
Strain Engineering of Cu2O@C2N for Enhanced Methane-to-Methanol Conversion


Abstract
1. Introduction
2. Results and Discussion
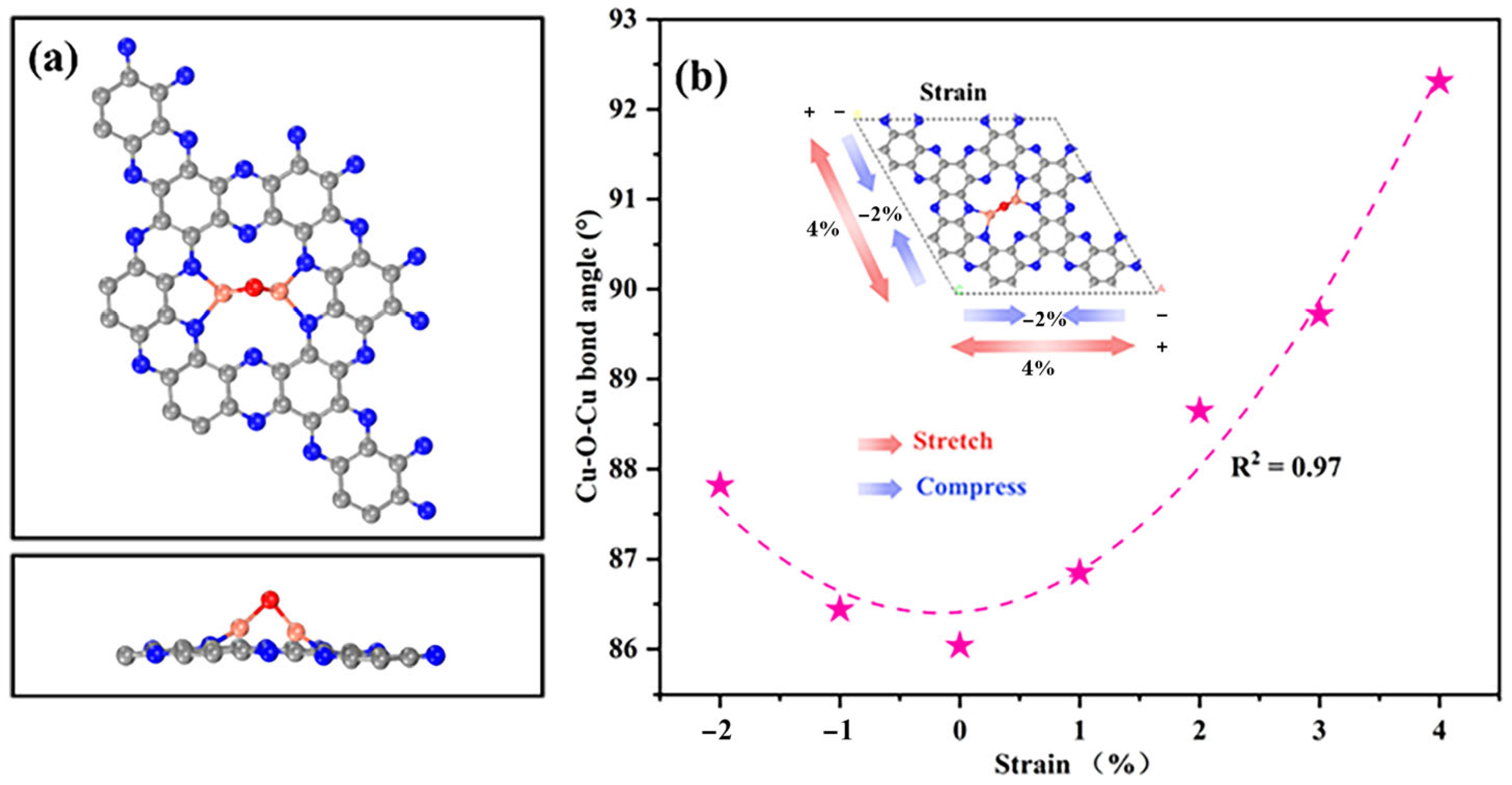
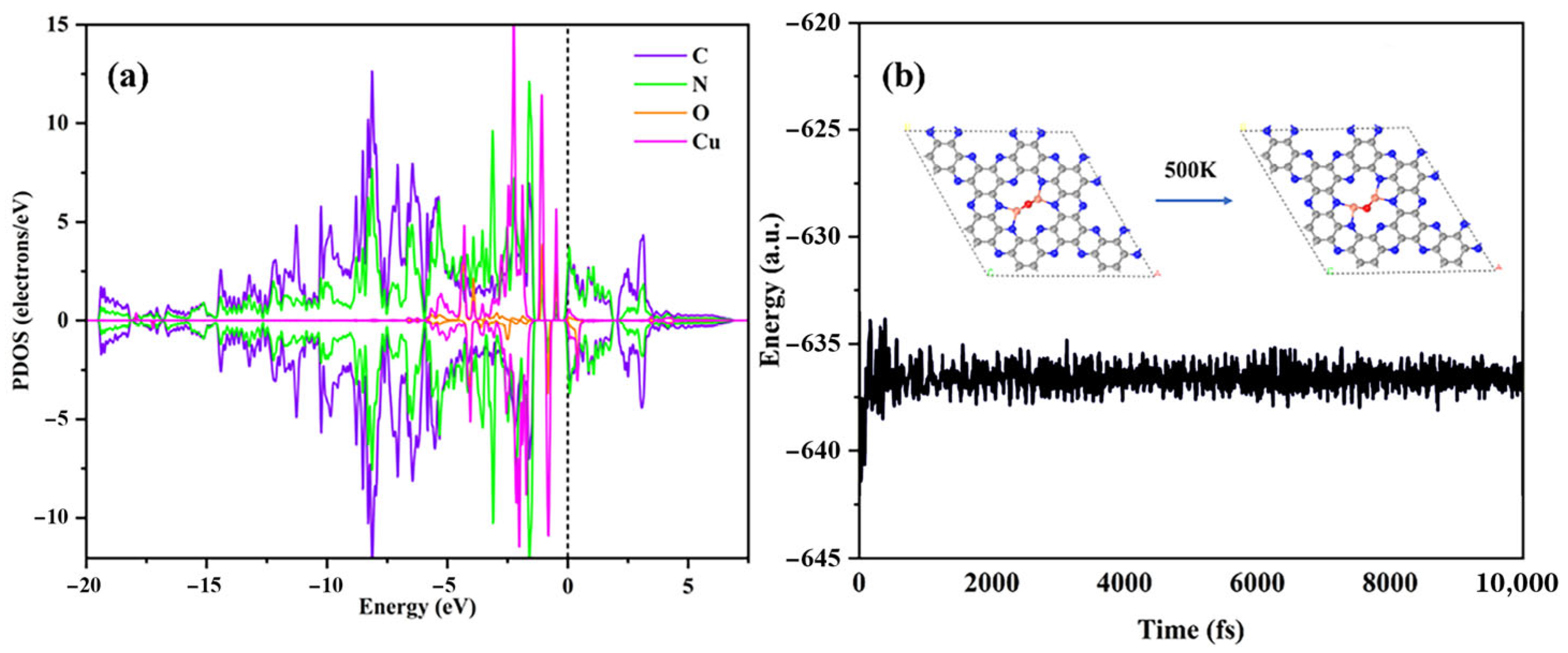
2.1. Structural and Electronic Properties of Cu2O@C2N
2.2. Catalytic Performance: Methane to Methanol Conversion
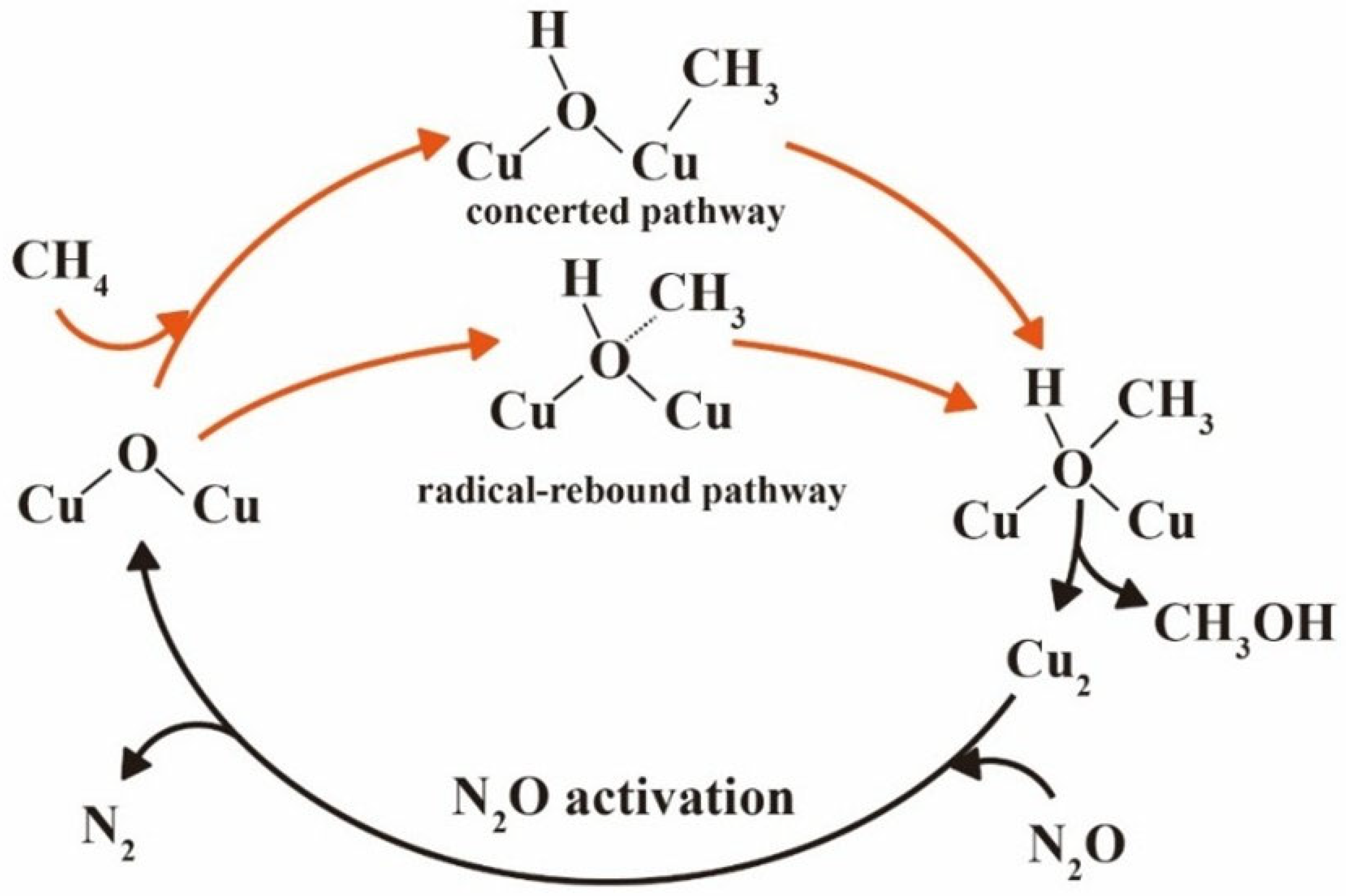
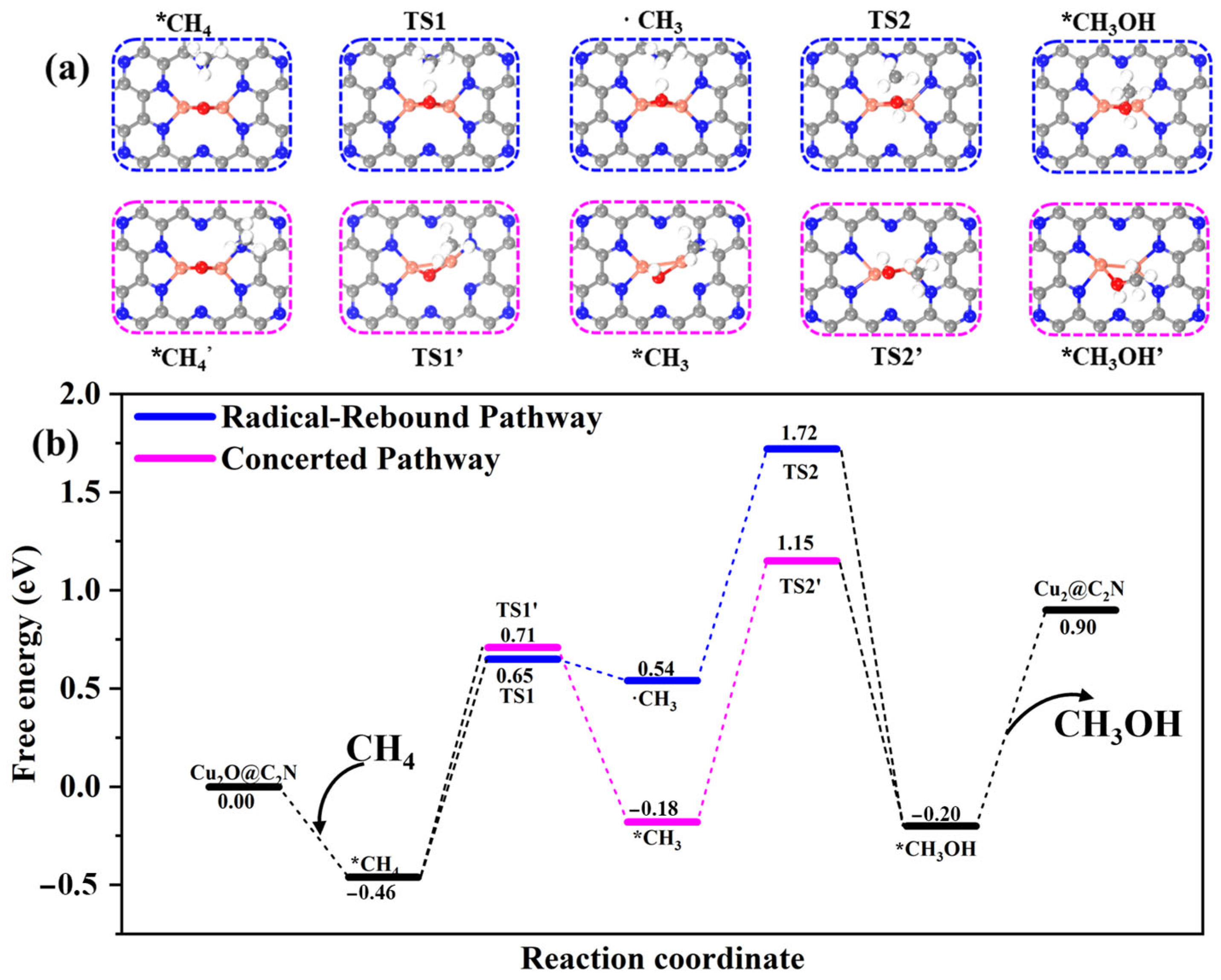
2.2.1. Reaction Mechanisms: Concerted vs. Radical–Rebound
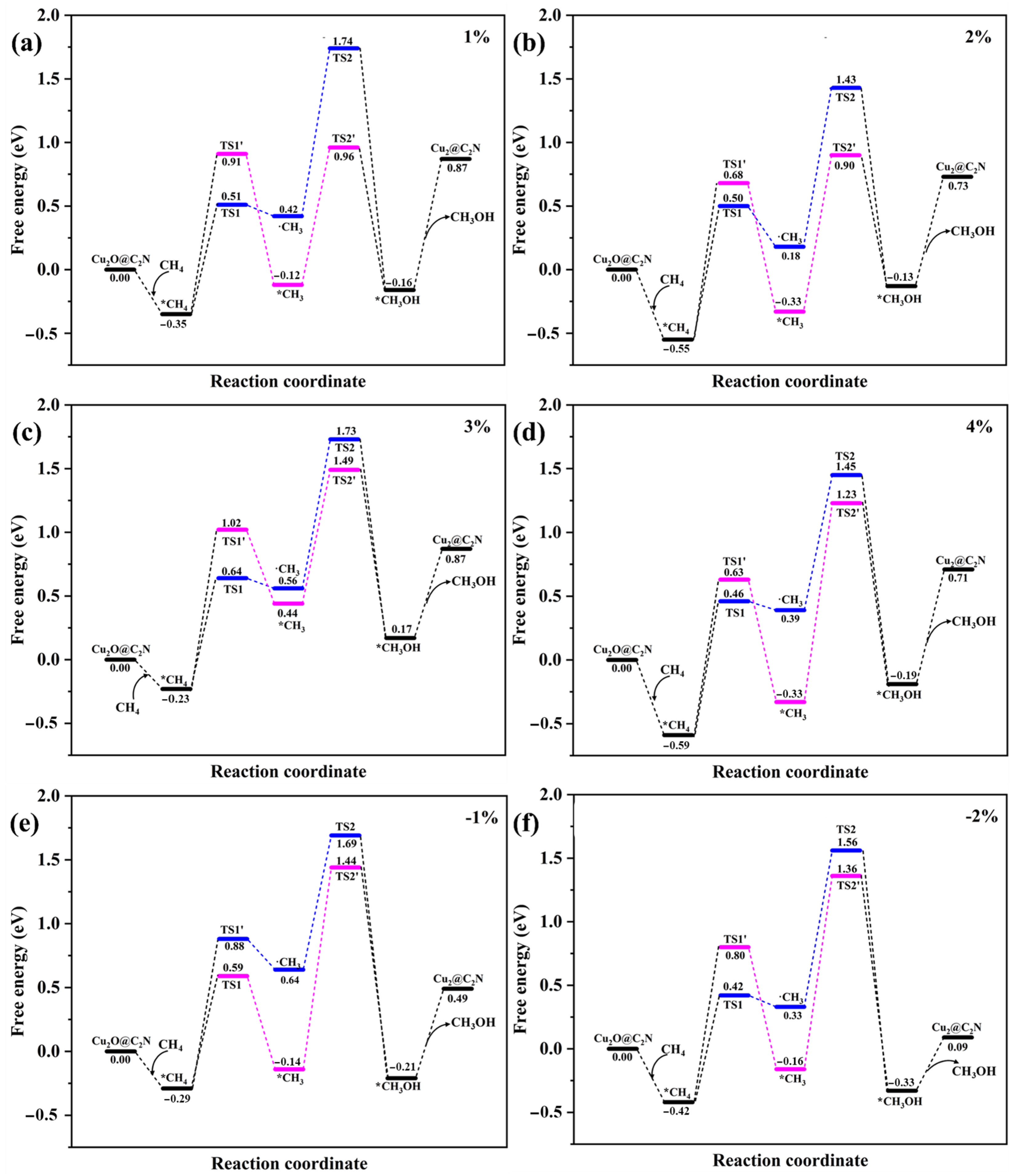
2.2.2. Effect of Strain on Mechanism Selectivity
2.3. Active Site Regeneration and Strain-Responsive Reactivation Behavior
2.4. Activity Origin and Electronic Descriptor Analysis
3. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yuan, J.; Zhang, W.; Li, X.; Yang, J. A High Performance Catalyst for Methane Conversion to Methanol: Graphene Supported Single Atom Co. Chem. Commun. 2018, 54, 2284–2287. [Google Scholar] [CrossRef] [PubMed]
- Baltrusaitis, J.; Jansen, I.; Christus, J.D.S. Renewable Energy Based Catalytic CH4 Conversion to Fuels. Catal. Sci. Technol. 2014, 4, 2397–2411. [Google Scholar] [CrossRef]
- Sushkevich, V.L.; Palagin, D.; Ranocchiari, M.; Van Bokhoven, J.A. Selective Anaerobic Oxidation of Methane Enables Direct Synthesis of Methanol. Science 2017, 356, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Li, S.; Sun, Y. Theoretical Study on the Dissociative Adsorption of CH4 on Pd-Doped Ni Surfaces. Chin. J. Catal. 2013, 34, 911–922. [Google Scholar] [CrossRef]
- Narsimhan, K.; Iyoki, K.; Dinh, K.; Román-Leshkov, Y. Catalytic Oxidation of Methane into Methanol over Copper-Exchanged Zeolites with Oxygen at Low Temperature. ACS Cent. Sci. 2016, 2, 424–429. [Google Scholar] [CrossRef] [PubMed]
- Horn, R.; Schlögl, R. Methane Activation by Heterogeneous Catalysis. Catal. Lett. 2015, 145, 23–39. [Google Scholar] [CrossRef]
- Kondratenko, E.V.; Peppel, T.; Seeburg, D.; Kondratenko, V.A.; Kalevaru, N.; Martin, A.; Wohlrab, S. Methane Conversion into Different Hydrocarbons or Oxygenates: Current Status and Future Perspectives in Catalyst Development and Reactor Operation. Catal. Sci. Technol. 2017, 7, 366–381. [Google Scholar] [CrossRef]
- Schwarz, H.; González-Navarrete, P.; Li, J.; Schlangen, M.; Sun, X.; Weiske, T.; Zhou, S. Unexpected Mechanistic Variants in the Thermal Gas-Phase Activation of Methane. Organometallics 2017, 36, 8–17. [Google Scholar] [CrossRef]
- Olivos-Suarez, A.I.; Szécsényi, À.; Hensen, E.J.; Ruiz-Martinez, J.; Pidko, E.A.; Gascon, J. Strategies for the Direct Catalytic Valorization of Methane Using Heterogeneous Catalysis: Challenges and Opportunities. ACS Catal. 2016, 6, 2965–2981. [Google Scholar] [CrossRef]
- Hammond, C.; Conrad, S.; Hermans, I. Oxidative Methane Upgrading. ChemSusChem 2012, 5, 1668–1686. [Google Scholar] [CrossRef] [PubMed]
- Gunsalus, N.J.; Koppaka, A.; Park, S.H.; Bischof, S.M.; Hashiguchi, B.G.; Periana, R.A. Homogeneous Functionalization of Methane. Chem. Rev. 2017, 117, 8521–8573. [Google Scholar] [CrossRef] [PubMed]
- Hartwig, J.F. Evolution of C-H Bond Functionalization from Methane to Methodology. J. Am. Chem. Soc. 2016, 138, 2–24. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Albarracín-Suazo, S.; Pagán-Torres, Y.; Nikolla, E. Advances in Methane Conversion Processes. Catal. Today 2017, 285, 147–158. [Google Scholar] [CrossRef]
- Mesters, C. A Selection of Recent Advances in C1 Chemistry. Annu. Rev. Chem. Biomol. Eng. 2016, 7, 223–238. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, H. Chemistry with Methane: Concepts Rather than Recipes. Angew. Chem. Int. Ed. 2011, 50, 10096–10115. [Google Scholar] [CrossRef] [PubMed]
- Taifan, W.; Baltrusaitis, J. CH4 Conversion to Value Added Products: Potential, Limitations and Extensions of a Single Step Heterogeneous Catalysis. Appl. Catal. B Environ. 2016, 198, 525–547. [Google Scholar] [CrossRef]
- Ma, L.; Ding, C.; Wang, J.; Xu, H.; Zhang, K. In Situ Surface Chemical Properties and Catalytic Performances of Pt/CeO2 Catalyst in Methane Partial Oxidation under Different Conditions. Appl. Surf. Sci. 2023, 609, 155195. [Google Scholar] [CrossRef]
- Liu, H.; Li, L.; Qin, Y.; Wang, H.; Zhu, X.; Ge, Q. Synergetic C-H Bond Activation and C-O Formation on CuOx Facilitates Facile Conversion of Methane to Methanol. Appl. Surf. Sci. 2023, 627, 157283. [Google Scholar] [CrossRef]
- Tomkins, P.; Ranocchiari, M.; van Bokhoven, J.A. Direct Conversion of Methane to Methanol under Mild Conditions over Cu-Zeolites and Beyond. Acc. Chem. Res. 2017, 50, 418–425. [Google Scholar] [CrossRef] [PubMed]
- Groothaert, M.H.; Smeets, P.J.; Sels, B.F.; Jacobs, P.A.; Schoonheydt, R.A. Selective Oxidation of Methane by the Bis(μ-oxo) Dicopper Core Stabilized on ZSM-5 and Mordenite Zeolites. J. Am. Chem. Soc. 2005, 127, 1394–1395. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.; Bell, A.T.; Head-Gordon, M.; Chakraborty, A.K. Density Functional Theory Investigations of the Direct Oxidation of Methane on an Fe-Exchanged Zeolite. J. Phys. Chem. B 2004, 108, 4362–4368. [Google Scholar] [CrossRef]
- Meng, H.; Han, B.; Li, F.; Zhao, J.; Chen, Z. Understanding the CH4 Conversion over Metal Dimers from First Principles. Nanomaterials 2022, 12, 1518. [Google Scholar] [CrossRef] [PubMed]
- Impeng, S.; Khongpracha, P.; Warakulwit, C.; Jansang, B.; Sirijaraensre, J.; Ehara, M.; Limtrakul, J. Direct Oxidation of Methane to Methanol on Fe-O Modified Graphene. RSC Adv. 2014, 4, 12572–12578. [Google Scholar] [CrossRef]
- Yu, H.Y.; Wang, X.C.; Zhang, S.X.A. Electron Reservoir Enhances Methane Conversion on Zn Dimer Supported by N Doped Graphene: A DFT Study. Appl. Surf. Sci. 2021, 563, 150328. [Google Scholar] [CrossRef]
- Wu, S.Y.; Lin, C.H.; Ho, J.J. Density-Functional Calculations of the Conversion of Methane to Methanol on Platinum-Decorated Sheets of Graphene Oxide. Phys. Chem. Chem. Phys. 2015, 17, 26191–26197. [Google Scholar] [CrossRef] [PubMed]
- Solomon, E.I.; Heppner, D.E.; Johnston, E.M.; Ginsbach, J.W.; Cirera, J.; Qayyum, M.; Tian, L. Copper Active Sites in Biology. Chem. Rev. 2014, 114, 3659–3853. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, R.L.; Rosenzweig, A.C. Crystal Structure of a Membrane-Bound Metalloenzyme that Catalyses the Biological Oxidation of Methane. Nature 2005, 434, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, R.; Smith, S.M.; Rawat, S.; Yatsunyk, L.A.; Stemmler, T.L.; Rosenzweig, A.C. Oxidation of Methane by a Biological Dicopper Centre. Nature 2010, 465, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Panov, G.I.; Sobolev, V.I.; Dubkov, K.A.; Parmon, V.N.; Ovanesyan, N.S.; Shilov, A.E.; Shteinman, A.A. Iron Complexes in Zeolites as a New Model of Methane Monooxygenase. React. Kinet. Catal. Lett. 1997, 61, 251–258. [Google Scholar] [CrossRef]
- Wang, G.; Huang, L.; Chen, W.; Zhou, J.; Zheng, A. Rationally Designing Mixed Cu–(μ-O)–M (M = Cu, Ag, Zn, Au) Centers over Zeolite Materials with High Catalytic Activity Towards Methane Activation. Phys. Chem. Chem. Phys. 2018, 41, 26522–26531. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, J.; Lee, E.K.; Jung, M.; Shin, D.; Jeon, I.Y.; Jung, S.M.; Baek, J.B. Nitrogenated Holey Two-Dimensional Structures. Nat. Commun. 2015, 6, 6486. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Li, W.; Li, G.; Ao, Z.; An, T. Density Functional Theory Investigation of the Enhanced Adsorption Mechanism and Potential Catalytic Activity for Formaldehyde Degradation on Al-decorated C2N Monolayer. Chin. J. Catal. 2019, 40, 664–672. [Google Scholar] [CrossRef]
- He, B.; Shen, J.; Tian, Z. Iron-embedded C2N monolayer: A promising Low-Cost and High-Activity Single-Atom Catalyst for CO Oxidation. Phys. Chem. Chem. Phys. 2016, 18, 24261–24269. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhong, W.; Cui, P.; Li, J.; Jiang, J. Design of Efficient Catalysts with Double Transition Metal Atoms on C2N Layer. J. Phys. Chem. Lett. 2016, 7, 1750–1755. [Google Scholar] [CrossRef] [PubMed]
- Xie, P.; Ding, J.; Yao, Z.; Pu, T.; Zhang, P.; Huang, Z.; Wang, C.; Zhang, J.; Zecher-Freeman, N.; Zong, H.; et al. Oxo Dicopper Anchored on Carbon Nitride for Selective Oxidation of Methane. Nat. Commun. 2022, 13, 1375. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ren, Z.; He, Z.; Ouyang, P.; Duan, Y.; Zhang, W.; Lv, K.; Dong, F. Crystallinity-Defect Matching Relationship of g-C3N4: Experimental and Theoretical Perspectives. Green Energy Environ. 2024, 9, 9623–9658. [Google Scholar] [CrossRef]
- Tian, Z.; López-Salas, N.; Liu, C.; Liu, T.; Antonietti, M. C2N: A Class of Covalent Frameworks with Unique Properties. Adv. Sci. 2020, 7, 2001767. [Google Scholar] [CrossRef] [PubMed]
- Sohail, U.; Ullah, F.; Arfan, N.H.B.Z.; Hamid, M.H.S.A.; Mahmood, T.; Sheikh, N.S.; Ayub, K. Transition Metal Sensing with Nitrogenated Holey Graphene: A First-Principles Investigation. Molecules 2023, 28, 4060. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Amin, B.; Gan, L.-Y.; Ahmad, I. Strain Engineering of Electronic Structures and Photocatalytic Responses of MXenes Functionalized by Oxygen. Phys. Chem. Chem. Phys. 2017, 19, 14738–14744. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-H.; Su, X.-Y.; Zhang, R.-Z. Strain-Induced Band Modulation of Surface F-Functionalized Two-Dimensional Sc2C. Appl. Surf. Sci. 2019, 491, 276–285. [Google Scholar] [CrossRef]
- Deng, Q.; Huang, R.; Shao, L.-H.; Mumyatov, A.V.; Troshin, P.A.; An, C.; Wu, S.; Gao, L.; Yang, B.; Hu, N. Atomic Understanding of the Strain-Induced Electrocatalysis from DFT Calculation: Progress and Perspective. Phys. Chem. Chem. Phys. 2023, 25, 12565–12586. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Liu, T.; Xiao, W.; Wang, W.; Zhang, Y.; Wang, G.; Luo, Z.; Liu, J.-C. Strain Engineering in Single-Atom Catalysts: GaPS4 for Bifunctional Oxygen Reduction and Evolution. Inorg. Chem. Front. 2022, 9, 4272–4280. [Google Scholar] [CrossRef]
- Liu, C.; Gui, L.; Zheng, J.-J.; Xu, Y.-Q.; Song, B.; Yi, L.; Jia, Y.; Taledaohan, A.; Wang, Y.; Gao, X. Intrinsic Strain-Mediated Ultrathin Ceria Nanoantioxidant. J. Am. Chem. Soc. 2023, 145, 19086–19097. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Guo, S. Strain-Controlled Electrocatalysis on Multimetallic Nanomaterials. Nat. Rev. Mater. 2017, 2, 1–13. [Google Scholar] [CrossRef]
- Lei, Q.; Huang, L.; Yin, J.; Davaasuren, B.; Yuan, Y.; Dong, X.; Wu, Z.P.; Wang, X.; Yao, K.X.; Lu, X. Structural Evolution and Strain Generation of Derived-Cu Catalysts During CO2 Electroreduction. Nat. Commun. 2022, 13, 4857. [Google Scholar] [CrossRef] [PubMed]
- Ma, N.; Li, N.; Wang, T.; Ma, X.; Fan, J. Strain Engineering in the Oxygen Reduction Reaction and Oxygen Evolution Reaction Catalyzed by Pt-Doped Ti2CF2. J. Mater. Chem. A 2022, 10, 1390–1401. [Google Scholar] [CrossRef]
- Ma, N.; Li, N.; Zhang, Y.; Wang, T.; Zhao, J.; Fan, J. Strain Adjustment Pt-Doped Ti2CO2 as an Efficient Bifunctional Catalyst for Oxygen Reduction Reactions and Oxygen Evolution Reactions by First-Principles Calculations. Appl. Surf. Sci. 2022, 590, 153149. [Google Scholar] [CrossRef]
- Zhao, J.; Zhao, J.; Li, F. Copper Dimer Supported on a C2N Layer as an Efficient Electrocatalyst for CO2 Reduction Reaction: A Computational Study. J. Phys. Chem. C 2018, 122, 19712–19721. [Google Scholar] [CrossRef]
- Lee, K.J.; Lee, M.J.; Hwang, H. High-Temperature Steam Electrolysis Combined with Methane Partial Oxidation by Solid Oxide Electrolyzer Cells. Appl. Surf. Sci. 2019, 473, 746–749. [Google Scholar] [CrossRef]
- Li, P.; Feng, L.; Li, G.; Bai, F. Effects of Electron Donating Ability of Substituents and Molecular Conjugation on the Electronic Structures of Organic Radicals. Chem. Res. Chin. Univ. 2023, 39, 202–207. [Google Scholar] [CrossRef]
- Huang, E.; Liu, P. Screening of Cu-Based Catalysts for Selective Methane to Methanol Conversion. J. Phys. Chem. C 2024, 128, 7876–7883. [Google Scholar] [CrossRef]
- Liu, H.; Qin, Y.; Li, L.; Wang, H.; Zhu, X.; Ge, Q. CH3 Radical-Mediated Direct Methane to Methanol Conversion over CuO Supported on Rutile Oxides. J. Catal. 2024, 431, 115388. [Google Scholar] [CrossRef]
- Yu, X.; Peng, Z.; Xu, L.; Shi, W.; Li, Z.; Meng, X.; He, X.; Wang, Z.; Duan, S.; Tong, L.; et al. Manipulating 2D Materials Through Strain Engineering. Small 2024, 20, 2402561. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmüller, J. Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmüller, J. Efficiency of Ab-Initio Total Energy Calculations for Metals and Semiconductors Using a Plane-Wave Basis Set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Blöchl, P.E. Projector Augmented-Wave Method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Barnett, R.N.; Landman, U. Born-Oppenheimer Molecular-Dynamics Simulations of Finite Systems: Structure and Dynamics of (H2O2). Phys. Rev. B Condens. Matter 1993, 48, 2081–2097. [Google Scholar] [CrossRef] [PubMed]
- Henkelman, G.; Uberuaga, B.P.; Jónsson, H. A Climbing Image Nudged Elastic Band Method for Finding Saddle Points and Minimum Energy Paths. J. Chem. Phys. 2000, 113, 9901–9904. [Google Scholar] [CrossRef]
- Henkelman, G.; Jónsson, H. Improved Tangent Estimate in the Nudged Elastic Band Method for Finding Minimum Energy Paths and Saddle Points. J. Chem. Phys. 2000, 113, 9978–9985. [Google Scholar] [CrossRef]
- Xiao, P.; Sheppard, D.; Rogal, J. Solid-State Dimer Method for Calculating Solid-Solid Phase Transitions. J. Chem. Phys. 2014, 140, 174104. [Google Scholar] [CrossRef] [PubMed]
- Dinh, K.T.; Sullivan, M.M.; Narsimhan, K.; Serna, P.; Meyer, R.J.; Dinca, M.; Roman-Leshkov, Y. Continuous Partial Oxidation of Methane to Methanol Catalyzed by Diffusion-Paired Copper Dimers in Copper-Exchanged Zeolites. J. Am. Chem. Soc. 2019, 141, 11641–11650. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain (%) | Concerted Pathway | Radical–Rebound Pathway |
|---|---|---|
| 4 | 1.82 | 2.04 |
| 3 | 1.72 | 1.96 |
| 2 | 1.45 | 1.98 |
| 1 | 1.31 | 2.09 |
| −1 | 1.73 | 1.98 |
| −2 | 1.78 | 1.98 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuai, S.; Li, B.; Liu, J. Strain Engineering of Cu2O@C2N for Enhanced Methane-to-Methanol Conversion. Molecules 2025, 30, 3073. https://doi.org/10.3390/molecules30153073
Kuai S, Li B, Liu J. Strain Engineering of Cu2O@C2N for Enhanced Methane-to-Methanol Conversion. Molecules. 2025; 30(15):3073. https://doi.org/10.3390/molecules30153073
Chicago/Turabian StyleKuai, Shuxin, Bo Li, and Jingyao Liu. 2025. "Strain Engineering of Cu2O@C2N for Enhanced Methane-to-Methanol Conversion" Molecules 30, no. 15: 3073. https://doi.org/10.3390/molecules30153073
APA StyleKuai, S., Li, B., & Liu, J. (2025). Strain Engineering of Cu2O@C2N for Enhanced Methane-to-Methanol Conversion. Molecules, 30(15), 3073. https://doi.org/10.3390/molecules30153073