
Pd/Ligand-Free Synthesis of 2-Alkynylated Pyrano[4,3-d]imidazol-4-ones via One-Pot Cu-Mediated Tandem Sonogashira Coupling/Regioselective 6-endo-dig Oxacyclization Reaction

 , , , and
, , , and
Abstract

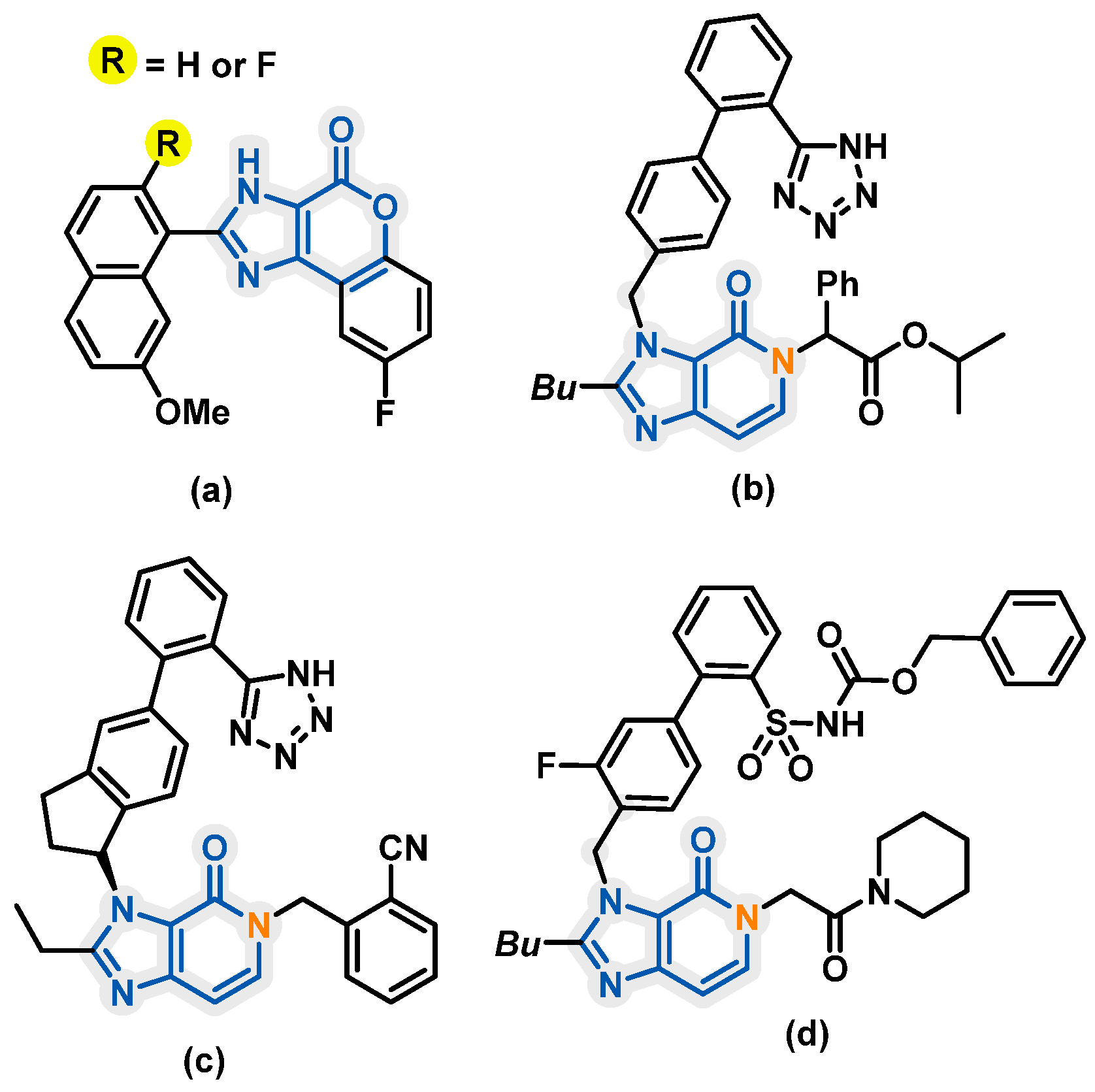
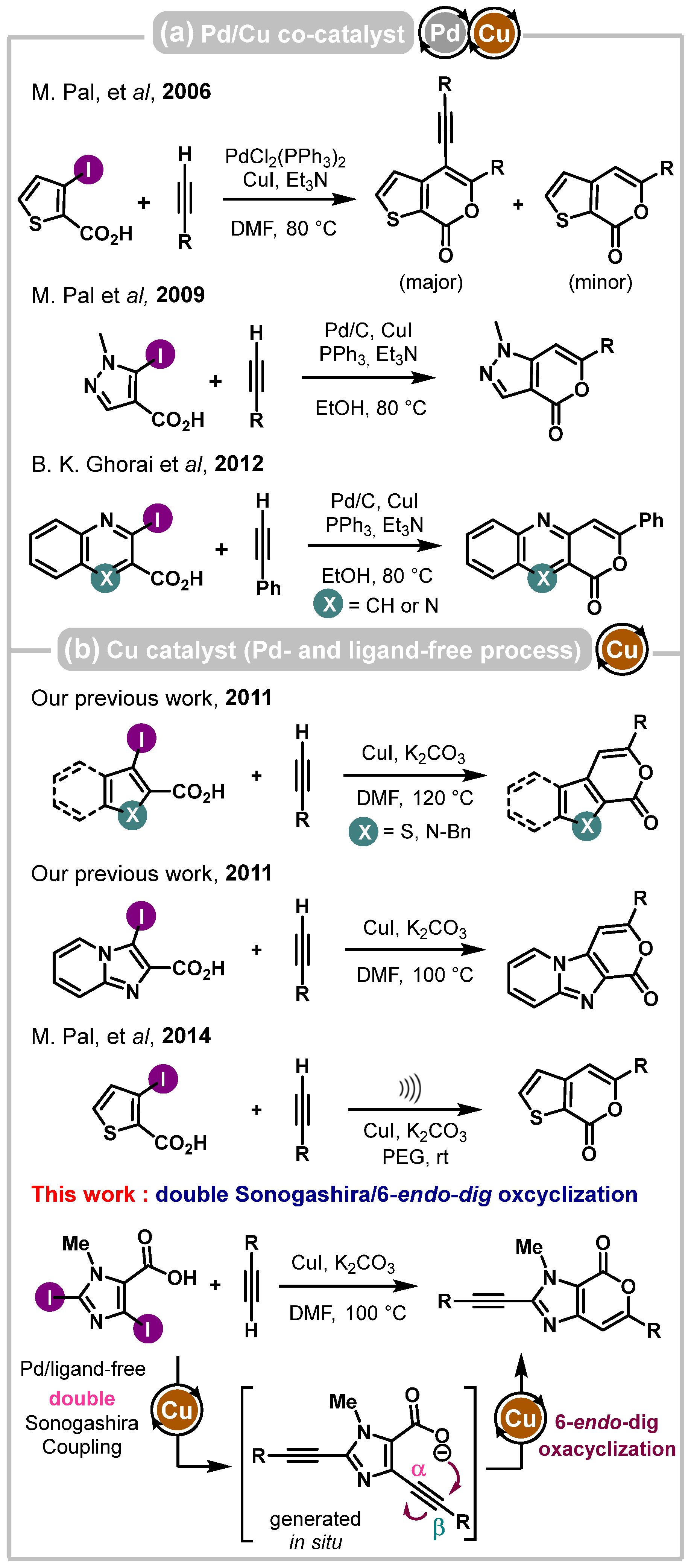
1. Introduction
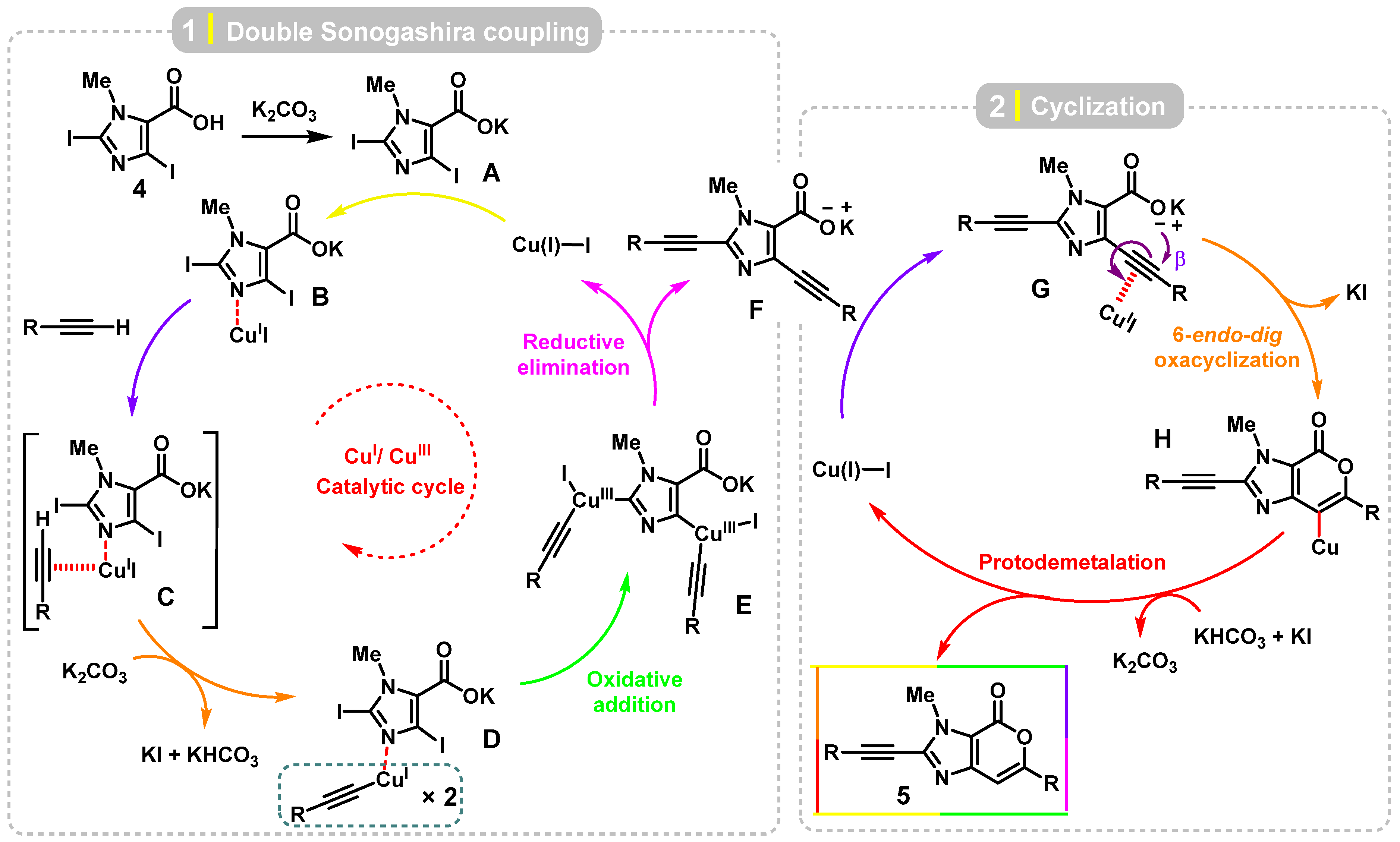
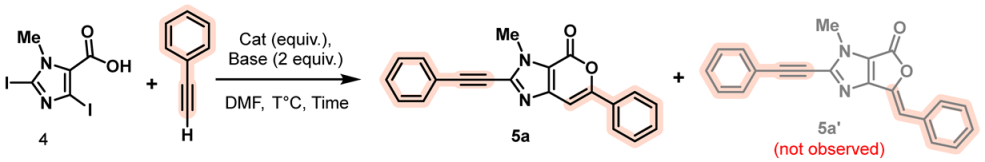
2. Results and Discussion
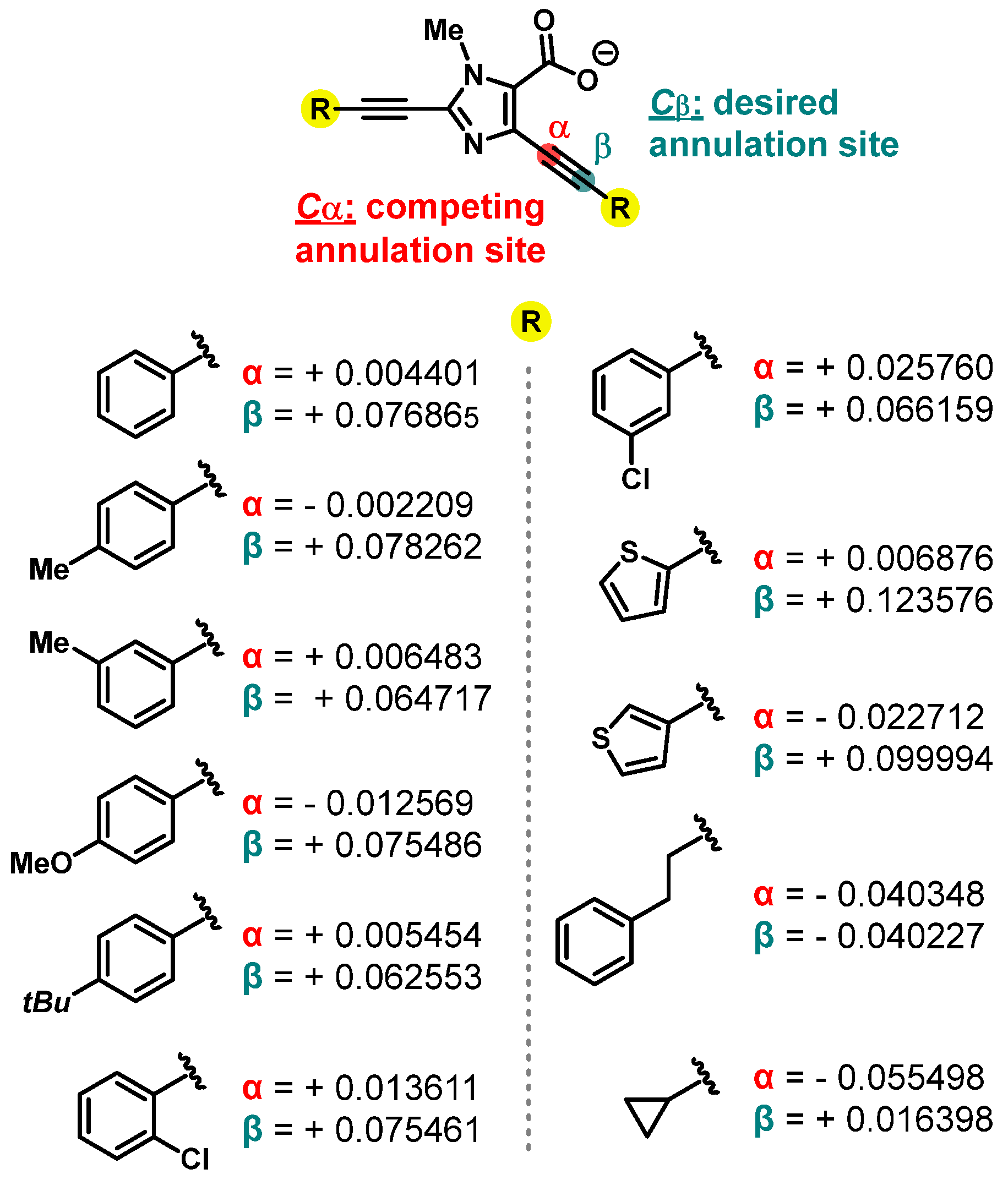
Theoretical Calculation (Computational Studies)
3. Materials and Methods
3.1. General Information
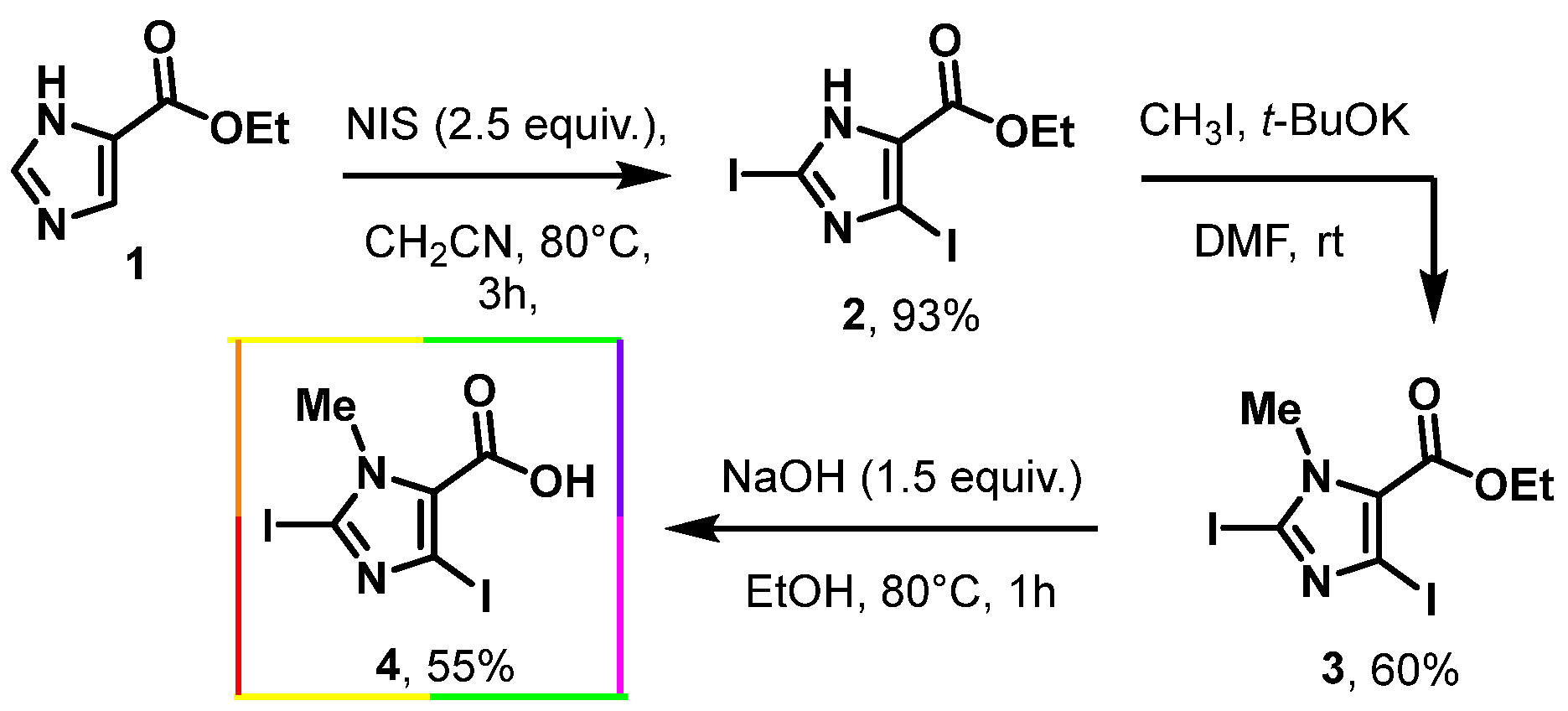
- Ethyl-2,4-diiodo-1H-imidazol-5-carboxylate (2)
- Ethyl-2,4-diiodo-1-methyl-1H-imidazol-5-carboxylate (3)
- 2,4-Diiodo-1-methyl-1H-imidazole-5-carboxylic acid (4)
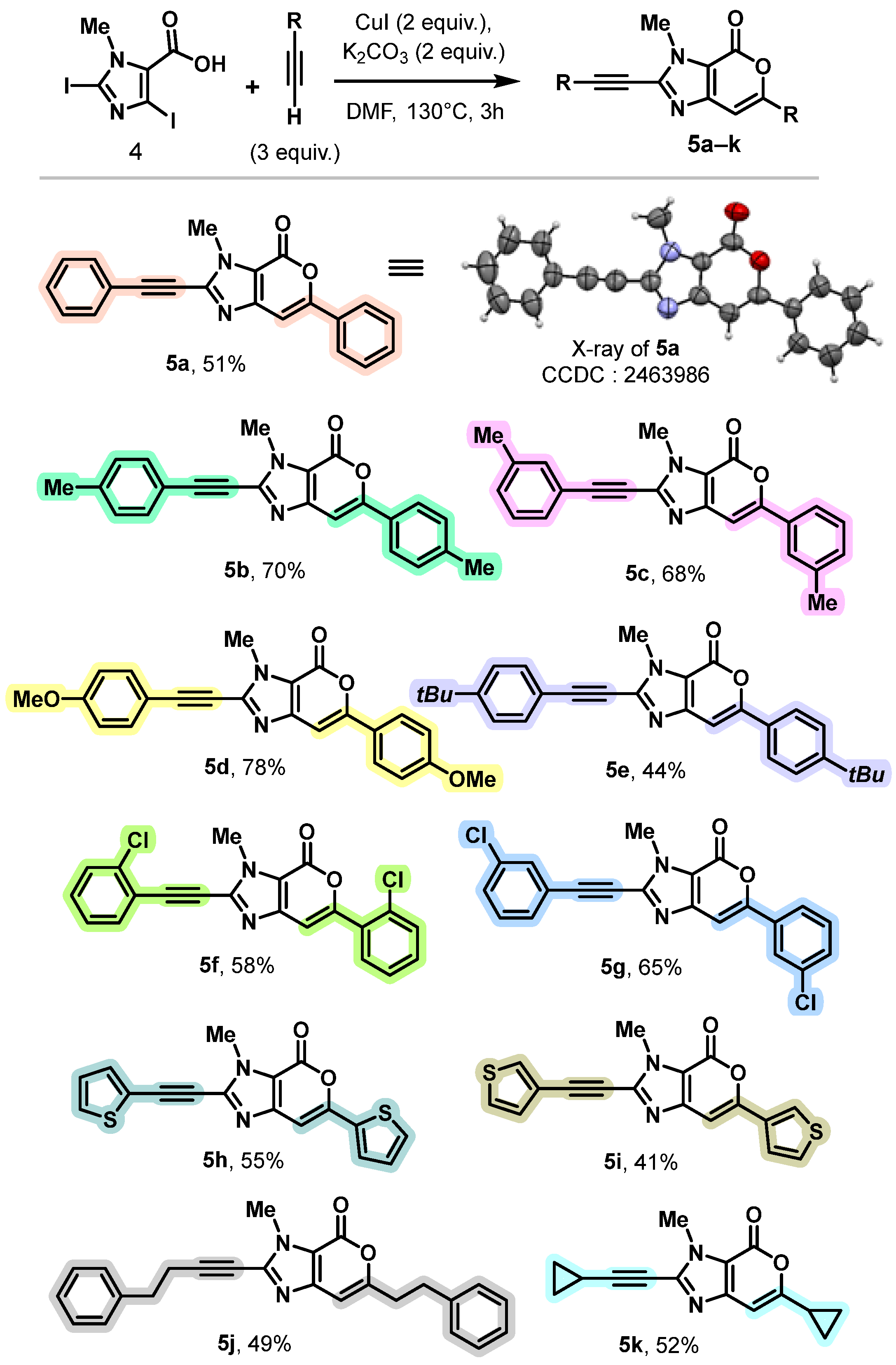
3.2. General Procedure for the Synthesis of 3-Methyl-6-aryl-2-alcynylpyrano[3,4-d]imidazole-4(3H)-one (5a–k)
- 3-Methyl-6-phenyl-2-(phenylethynyl)pyrano[3,4-d]imidazol-4(3H)-one (5a)
- 3-Methyl-6-(4-methylphenyl)-2-[(4-methylphenyl)ethynyl]pyrano[3,4-d]imidazol-4(3H)-one (5b)
- 3-Methyl-6-(3-methylphenyl)-2-[(3-methylphenyl)ethynyl]pyrano[3,4-d]imidazol-4(3H)-one (5c)
- 6-(4-Methoxyphenyl)-2-[(4-methoxyphenyl)ethynyl]-3-methylpyrano[3,4-d]imidazol-4(3H)-one (5d)
- 6-(4-(Tert-butyl)phenyl)-2-((4-(tert-butyl)phenyl)ethynyl)-3-methylpyrano[3,4-d]imidazol-4(3H)-one (5e)
- 6-(2-Chlorophenyl)-2-[(2-chlorophenyl)ethynyl]-3-methylpyrano[3,4-d]imidazol-4(3H)-one (5f)
- 6-(3-Chlorophenyl)-2-[(3-chlorophenyl)ethynyl]-3-methylpyrano[3,4-d]imidazol-4(3H)-one (5g)
- 3-Methyl-6-(thiophen-2-yl)-2-(thiophen-2-ylethynyl)pyrano[3,4-d]imidazol-4(3H)-one (5h)
- 3-Methyl-6-(thiophen-3-yl)-2-(thiophen-3-ylethynyl)pyrano[3,4-d]imidazol-4(3H)-one (5i)
- 3-Methyl-6-phenethyl-2-(4-phenylbut-1-yn-1-yl)pyrano[3,4-d]imidazol-4(3H)-one (5j)
- 6-Cyclopropyl-2-(cyclopropylethynyl)-3-methylpyrano[3,4-d]imidazol-4(3H)-one (5k)
- Ethyl 1-methyl-2,4-bis(phenylethynyl)-1H-imidazole-5-carboxylate (6)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kishore, D.R.; Sreenivasulu, C.; Satyanarayana, G. 2H-Pyran-2-ones’ Synthesis: State-of-the-Art Methods and Applications. Asian J. Org. Chem. 2025, 14, e202400726. [Google Scholar] [CrossRef]
- Gogoi, N.; Parhi, R.; Tripathi, R.K.P.; Pachuau, L.; Kaishap, P.P. Recent advances in synthesis of isocoumarins: An overview. Tetrahedron 2024, 150, 133740. [Google Scholar] [CrossRef]
- Ram, V.J.; Goel, A.; Shukla, P.K.; Kapil, A. Synthesis of thiophenes and thieno[3,2-c]pyran-4-ones as antileishmanial and antifungal agents. Bioorg. Med. Chem. Lett. 1997, 7, 3101. [Google Scholar] [CrossRef]
- Thabet, H.K.; Althagbi, H.I.; Al Zahrani, N.A.; Helal, M.H.; Gouda, M.A. Recent Advances in the Chemistry of Thienopyranone Heterocycles: Synthesis, Reactivity, and Application. Mini-Rev. Org. Chem. 2025, 1875–6298. [Google Scholar] [CrossRef]
- Nakhi, A.; Adepu, R.; Rambabu, D.; Kishore, R.; Vanaja, G.R.; Kalle, A.M.; Pal, M. Thieno[3,2-c]pyran-4-one based novel small molecules: Their synthesis, crystal structure analysis and in vitro evaluation as potential anticancer agents. Bioorg. Med. Chem. Lett. 2012, 22, 4418–4427. [Google Scholar] [CrossRef]
- Ombrato, R.; Garofalo, B.; Mangano, G.; Capezzone de Joannon, A.; Corso, G.; Cavarishia, C.; Furlotti, G.; Iacoangeli, T. Antibacterial Compounds Having Broad Spectrum of Activity. US10221144B2, 5 March 2019. [Google Scholar]
- Mederski, W.W.K.R.; Dorsch, D.; Osswald, M.; Beier, N.; Lues, I.; Minck, K.-O.; Schelling, P.; Ladstetter, B.J. 4,5-Dihydro-4-oxo-3H-imidazo[4,5-c]pyridines: Potent arylacetic acid-derived AT1 antagonists with improved affinity for the AT2 receptor. Bioorg. Med. Chem. Lett. 1995, 5, 2665–2670. [Google Scholar] [CrossRef]
- Mederski, W.W.K.R.; Dorsch, D.; Osswald, M.; Schwartz, H.; Beier, N.; Christadler, M.; Minck, K.O.; Schelling, P.; Schmitges, C.J. Novel 4,5-dihydro-4-oxo-3H-imidazo[4,5-c]pyridines. Potent angiotensin II receptor antagonists with high affinity for both the AT1 and AT2 subtypes. Eur. J. Med. Chem. 1997, 32, 479–491. [Google Scholar] [CrossRef]
- Casimiro-Garcia, A.; Heemstra, R.J.; Bigge, C.F.; Chen, J.; Ciske, F.A.; Davis, J.A.; Ellis, T.; Esmaeil, N.; Flyn, D.; Han, S.; et al. PowellDesign, synthesis, and evaluation of imidazo[4,5-c]pyridin-4-one derivatives with dual activity at angiotensin II type 1 receptor and peroxisome proliferator-activated receptor-γ. Bioorg. Med. Chem. Lett. 2013, 23, 767–772. [Google Scholar] [CrossRef]
- Raju, S.; Batchu, V.R.; Swamy, N.K.; Dev, R.V.; Babu, J.M.; Kumar, P.R.; Mukkanti, K.; Pal, M. Palladium-mediated synthesis of 5-substituted 4-alkynylthieno[2,3-c]pyran-7-ones. Tetrahedron Lett. 2006, 47, 83–88. [Google Scholar] [CrossRef]
- Gorja, D.R.; Batchu, V.R.; Ettam, A.; Pal, M. Pd/C-mediated synthesis of α-pyrone fused with a five-membered nitrogen heteroaryl ring: A new route to pyrano[4,3-c]pyrazol-4(1H)-ones. Beilstein J. Org. Chem. 2009, 5, 64. [Google Scholar] [CrossRef]
- Roy, P.; Ghorai, B.K. One-pot synthesis of pyrano[4,3-b]quinolinones from 2-alkynyl-3-formylquinolines via oxidative 6-endo-dig ring closure. Tetrahedron Lett. 2012, 53, 235–238. [Google Scholar] [CrossRef]
- Rao, M.S.; Haritha, M.; Chandrasekhar, N.; Rao, M.V.B.; Pal, M. Pd/ligand-free synthesis of thienopyranones via Cu-catalyzed coupling-cyclization in PEG 400 under ultrasound. Tetrahedron Lett. 2014, 55, 1660–1663. [Google Scholar] [CrossRef]
- Ngi, S.I.; Guilloteau, V.; Abarbri, M.; Thibonnet, J. Regioselective Copper-Mediated Synthesis of Thieno[2,3-c]pyrane-7-one, Indolo[2,3-c]pyrane-1-one, and Indolo[3,2-c]pyrane-1-one. J. Org. Chem. 2011, 76, 8347–8354. [Google Scholar] [CrossRef]
- Bahlaouan, Z.; Abarbri, M.; Duchêne, A.; Thibonnet, J.; Henry, N.; Enguehard-Gueiffier, C.; Gueiffier, A. Copper(I)-mediated preparation of new pyrano[3′,4′:4,5]imidazo[1,2-a]pyridin-1-one compounds under mild palladium-free conditions. Org. Biomol. Chem. 2011, 9, 1212–1218. [Google Scholar] [CrossRef] [PubMed]
- Kanwal, I.; Mujahid, A.; Rasool, N.; Rizwan, K.; Malik, A.; Ahmad, G.; Shah, S.A.A.; Rashid, U.; Nadiah, M.N. Palladium and Copper Catalyzed Sonogashira cross Coupling an Excellent Methodology for C-C Bond Formation over 17 Years: A Review. Catalysts 2020, 10, 443. [Google Scholar] [CrossRef]
- Mohajer, F.; Heravi, M.M.; Zadsirjan, V.; Poormohammad, N. Copper-free Sonogashira cross-coupling reactions: An overview. RSC Adv. 2021, 11, 6885–6925. [Google Scholar] [CrossRef]
- Gazvoda, M.; Virant, M.; Pinter, B.; Košmrlj, J. Mechanism of copper-free Sonogashira reaction operates through palladium-palladium transmetallation. Nat. Commun. 2018, 9, 4814. [Google Scholar] [CrossRef]
- Thomas, A.M.; Sujathaa, A.; Anilkumar, G. Recent advances and perspectives in copper-catalyzed Sonogashira coupling reactions. RSC Adv. 2014, 4, 21688–21698. [Google Scholar] [CrossRef]
- Brasholz, M.; Luan, X.; Reissig, H.U. Towards the Rubromycins: An Efficient Synthesis of a Suitable Isocoumarin Precursor, its Lactam Analogue, and Palladium-Catalyzed Couplings. Synthesis 2005, 20, 3571–3580. [Google Scholar] [CrossRef]
- Brasholz, M.; Reissig, H.U. An Expedient and Short Synthesis of a 6-Iodo Isocoumarin Building Block for the Rubromycin Family and its First Palladium-Catalyzed Coupling. Synlett 2004, 15, 2736–2738. [Google Scholar] [CrossRef]
- Waters, S.P.; Kozlowski, M.C. Synthesis of the isocoumarin portion of the rubromycins. Tetrahedron Lett. 2001, 42, 3567–3570. [Google Scholar] [CrossRef]
- CCDC 2463986 [For Compound 5a] Contains the Supplementary Crystallographic Data for this Paper. These Data Can Be Obtained Free of Charge from the Cambridge Crystallographic Data Centre. Available online: https://www.ccdc.cam.ac.uk/ (accessed on 13 June 2025).
- Monnier, F.; Turtaut, F.; Duroure, L.; Taillefer, M. Copper-Catalyzed Sonogashira-Type Reactions Under Mild Palladium-Free Conditions. Org. Lett. 2008, 10, 3203–3206. [Google Scholar] [CrossRef] [PubMed]
- Kotovshchikov, Y.N.; Latyshev, G.V.; Lukashev, N.V.; Beletskaya, I.P. Alkynylation of steroids via Pd-free Sonogashira coupling. Org. Biomol. Chem. 2015, 13, 5542–5555. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Paul, S.; Mandal, K.K.; Anoop, A.; Nanda, S. Asymmetric Total Synthesis of Naturally Occurring (R)-2′-Methoxydihydroartemidin, (R)-(E)-3′-Hydroxyartemidin, and Its Structural Congeners: Method Optimization and Mechanistic Analysis. J. Org. Chem. 2024, 89, 15764–15776. [Google Scholar] [CrossRef] [PubMed]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminf. 2011, 3, 33. [Google Scholar] [CrossRef]
- Bannwarth, C.; Caldeweyher, E.; Ehlert, S.; Hansen, A.; Pracht, P.; Seibert, J.; Spicher, S.; Grimme, S. Extended tight-binding quantum chemistry methods. WIREs Comput. Mol. Sci. 2020, 11, e01493. [Google Scholar] [CrossRef]
- Neese, F. The ORCA program system. WIREs Comput Mol Sci 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Raghavachari, K. Perspective on “Density functional thermochemistry. III. The role of exact exchange”. Theor. Chem. Acc. 2000, 103, 361–363. [Google Scholar] [CrossRef]
- Arnim, H.; Dmitrij, R. Development of new auxiliary basis functions of the Karlsruhe segmented contracted basis sets including diffuse basis functions (def2-SVPD, def2-TZVPPD, and def2-QVPPD) for RI-MP2 and RI-CC calculations. Phys. Chem. Chem. Phys. 2015, 17, 1010–1017. [Google Scholar]
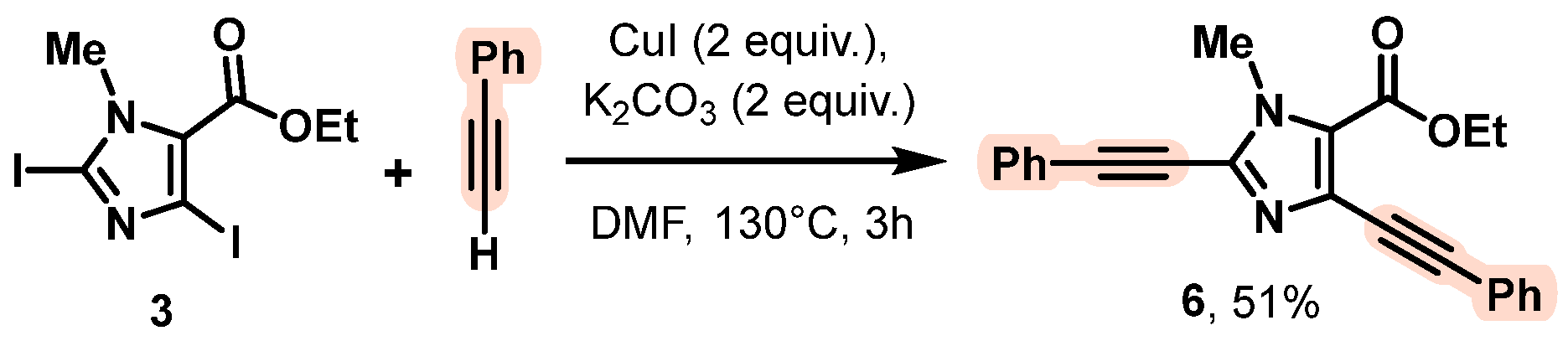

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Base | Catalyst (equiv.) | Alkyne (equiv.) | T (°C) | Time (h) | Ratio (%) a5a/4 |
| 1 | Et3N | CuI/PdCl2(PPh3)2 (0.2/0.1) | 3 | 130 | 3 | 47/53 |
| 2 | Et3N | CuI/PdCl2(PPh3)2 (1/0.1) | 3 | 130 | 3 | 70/30 |
| 3 | K2CO3 | CuI/PdCl2(PPh3)2 (1/0.1) | 3 | 130 | 3 | 100/0 (44%) b |
| 4 | K2CO3 | CuI (1) | 3 | 130 | 24 | 100/0 (27%) b |
| 5 | K2CO3 | CuI (2) | 3 | 130 | 3 | 100/0 (51%) b |
| 6 | K2CO3 | CuI (2.5) | 3 | 130 | 3 | 100/0 (47%) b |
| 7 | K2CO3 | CuI (2) | 2.05 | 130 | 3 | 100/0 (24%) b |
| 8 | K2CO3 | CuI (2) | 1.05 | 130 | 3 | 100/0 (12%) b |
| 9 | K2CO3 | CuI (2) | 3 | 150 | 3 | 100/0 (44%) b |
| 10 | K2CO3 | CuI (2) | 3 | 100 | 24 | 100/0 (12%) b |
| 11 | Na2CO3 | CuI (2) | 3 | 130 | 3 | 100/0 (9%) b |
| 12 | K3PO4 | CuI (2) | 3 | 130 | 3 | 55/45 |
| 13 | Cs2CO3 | CuI (2) | 3 | 130 | 3 | 52/48 |
| 14 | Et3N | CuI (2) | 3 | 130 | 3 | 55/45 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ayachi, A.; Tikad, A.; Lazeran, V.; Allouchi, H.; Bletry, M.; Besbes, R.; Abarbri, M.; Jismy, B. Pd/Ligand-Free Synthesis of 2-Alkynylated Pyrano[4,3-d]imidazol-4-ones via One-Pot Cu-Mediated Tandem Sonogashira Coupling/Regioselective 6-endo-dig Oxacyclization Reaction. Molecules 2025, 30, 3045. https://doi.org/10.3390/molecules30143045
Ayachi A, Tikad A, Lazeran V, Allouchi H, Bletry M, Besbes R, Abarbri M, Jismy B. Pd/Ligand-Free Synthesis of 2-Alkynylated Pyrano[4,3-d]imidazol-4-ones via One-Pot Cu-Mediated Tandem Sonogashira Coupling/Regioselective 6-endo-dig Oxacyclization Reaction. Molecules. 2025; 30(14):3045. https://doi.org/10.3390/molecules30143045
Chicago/Turabian StyleAyachi, Abir, Abdellatif Tikad, Vincent Lazeran, Hassan Allouchi, Marc Bletry, Rafâa Besbes, Mohamed Abarbri, and Badr Jismy. 2025. "Pd/Ligand-Free Synthesis of 2-Alkynylated Pyrano[4,3-d]imidazol-4-ones via One-Pot Cu-Mediated Tandem Sonogashira Coupling/Regioselective 6-endo-dig Oxacyclization Reaction" Molecules 30, no. 14: 3045. https://doi.org/10.3390/molecules30143045
APA StyleAyachi, A., Tikad, A., Lazeran, V., Allouchi, H., Bletry, M., Besbes, R., Abarbri, M., & Jismy, B. (2025). Pd/Ligand-Free Synthesis of 2-Alkynylated Pyrano[4,3-d]imidazol-4-ones via One-Pot Cu-Mediated Tandem Sonogashira Coupling/Regioselective 6-endo-dig Oxacyclization Reaction. Molecules, 30(14), 3045. https://doi.org/10.3390/molecules30143045