. Vol 1. The Spectra of Diatomic Molecules, 2nd ed.; D. Van Nostrand: New York, NY, USA, 1950. [Google Scholar]
- Wong, C.-H.; Schomaker, V. An Electron Diffraction Investigation of the Structure of Cuprous Chloride Trimer. J. Phys. Chem. 1957, 61, 358–360. [Google Scholar] [CrossRef]
- Hilden, D.L.; Gregory, N.W. Vapor-Phase Absorbance and Thermodynamic Properties of Cuprous Chloride and Cuprous Bromide. J. Phys. Chem. 1972, 76, 1632–1637. [Google Scholar] [CrossRef]
- Andersson, S.; Jagner, S. Crystal Structures of Tetraphenylarsonium Dichlorocuprate(I), Tetraphenylphosphonium Dichlorocuprate(I) and Tetraphenylphosphonium Dibromocuprate(I). Acta Chem. Scand. A 1985, 39, 297–305. [Google Scholar] [CrossRef]
- Persson, I.; Sandstrom, M.; Steel, A.T.; Zapatero, M.J.; Akesson, R. A Large-Angle X-ray Scattering, XAFS, and Vibrational Spectroscopic Study of Copper(I) Halide Complexes in Dimethyl Sulfoxide, Acetonitrile, Pyridine, and Aqueous Solutions. Inorg. Chem. 1991, 30, 4075–4081. [Google Scholar] [CrossRef]
- Bowmaker, G.A.; Brockliss, L.D.; Whiting, R. Bonding in d10 Transition Metal Complexes. I. Infrared, Raman, and N.Q.R. Studies of Some Dihalocuprate(I) Complexes. Aust. J. Chem. 1973, 26, 29–42. [Google Scholar] [CrossRef]
- Pople, J.A.; Scott, A.P.; Wong, M.W.; Radom, L. Scaling Factors for Obtaining Fundamental Vibrational Frequencies and Zero-Point Energies from HF/6-31G* and MP2/6-31G* Harmonic Frequencies. Israel J. Chem. 1993, 33, 345–350. [Google Scholar] [CrossRef]
- Andersson, S.; Jagner, S. Coordination of Copper(I) in Two Novel Chlorocuprate(I) Anions; Structures of Tetramethylphosphonium catena-μ-Chloro-μ3-chloro-[μ-chloro-dicuprate(I)] and Bis(tetramethylphosphonium) Trichlorocuprate(I). Acta Chem. Scand. A 1988, 42, 691–697. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Cryst. B 2016, 72, 171–179. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Zakrzewski, V.G.; Montgomery, J.A., Jr.; Stratmann, R.E.; Burant, J.C.; et al. Gaussian 98, Revision A.9 [Computer Program]; Gaussian, Inc.: Pittsburgh, PA, USA, 1998. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A., Jr.; Vreven, T.; Kudin, K.N.; Burant, J.C.; et al. Gaussian 03, Revision D.02 [Computer Program]; Gaussian, Inc.: Wallingford, CT, USA, 2004. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision A.03 [Computer Program]; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species (n, Label) | HF | B3LYP | MP2 | |||
|---|---|---|---|---|---|---|
| 6-31+G* | 6-311+G* | 6-31+G* | 6-311+G* | 6-31+G* | 6-311+G* | |
| 0, C∞v | 2.1746 | 2.1834 | 2.0937 | 2.1023 | 2.0550 | 2.0638 |
| 0, Cu3Cl3, D3h | 2.3007 | 2.3098 | 2.2067 | 2.2120 | 2.1419 | 2.1494 |
| 1, Cs | 2.1713 | 2.1809 | 2.0922 | 2.0992 | 2.0422 | 2.0484 |
| 2, C1 #1 [2 + 1] | 2.1749 | 2.1865 | 2.0975 | 2.1054 | 2.0458 | 2.0528 |
| 2, C1 #2 [2 + 1] | 2.2084 | 2.2226 | 2.0972 | 2.1049 | n/a | n/a |
| 3, C1 #3 [2 + 2] | 2.2001 | 2.2135 | 2.1181 | 2.1256 | 2.0636 | 2.0712 |
| 4, C1 #4 [2 + 2 + 1] | 2.1987 | 2.2123 | 2.1189 | 2.1273 | 2.0642 | 2.0725 |
| 5, Cs #1 [2 + 4] | 2.2247 | 2.2416 | 2.1341 | 2.1427 | 2.0767 | 2.0849 |
| 7, Cs #1 [2 + 6] | 2.2253 | 2.2416 | 2.1401 | 2.1496 | 2.0812 | 2.0911 |
| Species (n, Label) | HF | B3LYP | MP2 | |||
|---|---|---|---|---|---|---|
| 6-31+G* | 6-311+G* | 6-31+G* | 6-311+G* | 6-31+G* | 6-311+G* | |
| 0, C∞v | 357 | 355 | 394 | 388 | 422 | 414 |
| 0, Cu3Cl3, D3h | 207, 239, 273, 300 | 210, 238, 276, 301 | 214, 294, 299, 368 | 213, 296, 302, 371 | 241, 335, 345, 417 | 235, 326, 340, 410 |
| 1, Cs | 260, 379 | 263, 380 | 327, 438 | 334, 446 | 362, 487 | 370, 490 |
| 2, C1 #1 [2 + 1] | 296, 390 | 298, 394 | 343, 458 | 354, 468 | 388, 505 | 386, 513 |
| 2, C1 #2 [2 + 1] | 276, 347 | 282, 345 | 341, 457 | 355, 469 | n/a | n/a |
| 3, C1 #3 [2 + 2] | 284, 377 | 285, 379 | 342, 459 | 341, 467 | 373, 495 | 370, 499 |
| 4, C1 #4 [2 + 2 + 1] | 275, 391 | 280, 395 | 336, 484 | 337, 491 | 386, 513 | 382, 520 |
| 5, Cs #1 [2 + 4] | 279, 392 | 279, 396 | 335, 483 | 331, 486 | 375, 525 | 364, 533 |
| 7, Cs #1 [2 + 6] | 284, 415 | 280, 422 | 336, 513 | 334, 514 | 375, 553 | 365, 559 |
| Species (n, Label) | HF | B3LYP | MP2 | |||
|---|---|---|---|---|---|---|
| 6-31+G* | 6-311+G* | 6-31+G* | 6-311+G* | 6-31+G* | 6-311+G* | |
| 0, D∞h | 2.2498 | 2.2620 | 2.1640 | 2.1709 | 2.0992 | 2.1059 |
| 1, Cs #1 | 2.257, 2.241 | 2.270, 2.253 | 2.167, 2.154 | 2.175, 2.161 | 2.102, 2.091 | 2.1025 |
| 2, C1 #1 | 2.256, 2.238 | 2.269, 2.250 | 2.168, 2.153 | 2.176, 2.160 | 2.103, 2.092 | 2.111, 2.098 |
| 3, C2 #4 | n/a | n/a | 2.1597 | n/a | 2.0929 | n/a |
| 3, C2 #5 | n/a | 2.2561 | n/a | n/a | n/a | 2.1022 |
| 3, C2 #6 | n/a | n/a | n/a | n/a | n/a | 2.0985 |
| 3, Cs #4 | n/a | n/a | 2.163, 2.152 | 2.172, 2.159 | n/a | n/a |
| 6, S6 #1 | 2.2557 | 2.2688 | 2.1620 | 2.1692 | 2.0970 | 2.1048 |
| 6, S6 #2 | 2.3630 | 2.3937 | 2.2516 | 2.2665 | 2.1674 | 2.1811 |
| 6, D3 #2 | 2.2557 | 2.2688 | 2.1620 | 2.1692 | 2.0970 | 2.1048 |
| 6, C3h #1 | 2.2558 | n/a | 2.1620 | 2.1692 | 2.0970 | n/a |
| Species (n, Label) | HF | B3LYP | MP2 | |||
|---|---|---|---|---|---|---|
| 6-31+G* | 6-311+G* | 6-31+G* | 6-311+G* | 6-31+G* | 6-311+G* | |
| 0, D∞h | 234, 332 | 232, 332 | 274, 376 | 271, 376 | 314, 432 | 305, 425 |
| 1, Cs #1 | 232, 334 | 233, 332 | 278, 383 | 272, 384 | 319, 436 | 311, 431 |
| 2, C1 #1 | 237, 336 | 234, 334 | 278, 383 | 275, 383 | 315, 436 | 306, 429 |
| 3, C2 #4 | n/a | n/a | 278, 384 | n/a | 323, 442 | n/a |
| 3, C2 #5 | n/a | 242, 334 | n/a | n/a | n/a | 312, 436 |
| 3, C2 #6 | n/a | n/a | n/a | n/a | n/a | 318, 437 |
| 3, Cs #4 | n/a | n/a | 287, 387 | 284, 387 | n/a | n/a |
| 6, S6 #1 | 243, 320/347 | 240, 313/341 | 286, 347/389 | 284, 346/390 | 318, 438 | 314, 430 |
| 6, S6 #2 | 202, 263 | 198, 253 | 240, 315 | 237, 312 | 274, 375 | 263, 364 |
| 6, D3 #2 | 243, 320/347 | 240, 313/342 | 286, 390 | 284, 390 | 318, 438 | 314, 430 |
| 6, C3h #1 | 243, 319/347 | n/a | 286, 390 | 284, 390 | 318, 438 | n/a |
| Species (n, Label) | HF | B3LYP | MP2 | |||
|---|---|---|---|---|---|---|
| 6-31+G* | 6-311+G* | 6-31+G* | 6-311+G* | 6-31+G* | 6-311+G* | |
| 0, D3h | 2.5402 | 2.5440 | 2.4035 | 2.4093 | 2.3019 | 2.3243 |
| 1, C2v | 2.479, 2.547 | 2.487, 2.548 | 2.343, 2.416 | 2.354, 2.419 | 2.262, 2.309 | 2.282, 2.333 |
| 2, C2v #1 | 2.480, 2.563 | 2.487, 2.558 | 2.346, 2.448 | 2.356, 2.447 | 2.267, 2.322 | 2.287, 2.348 |
| 2, Cs #1 | n/a | n/a | n/a | n/a | 2.266, 2.326 | 2.285, 2.351 |
| 2, C2v #2 | 2.431, 2.559 | 2.442, 2.554 | 2.301, 2.433 | 2.313, 2.433 | 2.230, 2.320 | 2.245, 2.340 |
| 3, D3h | 2.489 | 2.4905 | 2.3670 | 2.3717 | 2.2761 | 2.2967 |
| 3, C3v | n/a | n/a | n/a | n/a | 2.2757 | 2.2966 |
| 3, Cs | 2.431, 2.484, 2.584 | 2.441, 2.486, 2.573 | 2.297, 2.341, 2.511 | 2.309, 2.350, 2.495 | 2.231, 2.270, 2.348 | 2.248, 2.286, 2.368 |
| 4, C2v #1 | 2.437, 2.498 | 2.443, 2.495 | 2.315, 2.384 | 2.324, 2.384 | 2.238, 2.288 | 2.257, 2.306 |
| 4, Cs #1 | n/a | n/a | n/a | n/a | 2.237, 2.289 | 2.258, 2.304 |
| 4, C2v #2 | 2.433, 2.609 | 2.440, 2.591 | 2.284, 2.622 | 2.300, 2.567 | 2.237, 2.366 | 2.255, 2.370 |
| 5, C2v | 2.444, 2.509 | 2.446, 2.500 | 2.326, 2.412 | 2.332, 2.403 | 2.166, 2.565 | 2.268, 2.307 |
| 6, D3h | 2.4513 | 2.4494 | 2.3446 | 2.3450 | 2.2587 | 2.2734 |
| 6, C3v | n/a | n/a | n/a | n/a | n/a | 2.2733 |
| Species (n, Label) | HF | B3LYP | MP2 | |||
|---|---|---|---|---|---|---|
| 6-31+G* | 6-311+G* | 6-31+G* | 6-311+G* | 6-31+G* | 6-311+G* | |
| 0, D3h | 125, 144 | 130, 146 | 152, 172 | 160, 175 | 207, 206 | 198, 199 |
| 1, C2v | 120, 143, 158 | 126, 147, 161 | 145, 174, 202 | 148, 175, 199 | 197, 209, 238 | 187, 202, 229 |
| 2, C2v #1 | 124, 159, 163 | 132, 162, 168 | 142, 191, 204 | 151, 192, 209 | 198, 223, 245 * | 189, 215, 237 * |
| 2, Cs #1 | n/a | n/a | n/a | n/a | 196, 224, 247 | 187, 215, 238 |
| 2, C2v #2 | 116, 148, 180 | 121, 152, 183 | 132, 175, 225 | 137, 178, 226 | 185, 210, 264 | 178, 206, 257 |
| 3, D3h | 155, 163 | 162, 167 | 184, 189 | 190, 192 | 223, 233 * | 216, 224 * |
| 3, C3v | n/a | n/a | n/a | n/a | 223, 234 | 216, 225 |
| 3, Cs | 119, 161, 185 | 127, 165, 189 | 120, 197, 235 | 129, 199, 236 | 185, 227, 270 | 179, 220, 262 |
| 4, C2v #1 | 147, 164, 181 | 157, 170, 186 | 172, 190, 220 | 180, 196, 222 | 220, 225, 262 * | 213, 219, 253 * |
| 4, Cs #1 | n/a | n/a | n/a | n/a | 220, 225, 262 | 214, 219, 252 |
| 4, C2v #2 | 116, 171, 196 | 125, 174, 201 | 102, 212, 262 | 111, 210, 260 | 179, 236, 280 | 180, 228, 269 |
| 5, C2v | 153, 174, 186 | 163, 179, 193 | 173, 204, 223 | 181, 207, 229 | 220, 238, 264 | 218, 231, 253 |
| 6, D3h | 177, 178 | 182, 187 | 204, 205 | 208, 214 | 235, 252 | 229, 245 * |
| 6, C3v | n/a | n/a | n/a | n/a | n/a | 229, 244 |
| Species (n, Label) | HF | B3LYP | MP2 | |||
|---|---|---|---|---|---|---|
| 6-31+G* | 6-311+G* | 6-31+G* | 6-311+G* | 6-31+G* | 6-311+G* | |
| 0, Td | 3.0931 | 3.1508 | 2.8015 | 2.8246 | 2.5907 | 2.6269 |
| 6, Td | 2.7513 | 2.7443 | 2.5760 | 2.5652 | 2.4450 | 2.4663 |
| Species (n, Label) | HF | B3LYP | MP2 | |||
|---|---|---|---|---|---|---|
| 6-31+G* | 6-311+G* | 6-31+G* | 6-311+G* | 6-31+G* | 6-311+G* | |
| 0, Td | 85i,61 | 87i,55 | 78i,88 | 91i,79 | 56i,88 | 64i,88 |
| 6, Td | 45,122 | 41,122 | 78,137 | 85,143 | 120,166 | 119,164 |
| Order | Point Group | Irreducible Representation | Subgroup |
|---|---|---|---|
| 2 | Cs | A” | C1 |
| Ci | Au | C1 | |
| C2 | B | C1 | |
| 3, 5, 7 | C3/5/7 | En | C1 |
| 4 | C4 | B | C2 |
| E | C1 | ||
| D2 | B1/2/3 | C2 (z/y/x) | |
| C2v | A2 | C2 | |
| B1/2 | Cs (σv(xz)/σv(yz)) | ||
| C2h | Bg | Ci | |
| Au | C2 | ||
| Bu | Cs | ||
| S4 | B | C2 | |
| E | C1 | ||
| 6 | C6 | B | C3 |
| E1 | C1 | ||
| E2 | C2 | ||
| D3 | A2 | C3 | |
| E | C2 | ||
| C3v | A2 | C3 | |
| E | Cs | ||
| C3h | E’ | Cs | |
| A” | C3 | ||
| E” | C1 | ||
| S6 | Eg | Ci | |
| Au | C3 | ||
| Eu | C1 | ||
| T | E | D2 | |
| T | C3 | ||
| 8 | C8 | B | C4 |
| E1/3 | C1 | ||
| E2 | C2 | ||
| D4 | A2 | C4 | |
| B1/2 | D2 (C2’/C2”) | ||
| E | C2 (C2’ or C2”) | ||
| C4v | A2 | C4 | |
| B1/2 | C2v (σv/d) | ||
| E | Cs (σv/d) | ||
| C4h | Bg | C2h | |
| Eg | Ci | ||
| Au | C4 | ||
| Bu | S4 | ||
| Eu | Cs | ||
| D2h | B1/2/3g | C2h (C2(z/y/x)) | |
| Au | D2 | ||
| B1/2/3u | C2v (C2(z/y/x)) | ||
| D2d | A2 | S4 | |
| B1 | D2 | ||
| B2 | C2v | ||
| E | C2 (C2’) or Cs | ||
| S8 | B | C4 | |
| E1/3 | C1 | ||
| E2 | C2 | ||
| 12 | D3h | A2’ | C3h |
| E’ | C2v | ||
| A1” | D3 | ||
| A2” | C3v | ||
| E” | C2 or Cs (σv) | ||
| D3d | A2g | S6 | |
| Eg | C2h | ||
| A1u | D3 | ||
| A2u | C3v | ||
| Eu | C2 or Cs |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Whynot, D.C.M.; Corbeil, C.R.; Mercer, D.J.W.; Pye, C.C. An Ab Initio Study of Aqueous Copper(I) Speciation in the Presence of Chloride. Molecules 2025, 30, 3147. https://doi.org/10.3390/molecules30153147
Whynot DCM, Corbeil CR, Mercer DJW, Pye CC. An Ab Initio Study of Aqueous Copper(I) Speciation in the Presence of Chloride. Molecules. 2025; 30(15):3147. https://doi.org/10.3390/molecules30153147
Chicago/Turabian StyleWhynot, Daniel C. M., Christopher R. Corbeil, Darren J. W. Mercer, and Cory C. Pye. 2025. "An Ab Initio Study of Aqueous Copper(I) Speciation in the Presence of Chloride" Molecules 30, no. 15: 3147. https://doi.org/10.3390/molecules30153147
APA StyleWhynot, D. C. M., Corbeil, C. R., Mercer, D. J. W., & Pye, C. C. (2025). An Ab Initio Study of Aqueous Copper(I) Speciation in the Presence of Chloride. Molecules, 30(15), 3147. https://doi.org/10.3390/molecules30153147