
Comparative Nitrene-Transfer Chemistry to Olefins Mediated by First-Row Transition Metal Catalysts Supported by a Pyridinophane Macrocycle with N4 Ligation

 , , , and
, , , and
Abstract

1. Introduction
2. Results and Discussion
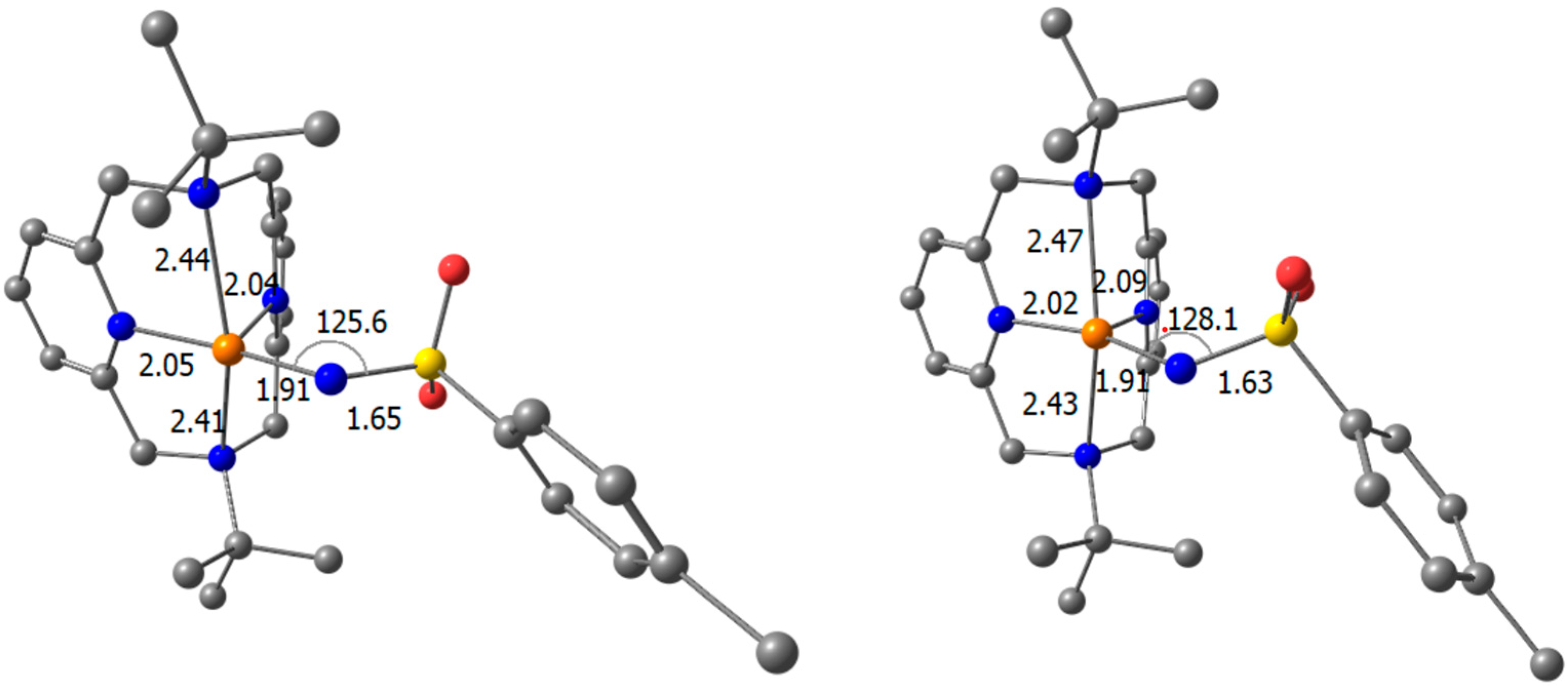
Computational Studies
3. Experimental Section
3.1. General Catalytic Olefin Aziridination Procedure
3.2. General Chemo-Selective Reaction Procedure
3.3. Catalytic Reaction Procedure for Hammett Analysis
3.4. Competitive Aziridinations of Deuterated Styrenes vs. Styrene (Evaluation of KIE)
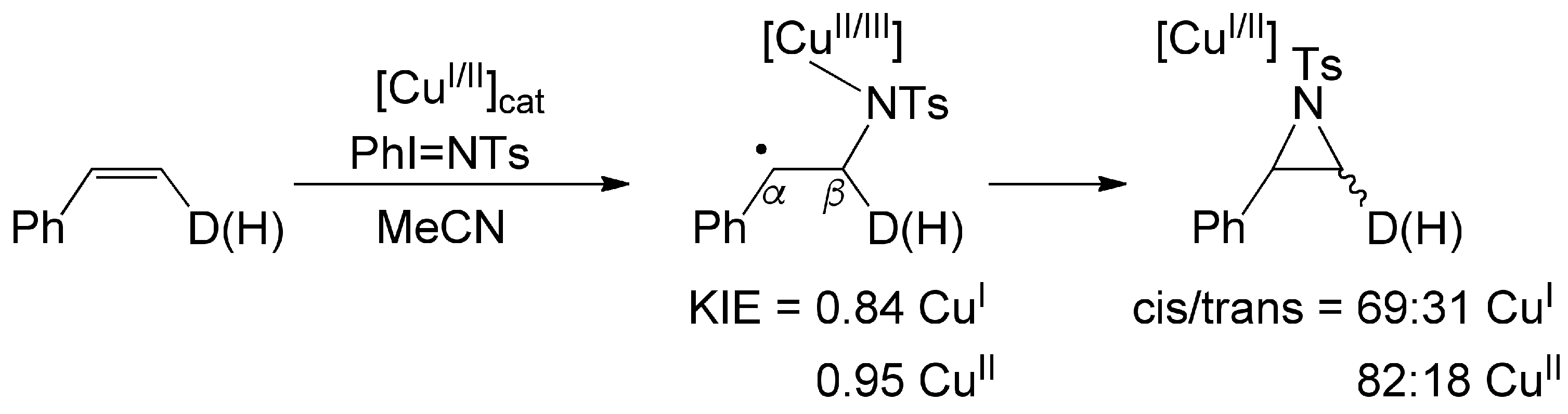
3.5. Stereochemical Scrambling in the Aziridination of cis-b-d1-Styrene
4. Conclusions
- (i)
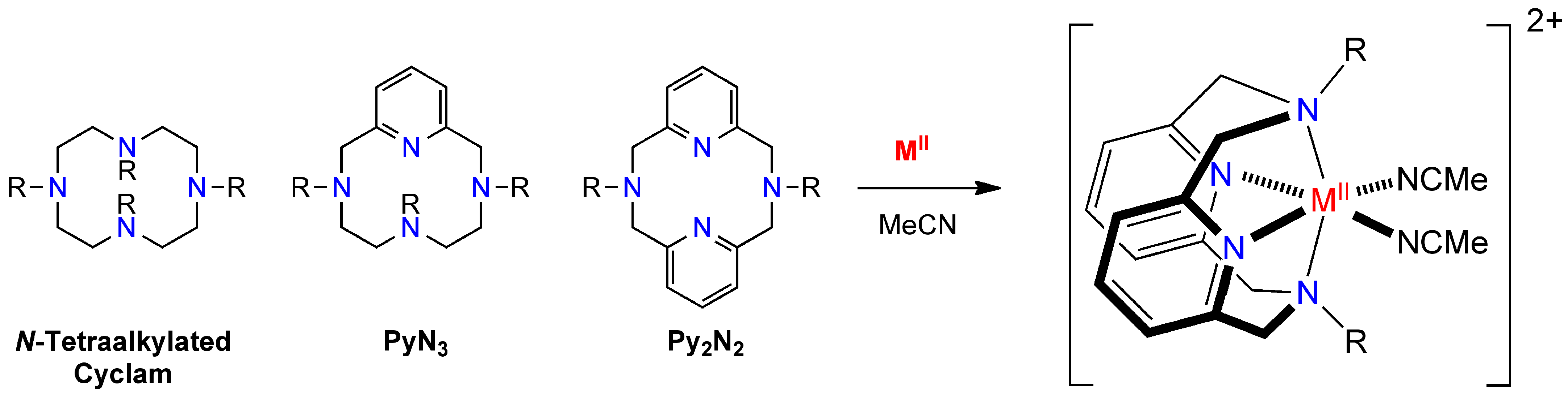
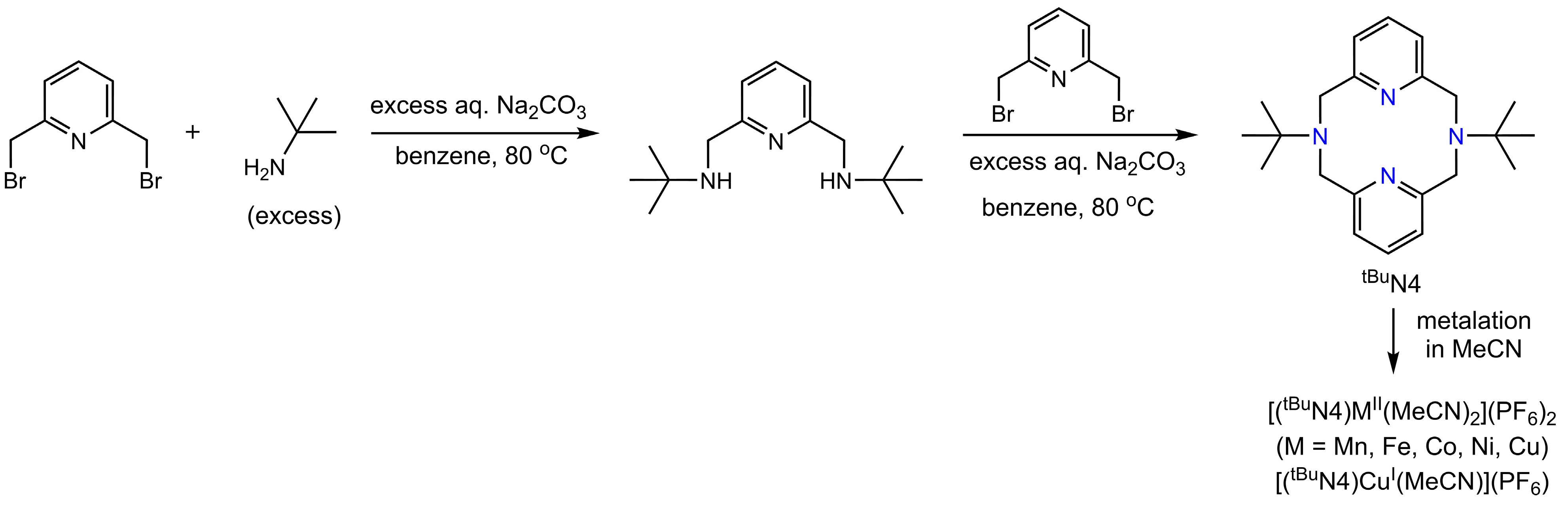
- A series of divalent compounds [(tBuN4)MII(CH3CN)2](PF6)2 (M = Mn, Fe, Co, Ni, and Cu) and monovalent [(tBuN4)CuI(CH3CN)](PF6) were synthesized with the known 12-membered Py2N2-type pyridinophane macrocycle tBuN4 and hexafluorophosphate as counter anion, for catalytic and mechanistic evaluation as mediators of olefin aziridinations via nitrene-transfer chemistry. The MnII and CuI congeners had been previously synthesized.
- (ii)
- For the aziridination of styrene and para-substituted styrenes using PhI=NTs, the Cu(I) reagent proved to be the most effective catalyst, with yields exceeding 80%, even for electron-withdrawing para substituents. The Cu(II) precatalyst is also highly competitive, with the exception of strongly electron-withdrawing para substituents (e.g., CF3 and NO2), for which yields drop to approximately 60%. All other divalent reagents mediate the same styrene aziridinations with modest yields (not exceeding 50%), demonstrating an approximate trend of Fe ≥ Mn ≥ Ni ≈ Co. As noted previously [63], these dicationic base metals give rise to metal-nitrene moieties that tend to decompose due to high electron deficiency, affording high TsNH2 yields.
- (iii)
- The best behaving Cu(I) and Cu(II) precatalysts also afford practicable aziridination yields with other styrenes, such electron-rich α-substituted congeners. However, bulky ortho- and/or β-substitution hamper yields, affecting turnover more prominently with the Cu(II) catalyst, presumably due to its less voluminous reaction cavity. Allylic aminations and other byproducts of aziridine-ring formation or cleavage are also observed, especially in chlorinated solvents that raise the acidity of the metal sites. As expected, the aziridination of aliphatic-substituted olefins is generally low-yielding. Notably, competitive aziridination of styrene vs. 1-hexene favors the former by a wide margin (27:1 for Cu(I)).
- (iv)
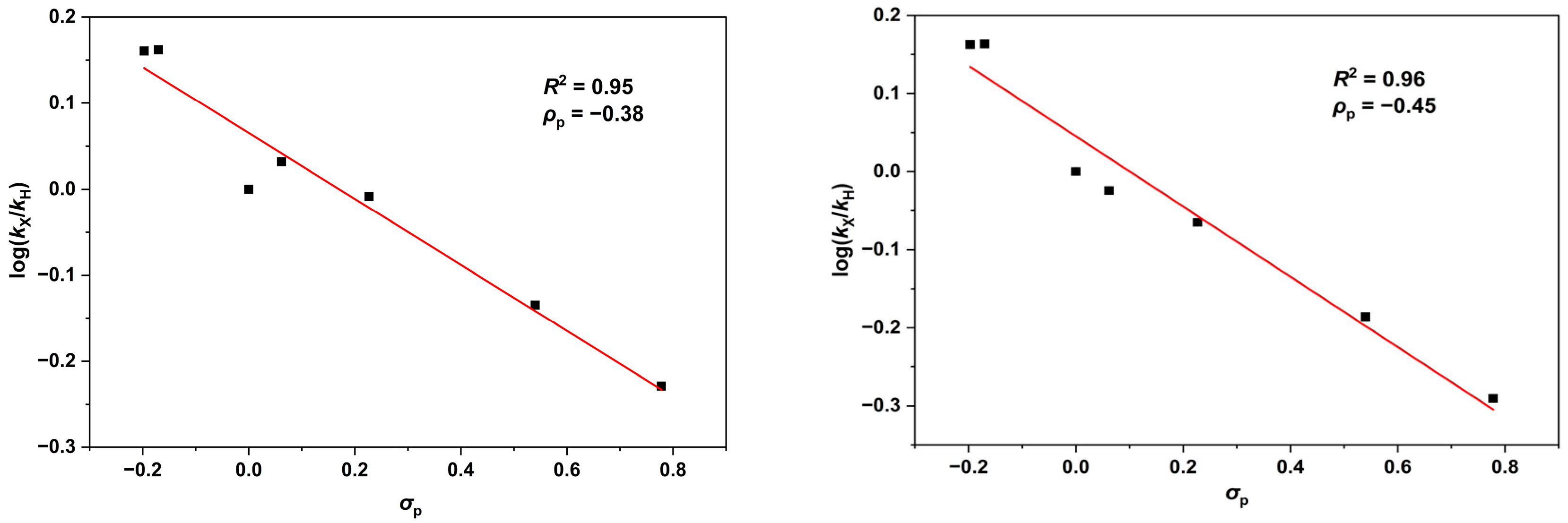
- Although Cu(I) or Cu(II) precatalysts have been shown to occasionally operate via the same catalytic cycle [73], the present study suggests that [(tBuN4)CuI]+ and [(tBuN4)CuII]2+ work via different catalytic manifolds. First, the Cu(I) congener mediates aziridination of unencumbered substrates, such as styrene, much faster than the Cu(II) analog. Although this may be due to the slower reaction of PhI=NTs with the Cu(II) site, other mechanistic indicators suggest that the Cu(I) and Cu(II) precatalysts may operate via distinct pathways. Hammett analysis and KIE parameters indicate that both precatalysts follow the general two-step mechanistic path (successive formation of N–Cβ and N–Cα bonds in the aziridination of styrene), but the Cu(I) catalyst is associated with a more modest positive charge at the styrenyl α-carbon as well as a larger inverse KIE value in the process of formation of the first N–Cβ bond, suggesting a more carboradical intermediate-like transition state. On the other hand, the Cu(II)-related manifold exhibits superior stereochemical integrity associated with the fast closure of the second, product-determining N–Cα bond.
- (v)
- Computational studies reveal that the triplet state of the putative five-coordinate [Cu] = NTs moiety (devoid of MeCN binding), generated from the Cu(I) precatalyst, is slightly (~2 kcal/mol) lower in energy than the broken-symmetry (BS) singlet. For the corresponding Cu(II) derived nitrene, the doublet and quartet states are almost degenerate. In all cases, significant spin density is localized on the nitrene N atom. Dual spin-state reactivity is thus possible for both mechanistic paths and may further complicate mechanistic analysis. Importantly, the fast-operating Cu(I)-mediated manifold is consistent with the superior electrophilicity of the Cu(I)-derived nitrene, as computed for all spin states.
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Thakur, M.S.; Singh, N.; Sharma, A.; Rana, R.; Syukor, A.R.A.; Naushad, M.; Kumar, S.; Kumar, M.; Singh, L. Metal coordinated macrocyclic complexes in different chemical transformations. Coord. Chem. Rev. 2022, 471, 214739. [Google Scholar] [CrossRef]
- Archibald, S.J. Coordination chemistry of macrocyclic ligands. Annu. Rep. Prog. Chem. Sect. A Inorg. Chem. 2009, 105, 297–332. [Google Scholar] [CrossRef]
- Joshi, T.; Graham, B.; Spiccia, L. Macrocyclic Metal Complexes for Metalloenzyme Mimicry and Sensor Development. Acc. Chem. Res. 2015, 48, 2366–2379. [Google Scholar] [CrossRef] [PubMed]
- Ruppel, J.V.; Fields, K.B.; Snyder, N.L.; Zhang, X.P. Metalloporphyrin-Catalyzed Asymmetric Atom/Group Transfer Reactions. In Handbook of Porphyrin Science; Kadish, K.M., Smith, K.M., Guilard, R., Eds.; World Scientific: Singapore, 2010; Volume 10, Chapter 43; pp. 1–182. [Google Scholar]
- Huang, X.; Bergsten, T.V.; Groves, J.T. Manganese-Catalyzed Late-Stage Aliphatic C–H Azidation. J. Am. Chem. Soc. 2015, 137, 5300–5303. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Huang, J.S.; Zhou, Z.Y.; Che, C.-M. Isolation and X-ray Crystal Structure of an Unusual Biscarbene Metal Complex and Its Reactivity toward Cyclopropanation and Allylic C–H Insertion of Unfunctionalized Alkenes. J. Am. Chem. Soc. 2001, 123, 4843–4844. [Google Scholar] [CrossRef] [PubMed]
- Key, H.M.; Dydio, P.; Liu, Z.; Rha, J.Y.; Nazarenko, A.; Seyedkazemi, V.; Clark, D.S.; Hartwig, J.F. Beyond Iron: Iridium-Containing P450 Enzymes for Selective Cyclopropanations of Structurally Diverse Alkenes. ACS Cent. Sci. 2017, 3, 302–308. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Lang, K.; Tao, J.; Marshall, M.K.; Cheng, Q.; Cui, X.; Wojtas, L.; Zhang, X.P. Next-Generation D2-Symmetric Chiral Porphyrins for Cobalt(II)-Based Metalloradical Catalysis: Catalyst Engineering by Distal Bridging. Angew. Chem. Int. Ed. 2019, 58, 2670–2674. [Google Scholar] [CrossRef] [PubMed]
- Fantauzzi, S.; Gallo, E.; Rose, E.; Raoul, N.; Caselli, A.; Issa, S.; Ragaini, F.; Cenini, S. Asymmetric Cyclopropanation of Olefins Catalyzed by Chiral Cobalt(II)-Binaphthyl Porphyrins. Organometallics 2008, 27, 6143–6151. [Google Scholar] [CrossRef]
- Zdilla, M.J.; Dexheimer, J.L.; Abu-Omar, M.M. Hydrogen Atom Transfer Reactions of Imido Manganese(V) Corrole: One Reaction with Two Mechanistic Pathways. J. Am. Chem. Soc. 2007, 129, 11505–11511. [Google Scholar] [CrossRef] [PubMed]
- Paradine, S.M.; Griffin, J.R.; Zhao, J.; Petronico, A.L.; Miller, S.M.; White, M.C. A manganese catalyst for highly reactive yet chemoselective intramolecular C(sp3)–H amination. Nat. Chem. 2015, 7, 987–994. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Gao, S.; Lal, R.G.; Hicks, M.H.; Oyala, P.H.; Arnold, F.H. Reaction Discovery Using Spectroscopy Insights from an Enzymatic C–H Amination Intermediate. J. Am. Chem. Soc. 2024, 146, 20556–20562. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Das, A.; Alfonzo, E.; Sicinski, K.M.; Rieger, D.; Arnold, F.H. Enzymatic Nitrogen Incorporation Using Hydroxylamine. J. Am. Chem. Soc. 2023, 145, 20196–20201. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Vargas, D.A.; Ma, P.; Sengupta, A.; Zhu, L.; Houk, K.N.; Fasan, R. Stereoselective construction of β-, γ- and δ-lactam rings via enzymatic C–H amidation. Nat. Catal. 2024, 7, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Steck, V.; Kolev, J.N.; Ren, X.; Fasan, R. Mechanism-Guided Design and Discovery of Efficient Cytochrome P450-Derived C–H Amination Biocatalysts. J. Am. Chem. Soc. 2020, 142, 10343–10357. [Google Scholar] [CrossRef] [PubMed]
- Jeong, D.; Valentine, J.S.; Cho, J. Bio-inspired mononuclear nonheme metal peroxo complexes: Synthesis, structures and mechanistic studies toward understanding enzymatic reactions. Coord. Chem. Rev. 2023, 480, 215021. [Google Scholar] [CrossRef]
- Collins, T.J.; Ryabov, A.D. Targeting of High-Valent Iron-TAML. Activators of Hydrocarbons and Beyond. Chem. Rev. 2017, 117, 9140–9162. [Google Scholar] [CrossRef] [PubMed]
- Somasundar, Y.; Shen, L.Q.; Hoane, A.G.; Tang, L.L.; Mills, M.R.; Burton, A.E.; Ryabov, A.D.; Collins, T.J. Structural, Mechanistic, and Ultradilute Catalysis Portrayal of Substrate Inhibition in the TAML−Hydrogen Peroxide Catalytic Oxidation of the Persistent Drug and Micropollutant, Propranolol. J. Am. Chem. Soc. 2018, 140, 12280–12289. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Li, X.-X.; Lee, Y.-M.; Jang, Y.; Seo, M.S.; Hong, S.; Cho, K.-B.; Fukuzumi, S.; Nam, W. Electron-Transfer and Redox Reactivity of High-Valent Iron Imido and Oxo Complexes with the Formal Oxidation States of Five and Six. J. Am. Chem. Soc. 2020, 142, 3891–3904. [Google Scholar] [CrossRef] [PubMed]
- van Leest, N.P.; Tepaske, M.A.; Venderbosch, B.; Oudsen, J.-P.H.; Tromp, M.; van der Vlugt, J.I.; de Bruin, B. Electronically Asynchronous Transition States for C–N Bond Formation by Electrophilic [CoIII(TAML)]-Nitrene Radical Complexes Involving Substrate-to-Ligand Single-Electron Transfer and a Cobalt-Centered Spin Shuttle. ACS Catal. 2020, 10, 7449–7463. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-W.; Chuang, Y.-W.; Lu, K.-Y.; Wang, Y.-H. First-Row Transition-Metal Complexes with Tetra-Amido Macrocyclic Ligands for Water and C(sp3)–H Bond Oxidation: Performance Benchmarking Using Free Energy Relationships. ChemCatChem 2024, 16, e202301375. [Google Scholar] [CrossRef]
- Chandrachud, P.P.; Bass, H.M.; Jenkins, D.M. Synthesis of Fully Aliphatic Aziridines with a Macrocyclic Tetracarbene Iron Catalyst. Organometallics 2016, 35, 1652–1657. [Google Scholar] [CrossRef]
- Isbill, S.B.; Chandrachud, P.P.; Kern, J.L.; Jenkins, D.M.; Roy, S. Elucidation of the Reaction Mechanism of C2 + N1 Aziridination from Tetracarbene Iron Catalysts. ACS Catal. 2019, 9, 6223–6233. [Google Scholar] [CrossRef] [PubMed]
- Barefield, E.K. Coordination chemistry of N-tetraalkylated cyclam ligand—A status report. Coord. Chem. Rev. 2010, 254, 1607–1627. [Google Scholar] [CrossRef]
- Tseberlidis, G.; Intrieri, D.; Caselli, A. Catalytic Applications of Pyridine-Containing Macrocyclic Complexes. Eur. J. Inorg. Chem. 2017, 3589–3603. [Google Scholar] [CrossRef]
- Haque, A.; Ilmi, R.; Al-Busaidi, I.J.; Khan, M.S. Coordination chemistry and application of mono- and oligopyridine-based macrocycles. Coord. Chem. Rev. 2017, 350, 320–339. [Google Scholar] [CrossRef]
- Zhu, W.; Wu, P.; Larson, V.A.; Kumar, A.; Li, X.-X.; Seo, M.S.; Lee, Y.-M.; Wang, B.; Lehnert, N.; Nam, W. Electronic Structure and Reactivity of Mononuclear Nonheme Iron–Peroxo Complexes as a Biomimetic Model of Rieske Oxygenases: Ring Size Effects of Macrocyclic Ligands. J. Am. Chem. Soc. 2024, 146, 250–262. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.; Sarangi, R.; Nam, W. Mononuclear Metal–O2 Complexes Bearing Macrocyclic N-Tetramethylated Cyclam Ligands. Acc. Chem. Res. 2012, 45, 1321–1330. [Google Scholar] [CrossRef] [PubMed]
- Zhou, A.; Prakash, J.; Rohde, G.T.; Klein, J.E.M.N.; Kleespies, S.T.; Draksharapu, A.; Fan, R.; Guo, Y.; Cramer, C.J.; Que, L., Jr. The Two Faces of Tetramethylcyclam in Iron Chemistry: Distinct Fe–O–M Complexes Derived from [FeIV(Oanti/syn)(TMC)]2+ Isomers. Inorg. Chem. 2017, 56, 518–527. [Google Scholar] [CrossRef] [PubMed]
- Bigelow, J.O.; England, J.; Klein, J.E.M.N.; Farquhar, E.R.; Frisch, J.R.; Martinho, M.; Mandal, D.; Münck, E.; Shaik, S.; Que, L., Jr. Oxoiron(IV) Tetramethylcyclam Complexes with Axial Carboxylate Ligands: Effect of Tethering the Carboxylate on Reactivity. Inorg. Chem. 2017, 56, 3287–3301. [Google Scholar] [CrossRef] [PubMed]
- Derrick, J.S.; Kim, Y.; Tak, H.; Park, K.; Cho, J.; Kim, S.H.; Lim, M.H. Stereochemistry of metal tetramethylcyclam complexes directed by an unexpected anion effect. Dalton Trans. 2017, 46, 13166–13170. [Google Scholar] [CrossRef] [PubMed]
- Panza, N.; Tseberlidis, G.; Caselli, A.; Vicente, R. Recent progress in the chemistry of 12-membered pyridine-containing tetraazamacrocycles: From synthesis to catalysis. Dalton Trans. 2022, 51, 10635–10657. [Google Scholar] [CrossRef] [PubMed]
- Jeong, D.; Kim, K.; Lee, Y.; Cho, J. Synthetic Advances for Mechanistic Insights: Metal–Oxygen Intermediates with a Macrocyclic Pyridinophane System. Acc. Chem. Res. 2024, 57, 120–130. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Moon, D.; Cho, J. Controlling Redox Potential of a Manganese(III)–Bis(hydroxo) Complex through Protonation and the Hydrogen-Atom Transfer Reactivity. J. Am. Chem. Soc. 2024, 146, 15796–15805. [Google Scholar] [CrossRef] [PubMed]
- Jeong, D.; Yan, J.J.; Noh, H.; Hedman, B.; Hodgson, K.O.; Solomon, E.I.; Cho, J. Oxidation of Naphthalene with a Manganese(IV) Bis(hydroxo) Complex in the Presence of Acid. Angew. Chem. Int. Ed. 2018, 57, 7764–7768. [Google Scholar] [CrossRef] [PubMed]
- Jeong, D.; Ohta, T.; Cho, J. Structure and Reactivity of a Mononuclear Nonheme Manganese(III)–Iodosylarene Complex. J. Am. Chem. Soc. 2018, 140, 16037–16041. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.-T.; Xu, S.; Dickie, D.A.; Smith, J.M. A Robust Mn Catalyst for H2O2 Disproportionation in Aqueous Solution. Eur. J. Inorg. Chem. 2013, 3867–3873. [Google Scholar] [CrossRef]
- Lee, W.-T.; Muñoz III, S.B.; Dickie, D.A.; Smith, J.M. Ligand Modification Transforms a Catalase Mimic into a Water Oxidation Catalyst. Angew. Chem. Int. Ed. 2014, 53, 9856–9859. [Google Scholar] [CrossRef] [PubMed]
- To, W.-P.; Chow, T.W.-S.; Tse, C.-W.; Guan, X.; Huang, J.-S.; Che, C.-M. Water oxidation catalysed by iron complex of N,N′-dimethyl-2,11-diaza[3,3](2,6)pyridinophane. Spectroscopy of iron–oxo intermediates and density functional theory calculations. Chem. Sci. 2015, 6, 5891–5903. [Google Scholar] [CrossRef] [PubMed]
- Jeong, D.; Kim, H.; Cho, J. Oxidation of Aldehydes into Carboxylic Acids by a Mononuclear Manganese(III) Iodosylbenzene Complex through Electrophilic C–H Bond Activation. J. Am. Chem. Soc. 2023, 145, 888–897. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Lee, Y.; Tripodi, G.L.; Roithová, J.; Lee, S.; Cho, J. Controlling Reactivity through Spin Manipulation: Steric Bulkiness of Peroxocobalt(III) Complexes. J. Am. Chem. Soc. 2024, 146, 20660–20667. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Cho, D.; Noh, H.; Ohta, T.; Baik, M.-H.; Cho, J. Controlled Regulation of the Nitrile Activation of a Peroxocobalt(III) Complex with Redox-Inactive Lewis Acidic Metals. J. Am. Chem. Soc. 2021, 143, 11382–11392. [Google Scholar] [CrossRef] [PubMed]
- Noh, H.; Jeong, D.; Ohta, T.; Ogura, T.; Valentine, J.S.; Cho, J. Distinct Reactivity of a Mononuclear Peroxocobalt(III) Species toward Activation of Nitriles. J. Am. Chem. Soc. 2017, 139, 10960–10963. [Google Scholar] [CrossRef] [PubMed]
- Khusnutdinova, J.R.; Luo, J.; Rath, N.P.; Mirica, L.M. Late First-Row Transition Metal Complexes of a Tetradentate Pyridinophane Ligand: Electronic Properties and Reactivity Implications. Inorg. Chem. 2013, 52, 3920–3932. [Google Scholar] [CrossRef] [PubMed]
- Khusnutdinova, J.R.; Rath, N.P.; Mirica, L.M. The Aerobic Oxidation of a Pd(II) Dimethyl Complex Leads to Selective Ethane Elimination from a Pd(III) Intermediate. J. Am. Chem. Soc. 2012, 134, 2414–2422. [Google Scholar] [CrossRef] [PubMed]
- Khusnutdinova, J.R.; Rath, N.P.; Mirica, L.M. The Conformational Flexibility of the Tetradentate Ligand tBuN4 is Essential for the Stabilization of (tBuN4)PdIII Complexes. Inorg. Chem. 2014, 53, 13112–13129. [Google Scholar] [CrossRef] [PubMed]
- Tang, F.; Zhang, Y.; Rath, N.P.; Mirica, L.M. Detection of Pd(III) and Pd(IV) Intermediates during the Aerobic Oxidative C–C Bond Formation from a Pd(II) Dimethyl Complex. Organometallics 2012, 31, 6690–6696. [Google Scholar] [CrossRef]
- Smith, S.M.; Planas, O.; Gómez, L.; Rath, N.P.; Rivas, X.; Mirica, L.M. Aerobic C–C and C–O bond formation reactions mediated by high-valent nickel species. Chem. Sci. 2019, 10, 10366–10372. [Google Scholar] [CrossRef] [PubMed]
- Raje, S.; Mani, K.; Kandasamy, P.; Butcher, R.J.; Angamuthu, R. Bioinspired Oxidative Cleavage of Aliphatic C–C Bonds Utilizing Aerial Oxygen by Nickel Acireductone Dioxygenase Mimics. Eur. J. Inorg. Chem. 2019, 2164–2167. [Google Scholar] [CrossRef]
- Na, H.; Wessel, A.J.; Kim, S.-T.; Baik, M.-H.; Mirica, L.M. Csp3–H bond activation mediated by a Pd(II) complex under mild conditions. Inorg. Chem. Front. 2024, 11, 4415–4423. [Google Scholar] [CrossRef]
- Sinha, S.; Mirica, L.M. Electrocatalytic O2 Reduction by an Organometallic Pd(III) Complex via a Binuclear Pd(III) Intermediate. ACS Catal. 2021, 11, 5202–5211. [Google Scholar] [CrossRef]
- Wang, P.; Liang, G.; Boyd, C.L.; Webster, C.E.; Zhao, X. Catalytic H2 Evolution by a Mononuclear Cobalt Complex with a Macrocyclic Pentadentate Ligand. Eur. J. Inorg. Chem. 2019, 2134–2139. [Google Scholar] [CrossRef]
- Patil, P.H.; Filonenko, G.A.; Lapointe, S.; Fayzullin, R.R.; Khusnutdinova, J.R. Interplay between the Conformational Flexibility and Photoluminescent Properties of Mononuclear Pyridinophanecopper(I) Complexes. Inorg. Chem. 2018, 57, 10009–10027. [Google Scholar] [CrossRef] [PubMed]
- Filonenko, G.A.; Fayzullin, R.R.; Khusnutdinova, J.R. Intramolecular non-covalent interactions as a strategy towards controlled photoluminescence in copper(I) complexes. J. Mater. Chem. C 2017, 5, 1638–1645. [Google Scholar] [CrossRef]
- Castano, B.; Guidone, S.; Gallo, E.; Ragaini, F.; Casati, N.; Macchi, P.; Sisti, M.; Caselli, A. Asymmetric cyclopropanation of olefins catalysed by Cu(I) complexes of chiral pyridine-containing macrocyclic ligands (Pc-L*). Dalton Trans. 2013, 42, 2451–2462. [Google Scholar] [CrossRef] [PubMed]
- Caselli, A.; Cesana, F.; Gallo, E.; Casati, N.; Macchi, P.; Sisti, M.; Celentano, G.; Cenini, S. Designing new ligands: Asymmetric cyclopropanation by Cu(I) complexes based on functionalized pyridine-containing macrocyclic ligands. Dalton Trans. 2008, 4202–4205. [Google Scholar] [CrossRef] [PubMed]
- Tseberlidis, G.; Caselli, A.; Vicente, R. Carbene X–H bond insertions catalyzed by copper(I) macrocyclic pyridine-containing ligand (PcL) complexes. J. Organomet. Chem. 2017, 835, 1–5. [Google Scholar] [CrossRef]
- Wessel, A.J.; Schultz, J.W.; Tang, F.; Duan, H.; Mirica, L.M. Improved synthesis of symmetrically & asymmetrically N-substituted pyridinophane derivatives. Org. Biomol. Chem. 2017, 15, 9923–9931. [Google Scholar] [CrossRef] [PubMed]
- Zheng, B.; Tang, F.; Luo, J.; Schultz, J.W.; Rath, N.P.; Mirica, L.M. Organometallic Nickel(III) Complexes Relevant to Cross-Coupling and Carbon-Heteroatom Bond Formation Reactions. J. Am. Chem. Soc. 2014, 136, 6499–6504. [Google Scholar] [CrossRef] [PubMed]
- Schultz, J.W.; Fuchigami, K.; Zheng, B.; Rath, N.P.; Mirica, L.M. Isolated Organometallic Nickel(III) and Nickel(IV) Complexes Relevant to Carbon-Carbon Bond Formation Reactions. J. Am. Chem. Soc. 2016, 138, 12928–12934. [Google Scholar] [CrossRef] [PubMed]
- Khusnutdinova, J.R.; Rath, N.P.; Mirica, L.M. Stable Mononuclear Organometallic Pd(III) Complexes and Their C–C Bond Formation Reactivity. J. Am. Chem. Soc. 2010, 132, 7303–7305. [Google Scholar] [CrossRef] [PubMed]
- Che, C.-M.; Li, Z.-Y.; Wong, K.-Y.; Poon, C.-K.; Mak, T.C.W.; Peng, S.-M. A Simple Synthetic Route to N,N′-Dialkyl-2,11-Diaza[3.3](2,6)-Pyridinophanes. Crystal Structures of N,N′-Di-Tert-Butyl-2,11-Diaza[3.3](2,6)Pyridiniphane and Its Copper(II) Complex. Polyhedron 1994, 13, 771–776. [Google Scholar] [CrossRef]
- Sahoo, S.K.; Harfmann, B.; Ai, L.; Wang, Q.; Mohapatra, S.; Choudhury, A.; Stavropoulos, P. Cationic Divalent Metal Sites (M = Mn, Fe, Co) Operating as Both Nitrene-Transfer Agents and Lewis Acids Toward Mediating the Synthesis of Three- and Five-Membered N-Heterocycles. Inorg. Chem. 2023, 62, 10743–10761. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, S.K.; Harfmann, B.; Bhatia, H.; Singh, H.; Balijapelly, S.; Choudhury, A.; Stavropoulos, P. A Comparative Study of Cationic Copper(I) Reagents Supported by Bipodal Tetramethylguanidinyl-Containing Ligands as Nitrene-Transfer Catalysts. ACS Omega 2024, 9, 15697–15708. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.; Jensen, M.P. Half-Sandwich Scorpionates as Nitrene Transfer Catalysts. Organometallics 2012, 31, 8055–8058. [Google Scholar] [CrossRef]
- Anderson, C.M.; Aboelenen, A.M.; Jensen, M.P. Competitive Intramolecular Amination as a Clock for Iron-Catalyzed Nitrene Transfer. Inorg. Chem. 2019, 58, 1107–1119. [Google Scholar] [CrossRef] [PubMed]
- Lakk-Bogáth, D.; Török, P.; Pintarics, D.; Kaizer, J. A Mechanistic Study on Iron-Based Styrene Aziridination: Understanding Epoxidation via Nitrene Hydrolysis. Molecules 2024, 29, 3470. [Google Scholar] [CrossRef] [PubMed]
- Al-Ajlouni, A.; Espenson, J.H. Epoxidation of Styrenes by Hydrogen Peroxide as Catalyzed by Methylrhenium Trioxide. J. Am. Chem. Soc. 1995, 117, 9243–9250. [Google Scholar] [CrossRef]
- Müller, P.; Baud, C.; Jacquier, Y.; Moran, M.; Nägeli, I. Rhodium(II)-Catalyzed Aziridinations and CH Insertions with [N-(p-Nitrobenzenesulfonyl)Imino]Phenyliodinane. J. Phys. Org. Chem. 1996, 9, 341–347. [Google Scholar] [CrossRef]
- Neuenschwander, U.; Hermans, I. The Conformations of Cyclooctene: Consequences for Epoxidation Chemistry. J. Org. Chem. 2011, 76, 10236–10240. [Google Scholar] [CrossRef] [PubMed]
- Bagchi, V.; Paraskevopoulou, P.; Das, P.; Chi, L.; Wang, Q.; Choudhury, A.; Mathieson, J.S.; Cronin, L.; Pardue, D.B.; Cundari, T.R.; et al. A Versatile Tripodal Cu(I) Reagent for C–N Bond Construction via Nitrene-Transfer Chemistry: Catalytic Perspectives and Mechanistic Insights on C–H Aminations/Amidinations and Olefin Aziridinations. J. Am. Chem. Soc. 2014, 136, 11362–11381. [Google Scholar] [CrossRef] [PubMed]
- Bagchi, V.; Kalra, A.; Das, P.; Paraskevopoulou, P.; Gorla, S.; Ai, L.; Wang, Q.; Mohapatra, S.; Choudhury, A.; Sun, Z.; et al. Comparative Nitrene-Transfer Chemistry to Olefinic Substrates Mediated by a Library of Anionic Mn(II) Triphenylamido-Amine Reagents and M(II) Congeners (M = Fe, Co, Ni) Favoring Aromatic over Aliphatic Alkenes. ACS Catal. 2018, 8, 9183–9206. [Google Scholar] [CrossRef]
- Evans, D.A.; Faul, M.M.; Bilodeau, M.T. Development of the Copper-Catalyzed Olefin Aziridination Reaction. J. Am. Chem. Soc. 1994, 116, 2742–2753. [Google Scholar] [CrossRef]
- Maestre, L.; Sameera, W.M.C.; Díaz-Requejo, M.M.; Maseras, F.; Pérez, P.J. A General Mechanism for the Copper- and Silver-Catalyzed Olefin Aziridination Reactions: Concomitant Involvement of the Singlet and Triplet Pathways. J. Am. Chem. Soc. 2013, 135, 1338–1348. [Google Scholar] [CrossRef] [PubMed]
- Plajer, A.J.; Colebatch, A.L.; Rizzuto, F.J.; Pröhm, P.; Bond, A.D.; García-Rodríguez, R.; Wright, D.S. How Changing the Bridgehead Can Affect the Properties of Tripodal Ligands. Angew. Chem. Int. Ed. 2018, 57, 6648–6652. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.-K. Establishment and Successful Application of the σJJ• Scale of Spin-Delocalization Substituent Constants. Acc. Chem. Res. 1997, 30, 283–289. [Google Scholar] [CrossRef]
- Pérez-Ruíz, J.; Martínez, A.R.; Díaz-Requejo, M.M.; Pérez, P.J. Introducing the Aziridination of Fluorinated Olefins by Metal-Catalyzed Nitrene Transfer. Angew. Chem. Int. Ed. 2025, 64, e202419188. [Google Scholar] [CrossRef] [PubMed]
- Neese, F. Software Update: The ORCA Program System. Version 5.0 Wiley. Interdiscip. Rev. Comput. Mol. Sci. 2022, 12, e1606. [Google Scholar] [CrossRef]
- Stahn, M.; Ehlert, S.; Grimme, S. Extended Conductor-like Polarizable Continuum Solvation Model (CPCM-X) for Semiempirical Methods. J. Phys. Chem. A 2023, 127, 7036–7043. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Xu, X.; Truhlar, D.G. Minimally Augmented Karlsruhe Basis Sets. Theor. Chem. Acc. 2011, 128, 295–305. [Google Scholar] [CrossRef]
- Mulliken, R.S. Electronic Popylation Analysis on LCAO-MO Molecular Wave Functions. I. J. Chem. Phys. 1951, 23, 1833–1840. [Google Scholar] [CrossRef]
- Jupp, A.R.; Johnstone, T.C.; Stephan, D.W. Improving the Global Electrophilicity Index (GEI) as a Measure of Lewis Acidity. Inorg. Chem. 2018, 57, 14764–14771. [Google Scholar] [CrossRef] [PubMed]
- Coin, G.; Patra, R.; Rana, S.; Biswas, J.P.; Dubourdeaux, P.; Clemancey, M.; de Visser, S.P.; Maiti, D.; Maldivi, P.; Latour, J.-M. Fe-Catalyzed Aziridination is Governed by the Electron Affinity of the Active Imido-Iron Species. ACS Catal. 2020, 10, 10010–10020. [Google Scholar] [CrossRef]
- Kalra, A.; Bagchi, V.; Paraskevopoulou, P.; Das, P.; Ai, L.; Sanakis, Y.; Raptopoulos, G.; Mohapatra, S.; Choudhury, A.; Sun, Z.; et al. Is the Electrophilicity of the Metal Nitrene the Sole Predictor of Metal-Mediated Nitrene Transfer to Olefins? Secondary Contributing Factors as Revealed by a Library of High-Spin Co(II) Reagents. Organometallics 2021, 40, 1974–1996. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Zheng, W.-H. Kinetic Resolution of Tertiary Alcohols by Chiral Organotin-Catalyzed O-Acylation. Org. Lett. 2022, 24, 2349–2353. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.Y.; Lee, C.G.; Kim, J.N. A practical synthesis of N-tosylimines of arylaldehydes. Tetrahedron Lett. 2003, 44, 1231–1234. [Google Scholar] [CrossRef]
- Bruker’s APEX3, SAINT and SHELXTL; Bruker AXS Inc.: Madison, WI, USA, 2017.
- Bruker’s SMART; Bruker AXS Inc.: Madison, WI, USA, 2002.
- Bruker’s SAINT, SADABS, SHELXTL-PLUS; Bruker AXS Inc.: Madison, WI, USA, 2008.
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. 2008, A64, 112. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M.; Hubshle, C.B.; Dittrich, B. Shelxle: A Qt graphical user interface for SHELXL. J. Appl. Cryst. 2011, 44, 1281–1284. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||||
|---|---|---|---|---|---|---|---|
| Entry No. | Product | tBuN4MnII Yield (%) | tBuN4FeII Yield (%) | tBuN4CoII Yield (%) | tBuN4NiII Yield (%) | tBuN4CuII Yield (%) | tBuN4CuI Yield (%) |
| 1. | X = H | 39 | 40 | 32 | 37 | 86 | 91 |
| 2. | X = Me | 23 | 40 | 22 | 28 | 79 | 89 |
| 3. | X = tBu | 30 | 47 | 33 | 28 | 86 | 91 |
| 4. | X = Cl | 38 | 47 | 32 | 27 | 97 | 92 |
| 5. | X = F | 30 | 46 | 28 | 25 | 98 | 84 |
| 6. | X = CF3 | 23 | 36 | 20 | 16 | 57 | 82 |
| 7. | X = NO2 | 28 | 48 | 11 | 11 | 63 | 89 |
| Entry No. | Substrate | Product | DCM Yield (%) | 1,2-DCE Yield (%) | MeCN Yield (%) | |
|---|---|---|---|---|---|---|
| 1. |  |  | X = H | 88 | 96 | 91 |
| 2. | X = Me | 85 | 90 | 89 | ||
| 3. | X = tBu | 94 | 91 | 91 | ||
| 4. | X = OMe | Polymer | Polymer | 57 | ||
| 5. | X = OtBu | Polymer | Polymer | 77 | ||
| 6. | X = Cl | 92 | 99 | 92 | ||
| 7. | X = F | 93 | 82 | 84 | ||
| 8. | X = CF3 | 90 | 99 | 82 | ||
| 9. | X = NO2 | 98 | 99 | 89 |
| Entry No. | Substrate | Products | DCM Yield (%) | 1,2-DCE Yield (%) | MeCN Yield (%) | |
|---|---|---|---|---|---|---|
| 1. |  | 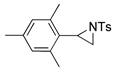 | 49 | 44 | 42 (30) b | |
| 2. | 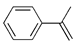 | 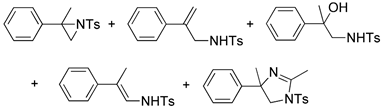 | 24, 9, 19 | 23, 8, 29 | 42, 5, 16, 4, 0 (28, 6, 12, 0, 17) b | |
| 3. | 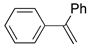 | 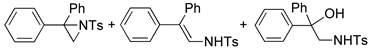 | 9, 50, 9 | 2, 52, 12 | 30, 30, 12 (2, 49, 22) b | |
| 4. | 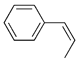 |  | 21, 27 | 24, 23 | 28, 29 (13, 11) b | |
| 5. |  | 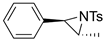 | 37 | 32 | 29 (18) b | |
| 6. | 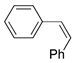 | 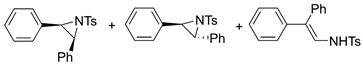 | 8, 26, 3 | 9, 26 | 14, 28 (6, 3, 9) b | |
| 7. | 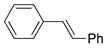 | 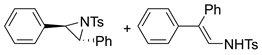 | 17, tr | 12, tr | 19 (5, tr) b | |
| 8. | 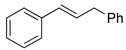 |  | 10, 32 | 11, 33 | 15, 26 | |
| 9. |  | 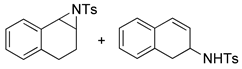 | 24, tr | 26, tr | 27, tr | |
| 10. | 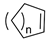 | 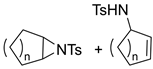 | n = 1 | 4, 8 | 4, 12 | 4, 16 |
| 11. | n = 2 | 4, 10 | 6, 12 | 6, 2 | ||
| 12. | n = 3 | 22 | 30 | 29 | ||
| 13. |  | 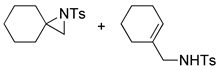 | 22, 6 | 15, 2 | 30, 3 | |
| 14 | 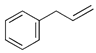 | 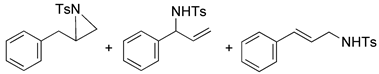 | 10, 8, 8 | 8, 7, 8 | 8, 2, 3 | |
| 15 | 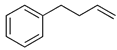 |  | 22 | 25 | 21 | |
| 16 | 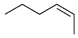 |  | 11, 4 | 11, 4 | 14, 3 | |
| 17. |  | 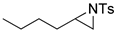 | 10 | 10 | 11 | |
| 18. | 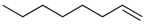 | 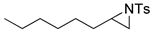 | 9 | 8 | 13 | |
| Catalyst | 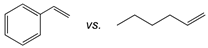 | 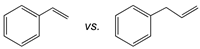 | 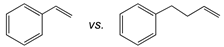 |
|---|---|---|---|
| [(tBuN4)CuI(MeCN)](PF6) | 27:1 | 21:1 | 18:1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhatia, H.; Adams, L.P.; Cordsiemon, I.; Sahoo, S.K.; Choudhury, A.; Cundari, T.R.; Stavropoulos, P. Comparative Nitrene-Transfer Chemistry to Olefins Mediated by First-Row Transition Metal Catalysts Supported by a Pyridinophane Macrocycle with N4 Ligation. Molecules 2025, 30, 3097. https://doi.org/10.3390/molecules30153097
Bhatia H, Adams LP, Cordsiemon I, Sahoo SK, Choudhury A, Cundari TR, Stavropoulos P. Comparative Nitrene-Transfer Chemistry to Olefins Mediated by First-Row Transition Metal Catalysts Supported by a Pyridinophane Macrocycle with N4 Ligation. Molecules. 2025; 30(15):3097. https://doi.org/10.3390/molecules30153097
Chicago/Turabian StyleBhatia, Himanshu, Lillian P. Adams, Ingrid Cordsiemon, Suraj Kumar Sahoo, Amitava Choudhury, Thomas R. Cundari, and Pericles Stavropoulos. 2025. "Comparative Nitrene-Transfer Chemistry to Olefins Mediated by First-Row Transition Metal Catalysts Supported by a Pyridinophane Macrocycle with N4 Ligation" Molecules 30, no. 15: 3097. https://doi.org/10.3390/molecules30153097
APA StyleBhatia, H., Adams, L. P., Cordsiemon, I., Sahoo, S. K., Choudhury, A., Cundari, T. R., & Stavropoulos, P. (2025). Comparative Nitrene-Transfer Chemistry to Olefins Mediated by First-Row Transition Metal Catalysts Supported by a Pyridinophane Macrocycle with N4 Ligation. Molecules, 30(15), 3097. https://doi.org/10.3390/molecules30153097