
Structural Modifications at the C3 and C30 Positions of the Lupane Skeleton with Carbon-Centered Nucleophiles



Abstract
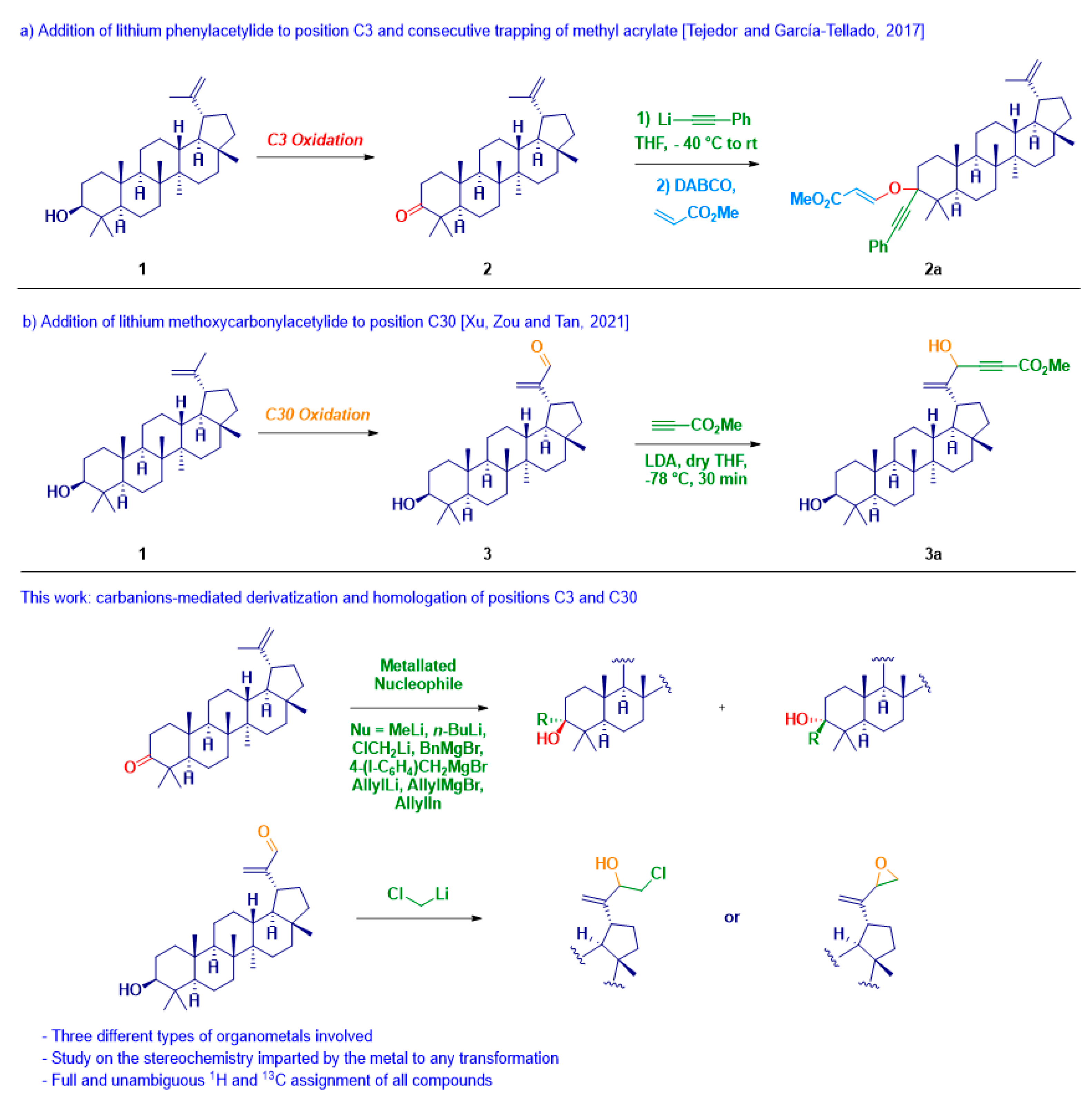
1. Introduction
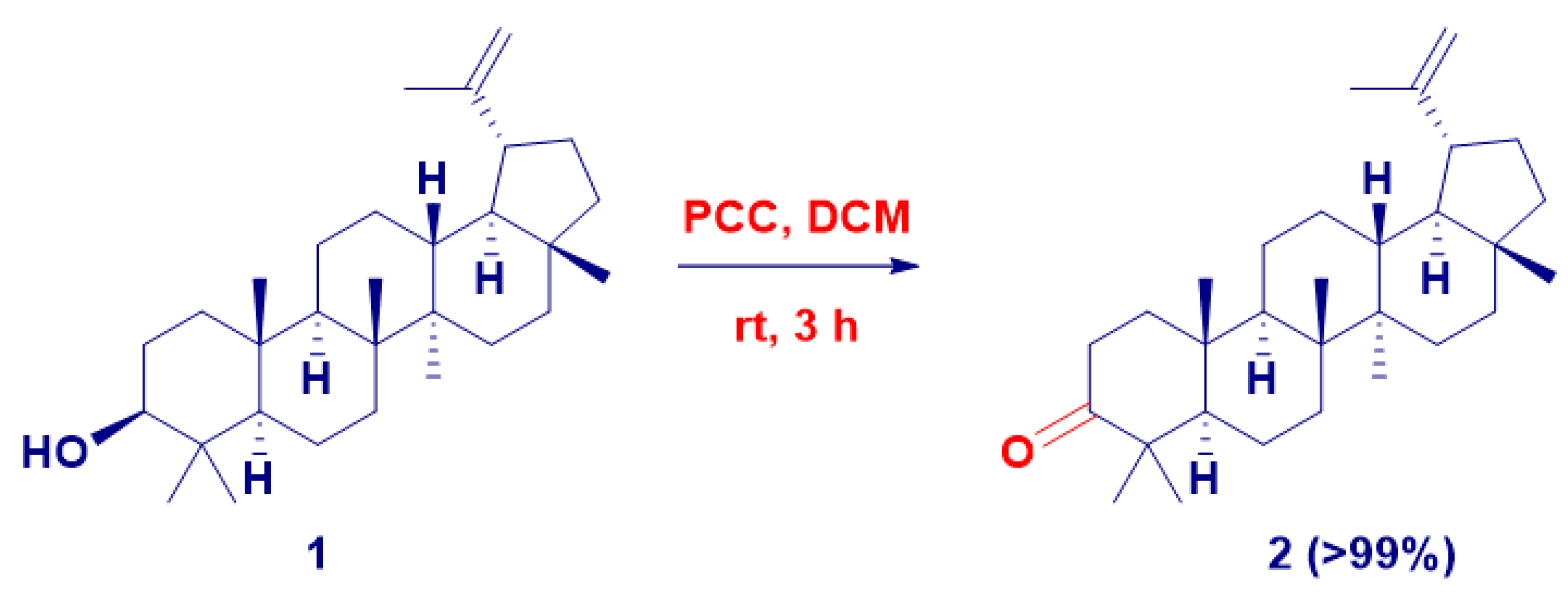
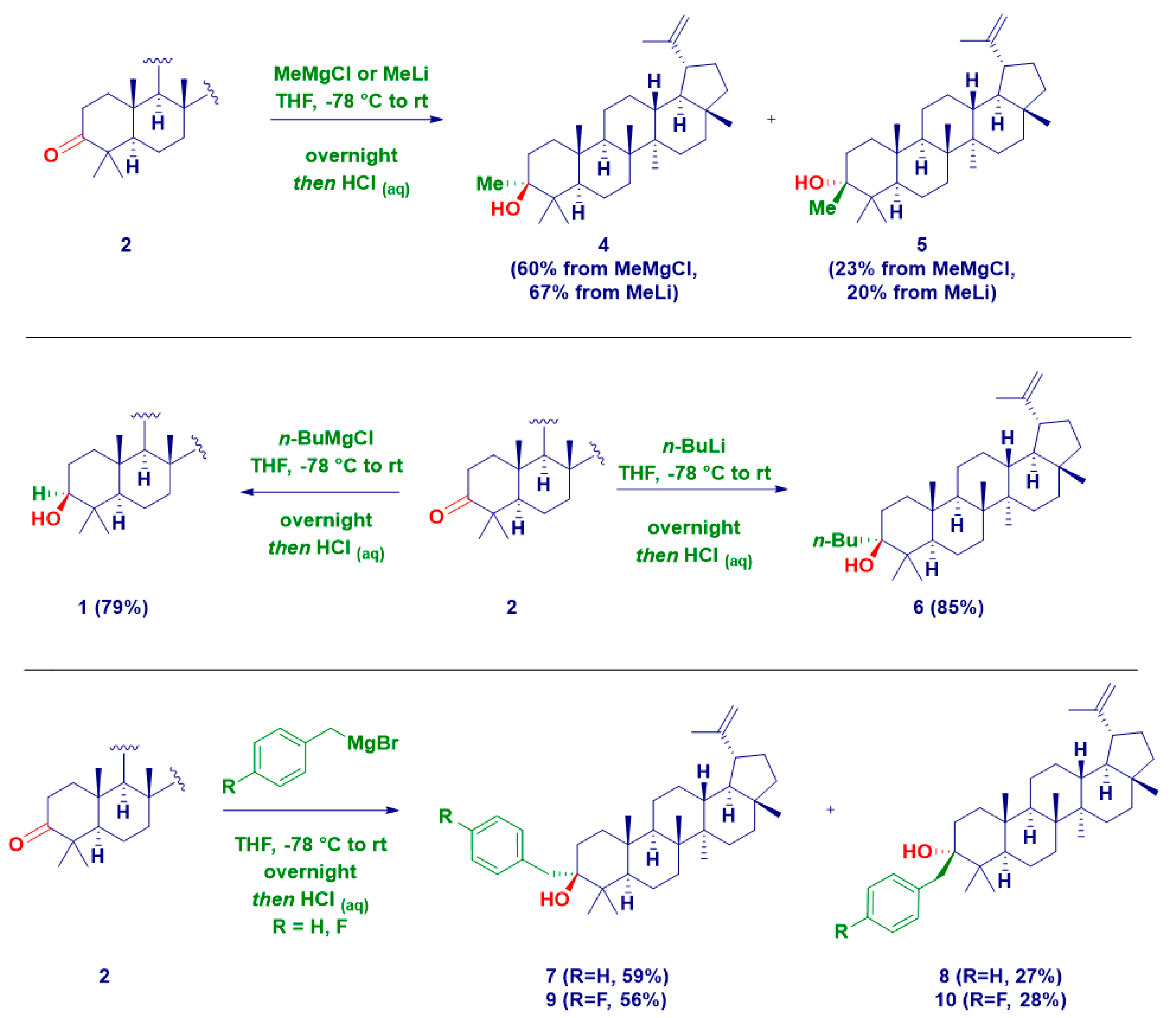
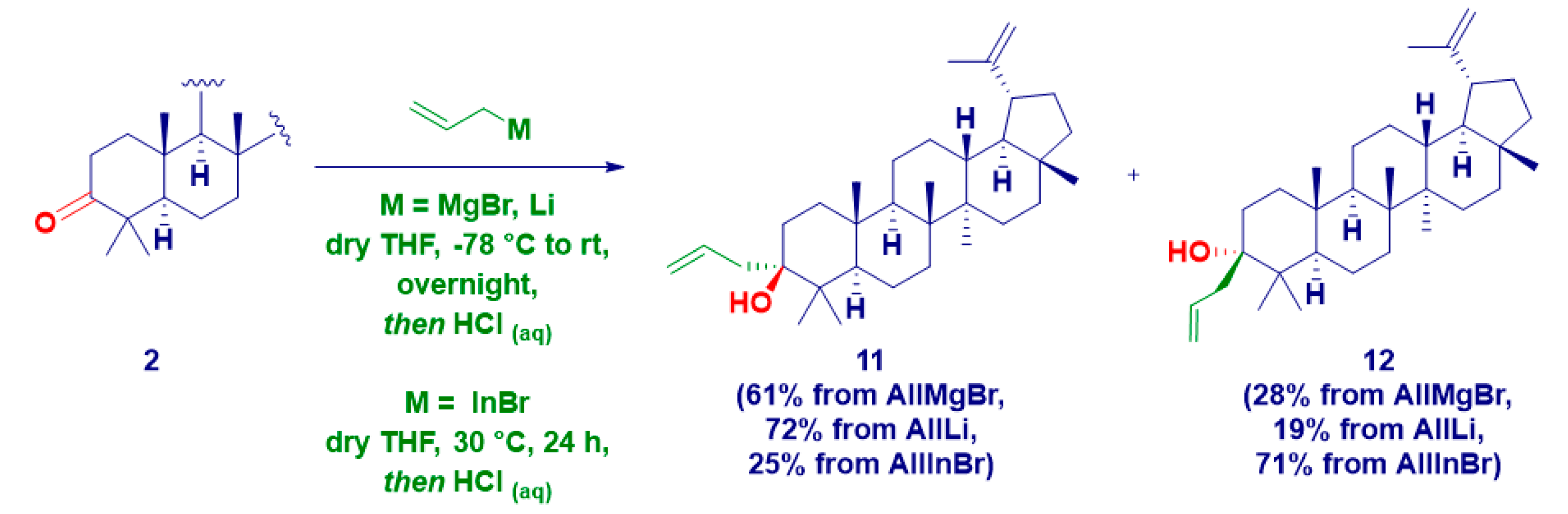
2. Results and Discussion
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| NMR | Nuclear magnetic resonance |
| HRMS | High-resolution mass spectrometry |
| THF | Tetrahydrofurane |
| DABCO | 1,4-diazabicyclo[2.2.2]octane |
| LDA | Lithium diisopropylamide |
| PCC | Pyridium chlorochromate |
| DCM | Dichloromethane |
| TS | Transition state |
| TGR5 | Takeda G protein-coupled receptor 5 |
References
- Patil, P.; Soujanya, B.; Kiran, K. A review on Lupeol: Superficial triterpenoid from horticulture crops. Int. J. Chem. Stud. 2018, 6, 3301–3305. [Google Scholar]
- Yoder, R.A.; Johnston, J.N. A case study in biomimetic total synthesis: Polyolefin carbocyclizations to terpenes and steroids. Chem. Rev. 2005, 105, 4730–4756. [Google Scholar] [CrossRef] [PubMed]
- Gallo, M.B.; Sarachine, M.J. Biological activities of lupeol. Int. J. Biomed. Pharm. Sci. 2009, 3, 46–66. [Google Scholar]
- Park, J.S.; Rehman, I.U.; Choe, K.; Ahmad, R.; Lee, H.J.; Kim, M.O. A triterpenoid lupeol as an antioxidant and anti-neuroinflammatory agent: Impacts on oxidative stress in Alzheimer’s disease. Nutrients 2023, 15, 3059. [Google Scholar] [CrossRef] [PubMed]
- Sudhahar, V.; Kumar, S.A.; Sudharsan, P.T.; Varalakshmi, P. Protective effect of lupeol and its ester on cardiac abnormalities in experimental hypercholesterolemia. Vasc. Pharmacol. 2007, 46, 412–418. [Google Scholar] [CrossRef] [PubMed]
- Malik, A.; Jamil, U.; Butt, T.T.; Waquar, S.; Gan, S.H.; Shafique, H.; Jafar, T.H. In silico and in vitro studies of lupeol and iso-orientin as potential antidiabetic agents in a rat model. Drug Des. Dev. Ther. 2019, 13, 1501–1513. [Google Scholar] [CrossRef] [PubMed]
- Lakshmi, V.; Mahdi, A.A.; Ahmad, M.K.; Agarwal, S.K.; Srivastava, A.K. Antidiabetic activity of lupeol and lupeol esters in streptozotocin-induced diabetic rats. Bangladesh Pharm. J. 2014, 17, 138–146. [Google Scholar] [CrossRef]
- Geetha, T.; Varalakshmi, P. Anti-inflammatory activity of lupeol and lupeol linoleate in rats. J. Ethnopharmacol. 2001, 76, 77–80. [Google Scholar] [CrossRef] [PubMed]
- Thirumalaisamy, R.; Ameen, F.; Subramanian, A.; Selvankumar, T.; Alwakeel, S.; Govarthanan, M. In-vitro and in-silico anti-inflammatory activity of Lupeol isolated from Crateva adansonii and its hidden molecular mechanism. Int. J. Pept. Res. Ther. 2020, 26, 2179–2189. [Google Scholar] [CrossRef]
- Saleem, M. Lupeol, a novel anti-inflammatory and anti-cancer dietary triterpene. Cancer Lett. 2009, 285, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Babu, T.S.; Michael, B.P.; Jerard, C.; Vijayakumar, N.; Ramachandran, R. Study on the anti metastatic and anticancer activity of triterpene compound lupeol in human lung cancer. Apoptosis 2019, 5, 16. [Google Scholar]
- Alam, P.; Al-Yousef, H.M.; Siddiqui, N.A.; Alhowiriny, T.A.; Alqasoumi, S.I.; Amina, M.; Hassan, W.H.B.; Abdelaziz, S.; Abdalla, R.H. Anticancer activity and concurrent analysis of ursolic acid, β-sitosterol and lupeol in three different Hibiscus species (aerial parts) by validated HPTLC method. Saudi Pharm. J. 2018, 26, 1060–1067. [Google Scholar] [CrossRef] [PubMed]
- Cháirez-Ramírez, M.; Moreno-Jiménez, M.; González-Laredo, R.; Gallegos-Infante, J.; Rocha-Guzmán, N.E. Lupane-type triterpenes and their anti-cancer activities against most common malignant tumors: A review. EXCLI J. 2016, 15, 758. [Google Scholar] [PubMed]
- Dwivedi, N.; Dwivedi, B.; Mishra, S.; Shukla, Y. Lupeol induced apoptosis in human lung cancer cell line: A flow cytometry study. Res. J. Pharmacol. Pharmacodyn. 2014, 6, 197. [Google Scholar]
- Malinowska, M.; Miroslaw, B.; Sikora, E.; Ogonowski, J.; Wojtkiewicz, A.M.; Szaleniec, M.; Pasikowska-Piwko, M.; Eris, I. New lupeol esters as active substances in the treatment of skin damage. PLoS ONE 2019, 14, e0214216. [Google Scholar] [CrossRef] [PubMed]
- Hodges, L.D.; Kweifio-Okai, G.; Macrides, T.A. Antiprotease effect of anti-inflammatory lupeol esters. Mol. Cell. Biochem. 2003, 252, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Fotie, J.; Bohle, D.S.; Leimanis, M.L.; Georges, E.; Rukunga, G.; Nkengfack, A.E. Lupeol long-chain fatty acid esters with antimalarial activity from Holarrhena f loribunda. J. Nat. Prod. 2006, 69, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Tamfu, A.N.; Munvera, A.M.; Botezatu, A.V.D.; Talla, E.; Ceylan, O.; Fotsing, M.T.; Mbafor, J.T.; Shaheen, F.; Dinica, R.M. Synthesis of benzoyl esters of β-amyrin and lupeol and evaluation of their antibiofilm and antidiabetic activities. Results Chem. 2022, 4, 100322. [Google Scholar] [CrossRef]
- Fontana, G.; Badalamenti, N.; Bruno, M.; Castiglione, D.; Notarbartolo, M.; Poma, P.; Spinella, A.; Tutone, M.; Labbozzetta, M. Synthesis, in vitro and in silico analysis of new oleanolic acid and lupeol derivatives against leukemia cell lines: Involvement of the NF-κB pathway. Int. J. Mol. Sci. 2022, 23, 6594. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Nicolás, F.; Gordillo-Román, B.; Oberti, J.C.; Estévez-Braun, A.; Ravelo, Á.G.; Joseph-Nathan, P. Synthesis and anti-HIV activity of lupane and olean-18-ene derivatives. Absolute configuration of 19, 20-epoxylupanes by VCD. J. Nat. Prod. 2012, 75, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Bednarczyk-Cwynar, B.; Wiȩcaszek, T.; Ruszkowski, P. Cytotoxic activity of some lupeol derivatives. Nat. Prod. Commun. 2016, 11, 1237–1238. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Xiao, Y. Synthesis and in vitro antitumour activities of lupeol derivatives. Nat. Prod. Res. 2018, 32, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Bhandari, P.; Patel, N.K.; Bhutani, K.K. Synthesis of new heterocyclic lupeol derivatives as nitric oxide and pro-inflammatory cytokine inhibitors. Bioorg. Med. Chem. Lett. 2014, 24, 3596–3599. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.F.; Mishra, D.P.; Ramakrishna, E.; Rawat, A.K.; Mishra, A.; Srivastava, A.K.; Maurya, R. Design and synthesis of lupeol analogues and their in vitro PTP-1B inhibitory activity. Med. Chem. Res. 2014, 23, 4156–4166. [Google Scholar] [CrossRef]
- Wal, A.; Wal, P.; Rai, A. Isolation and modification of pseudohybrid plant (Lupeol). J. Pharm. Sci. Res. 2010, 2, 13. [Google Scholar]
- Saini, M.; Khan, M.F.; Sangwan, R.; Khan, M.A.; Kumar, A.; Verma, R.; Ahamad, T.; Jain, S. Design, synthesis and in-vitro antitumor activity of lupeol derivatives via modification at C-3 and C-30 positions. ChemistrySelect 2019, 4, 1800–1805. [Google Scholar] [CrossRef]
- Rárová, L.; Pakulski, Z.; Strnad, M.; Kvasnicová, M.; Štenclová, T.; Cmoch, P. Effect of modification of betulinic acid at the C3-carbon atom of homolupane triterpenoids on the antiproliferative activity in vitro. J. Steroid Biochem. Mol. Biol. 2022, 224, 106161. [Google Scholar] [CrossRef] [PubMed]
- Genet, C.; Schmidt, C.; Strehle, A.; Schoonjans, K.; Auwerx, J.; Saladin, R.; Wagner, A. Redefining the TGR5 Triterpenoid Binding Pocket at the C-3 Position. ChemMedChem 2010, 5, 1983–1988. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Chang, L.-C.; Hsieh, K.-Y.; Hsu, P.-L.; Capuzzi, S.J.; Zhang, Y.-C.; Li, K.-P.; Morris-Natschke, S.L.; Goto, M.; Lee, K.-H. Design, synthesis and evaluation of antiproliferative activity of fluorinated betulinic acid. Bioorg. Med. Chem. 2019, 27, 2871–2882. [Google Scholar] [CrossRef] [PubMed]
- Fontana, G.; Bruno, M.; Notarbartolo, M.; Labbozzetta, M.; Poma, P.; Spinella, A.; Rosselli, S. Cytotoxicity of oleanolic and ursolic acid derivatives toward hepatocellular carcinoma and evaluation of NF-κB involvement. Bioorg. Chem. 2019, 90, 103054. [Google Scholar] [CrossRef] [PubMed]
- Tejedor, D.; Delgado-Hernández, S.; Carballo, R.M.; Dapueto, R.; Mena-Rejón, G.J.; García-Tellado, F. Diversifying Complexity by Domino Benzannulation of Polycyclic Natural Products. J. Org. Chem. 2017, 82, 5328–5336. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; Peng, W.; Wang, H.; Zhang, X.; Zhang, J.; Tan, G.; Xu, K.; Zou, Z.; Tan, H. Total syntheses of melodienones by redox isomerization of propargylic alcohols. Org. Biomol. Chem. 2021, 19, 5077–5081. [Google Scholar] [CrossRef] [PubMed]
- Burns, D.; Reynolds, W.F.; Buchanan, G.; Reese, P.B.; Enriquez, R.G. Assignment of 1H and 13C spectra and investigation of hindered side-chain rotation in lupeol derivatives. Magn. Reson. Chem. 2000, 38, 488–493. [Google Scholar] [CrossRef]
- Surendra, K.; Corey, E. A short enantioselective total synthesis of the fundamental pentacyclic triterpene lupeol. J. Am. Chem. Soc. 2009, 131, 13928–13929. [Google Scholar] [CrossRef] [PubMed]
- Rimoldi, I.; Pellizzoni, M.; Facchetti, G.; Molinari, F.; Zerla, D.; Gandolfi, R. Chemo- and biocatalytic strategies to obtain phenylisoserine, a lateral chain of Taxol by asymmetric reduction. Tetrahedron Asymmetry 2011, 22, 2110–2116. [Google Scholar] [CrossRef]
- Bellomo, A.; Daniellou, R.; Plusquellec, D. Allylation of cyclohexanones in aqueous media and influence of facial amphiphilic fructopyranosides. Tetrahedron Lett. 2010, 51, 4934–4936. [Google Scholar] [CrossRef]
- Ashby, E.; Laemmle, J. Stereochemistry of organometallic compound addition to ketones. Chem. Rev. 1975, 75, 521–546. [Google Scholar] [CrossRef]
- Bartolo, N.D.; Read, J.A.; Valentín, E.M.; Woerpel, K. Reactions of allylmagnesium halides with carbonyl compounds: Reactivity, structure, and mechanism. Synthesis 2017, 49, 3237–3246. [Google Scholar] [CrossRef]
- Bowyer, W.J.; Singaram, B.; Sessler, A.M. Nature of the intermediates formed during indium mediated allylation under Barbier conditions. Spectroscopic and experimental data on allylindium species. Tetrahedron 2011, 67, 7449–7460. [Google Scholar] [CrossRef]
- Corey, E.; Chaykovsky, M. Dimethyloxosulfonium methylide ((CH3) 2SOCH2) and dimethylsulfonium methylide ((CH3) 2SCH2). Formation and application to organic synthesis. J. Am. Chem. Soc. 1965, 87, 1353–1364. [Google Scholar] [CrossRef]
- Beutner, G.L.; George, D.T. Opportunities for the application and advancement of the Corey–Chaykovsky cyclopropanation. Org. Process Res. Dev. 2022, 27, 10–41. [Google Scholar] [CrossRef]
- Malik, M.; Senatore, R.; Castiglione, D.; Roller-Prado, A.; Pace, V. Highly chemoselective homologative assembly of the α-substituted methylsulfinamide motif from N-sulfinylamines. Chem. Commun. 2023, 59, 11065–11068. [Google Scholar] [CrossRef] [PubMed]
- Miele, M.; Castiglione, D.; Holzer, W.; Castoldi, L.; Pace, V. Chemoselective homologative preparation of trisubstituted alkenyl halides from carbonyls and carbenoids. Chem. Commun. 2025, 61, 1180–1183. [Google Scholar] [CrossRef] [PubMed]
- Ielo, L.; Pillari, V.; Miele, M.; Castiglione, D.; Pace, V. Carbenoid-Mediated Homologation Tactics for Assembling (Fluorinated) Epoxides and Aziridines. Synlett 2021, 32, 551–560. [Google Scholar] [CrossRef]
- Pace, V.; Castoldi, L.; Mamuye, A.D.; Langer, T.; Holzer, W. Chemoselective Addition of Halomethyllithiums to Functionalized Isatins: A Straightforward Access to Spiro-Epoxyoxindoles. Adv. Synth. Catal. 2016, 358, 172–177. [Google Scholar] [CrossRef]
- Monticelli, S.; Castoldi, L.; Touqeer, S.; Miele, M.; Urban, E.; Pace, V. Recent advances in the synthesis and reactivity of spiro-epoxyoxindoles. Chem. Heterocycl. Compd. 2018, 54, 389–393. [Google Scholar] [CrossRef]
- Parisi, G.; Colella, M.; Monticelli, S.; Romanazzi, G.; Holzer, W.; Langer, T.; Degennaro, L.; Pace, V.; Luisi, R. Exploiting a “beast” in carbenoid chemistry: Development of a straightforward direct nucleophilic fluoromethylation strategy. J. Am. Chem. Soc. 2017, 139, 13648–13651. [Google Scholar] [CrossRef] [PubMed]
- Monticelli, S.; Colella, M.; Pillari, V.; Tota, A.; Langer, T.; Holzer, W.; Degennaro, L.; Luisi, R.; Pace, V. Modular and chemoselective strategy for the direct access to α-fluoroepoxides and aziridines via the addition of fluoroiodomethyllithium to carbonyl-like compounds. Org. Lett. 2019, 21, 584–588. [Google Scholar] [CrossRef] [PubMed]
- Ke, Z.; Zhou, Y.; Gao, H.; Zhao, C.; Phillips, D.L. On the Mechanism and Stereochemistry of Chiral Lithium-Carbenoid-Promoted Cyclopropanation Reactions. Chem.–A Eur. J. 2007, 13, 6724–6731. [Google Scholar] [CrossRef] [PubMed]
- Colella, M.; Tota, A.; Großjohann, A.; Carlucci, C.; Aramini, A.; Sheikh, N.S.; Degennaro, L.; Luisi, R. Straightforward chemo-and stereoselective fluorocyclopropanation of allylic alcohols: Exploiting the electrophilic nature of the not so elusive fluoroiodomethyllithium. Chem. Commun. 2019, 55, 8430–8433. [Google Scholar] [CrossRef] [PubMed]
- Pace, V.; Castoldi, L.; Holzer, W. Synthesis of α, β-Unsaturated α′-Haloketones through the Chemoselective Addition of Halomethyllithiums to Weinreb Amides. J. Org. Chem. 2013, 78, 7764–7770. [Google Scholar] [CrossRef] [PubMed]
- Pace, V.; Castoldi, L.; Monticelli, S.; Rui, M.; Collina, S. New perspectives in lithium carbenoid mediated homologations. Synlett 2017, 28, 879–888. [Google Scholar] [CrossRef]
- Pace, V.; Castoldi, L.; Holzer, W. Chemoselective additions of chloromethyllithium carbenoid to cyclic enones: A direct access to chloromethyl allylic alcohols. Adv. Synth. Catal. 2014, 356, 1761–1766. [Google Scholar] [CrossRef]
- Aquilina, J.M.; Smith, M.W. Symmetry-Driven Total Synthesis of Myrioneurinol. J. Am. Chem. Soc. 2022, 144, 11088–11093. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
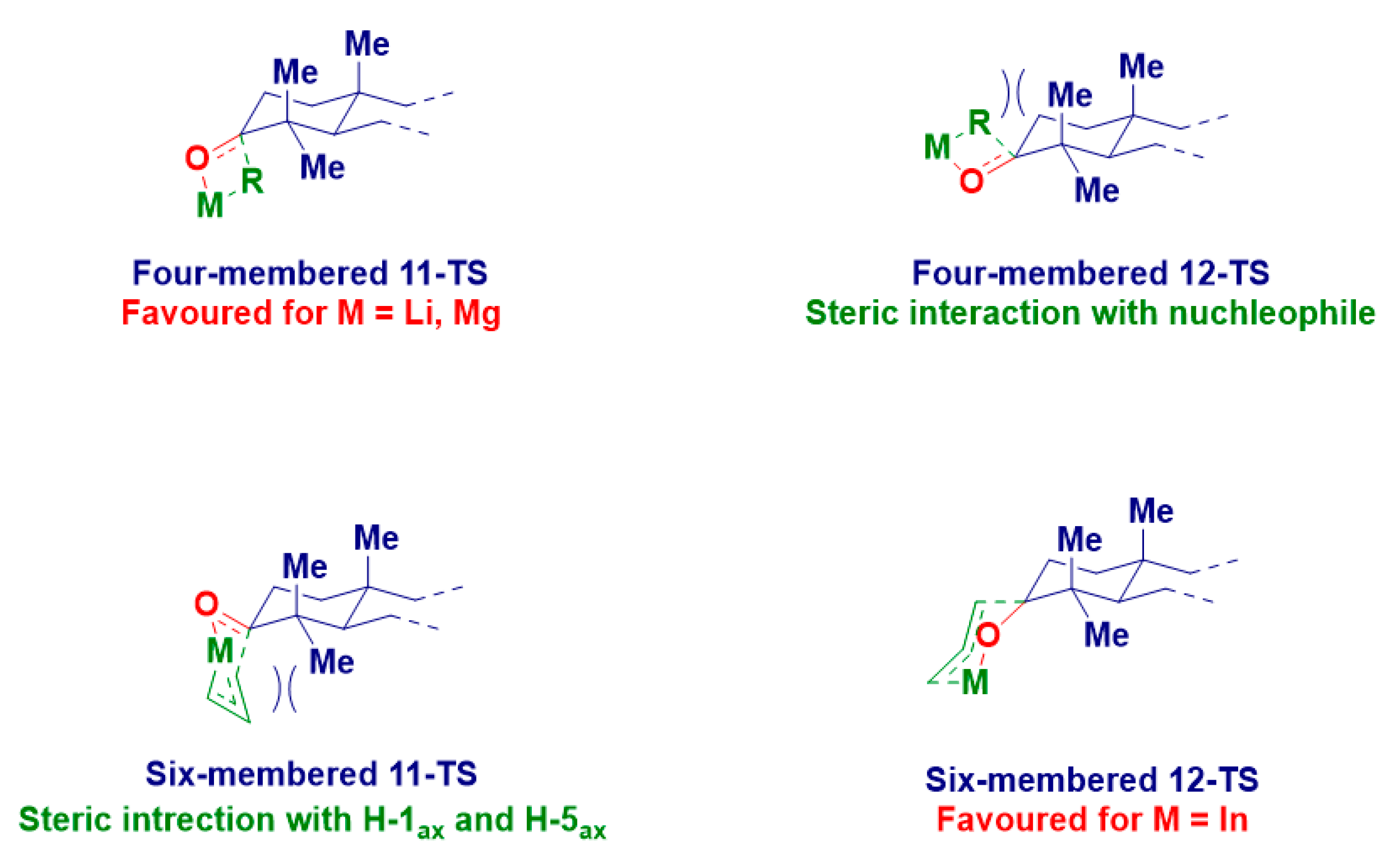
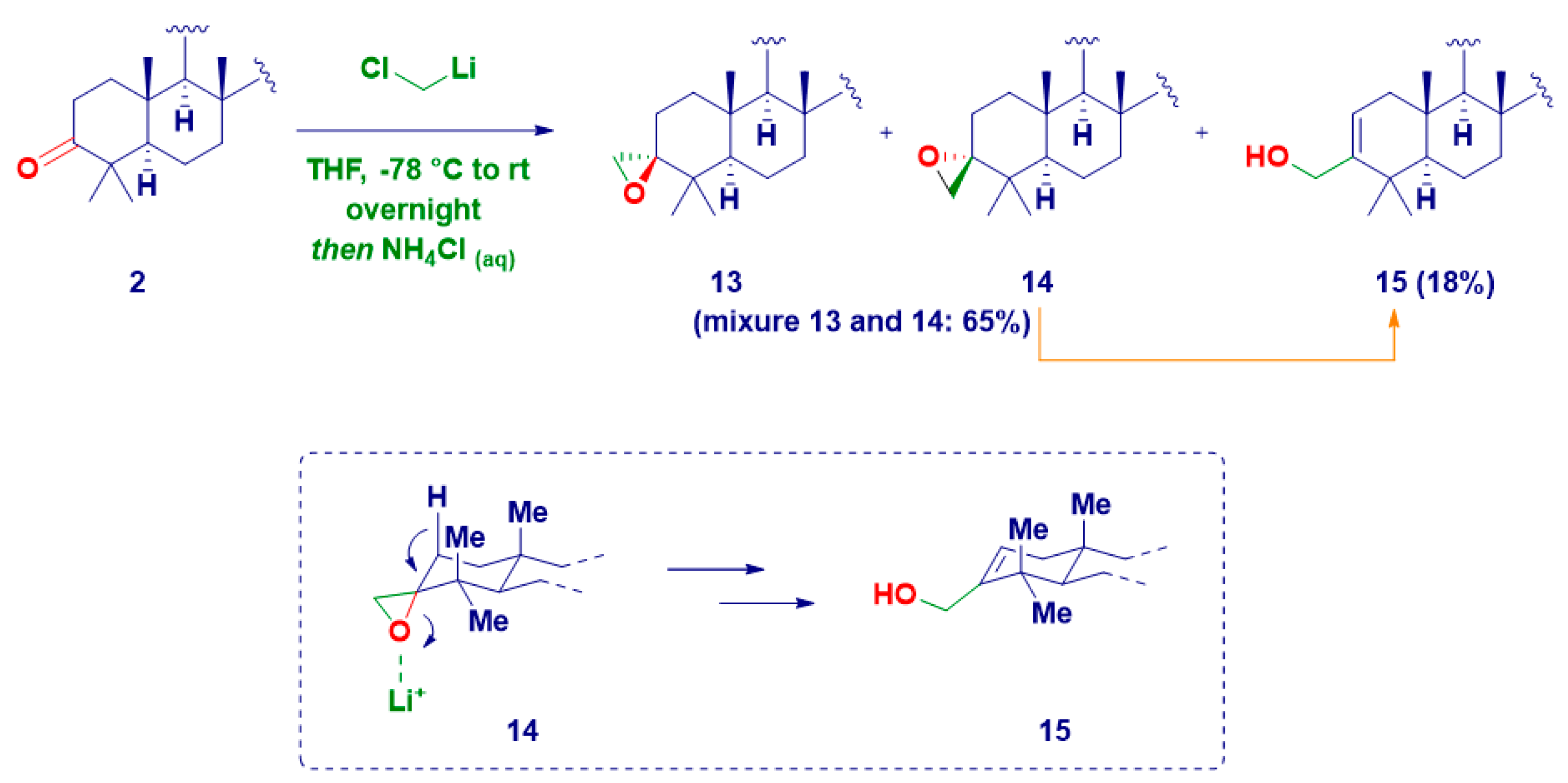
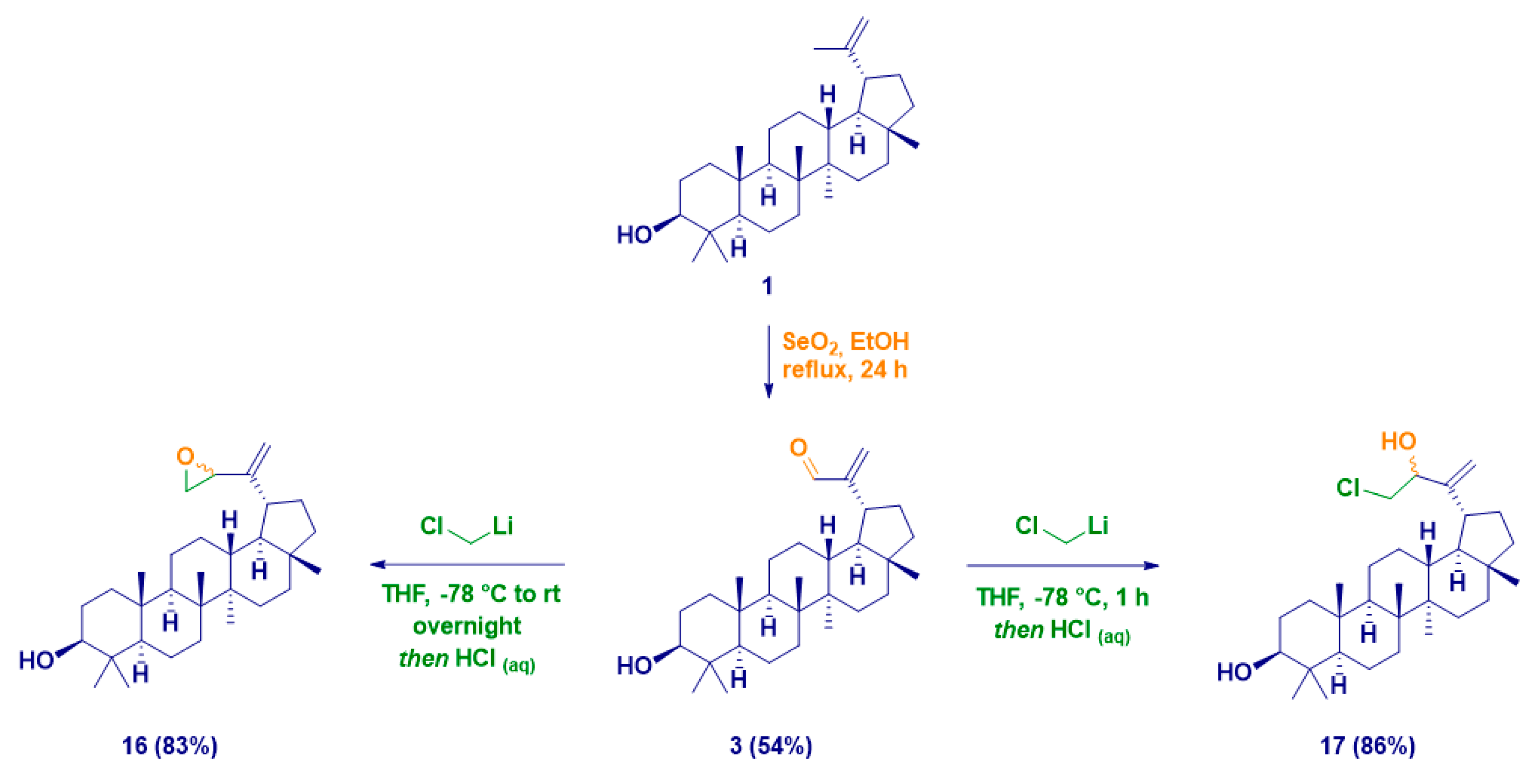
| Entry | Product | R | M | Temp. | Yield% | d.e.% |
|---|---|---|---|---|---|---|
| 1 | 4 | Me | Li | −78 °C to rt | 67 | 47 |
| 2 | 5 | 20 | ||||
| 3 | 4 | MgCl | 60 | 37 | ||
| 4 | 5 | 23 | ||||
| 5 | 6 | n-Bu | Li | 85 | >99 | |
| 6 | 1 | MgX | 79 | >99 | ||
| 7 | 7 | benzyl | 59 | 32 | ||
| 8 | 8 | 27 | ||||
| 9 | 9 | 4-F-benzyl | 56 | 28 | ||
| 10 | 10 | 28 | ||||
| 11 | 11 | allyl | Li | 72 | 53 | |
| 12 | 12 | 19 | ||||
| 13 | 11 | MgBr | 61 | 33 | ||
| 14 | 12 | 28 | ||||
| 15 | 11 | In | rt | 25 | 46 | |
| 16 | 12 | 71 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castiglione, D.; Fontana, G.; Castoldi, L.; Pace, V. Structural Modifications at the C3 and C30 Positions of the Lupane Skeleton with Carbon-Centered Nucleophiles. Molecules 2025, 30, 3064. https://doi.org/10.3390/molecules30153064
Castiglione D, Fontana G, Castoldi L, Pace V. Structural Modifications at the C3 and C30 Positions of the Lupane Skeleton with Carbon-Centered Nucleophiles. Molecules. 2025; 30(15):3064. https://doi.org/10.3390/molecules30153064
Chicago/Turabian StyleCastiglione, Davide, Gianfranco Fontana, Laura Castoldi, and Vittorio Pace. 2025. "Structural Modifications at the C3 and C30 Positions of the Lupane Skeleton with Carbon-Centered Nucleophiles" Molecules 30, no. 15: 3064. https://doi.org/10.3390/molecules30153064
APA StyleCastiglione, D., Fontana, G., Castoldi, L., & Pace, V. (2025). Structural Modifications at the C3 and C30 Positions of the Lupane Skeleton with Carbon-Centered Nucleophiles. Molecules, 30(15), 3064. https://doi.org/10.3390/molecules30153064