
Design and Synthesis of Boronic Chalcones with Dual Anticancer and Anti-Inflammatory Activity
 , , ,
, , ,  ,
,  , , ,
, , , 
Abstract

1. Introduction
2. Results
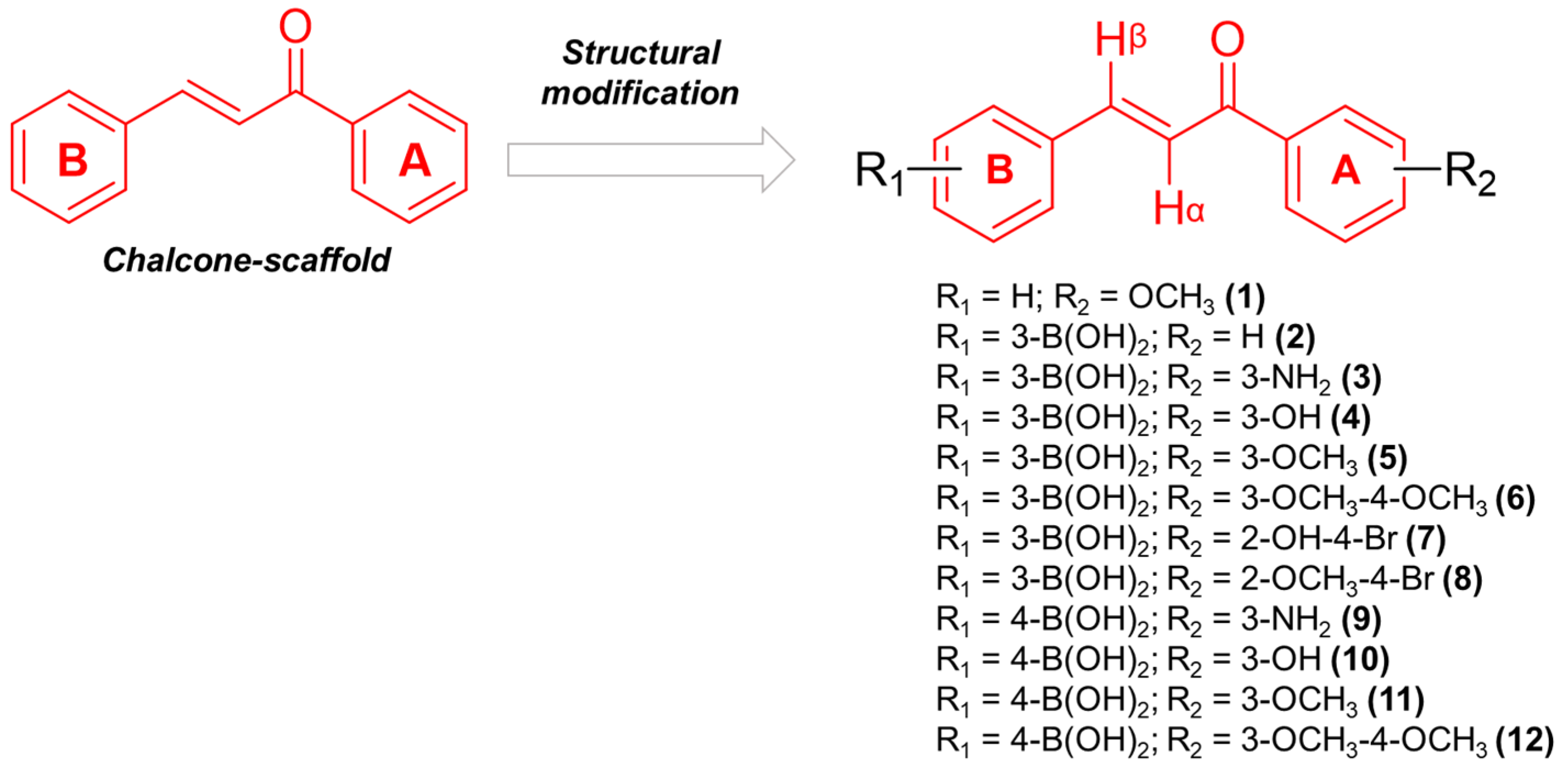
2.1. Design and Synthesis of Boronic Chalcones Derivatives
2.2. Determination of Log P and In Vitro Cytotoxicity in Cancer Models
2.2.1. Assessment of Log P and In Vitro Activity in SCC-25 and NOK-si Cells
2.2.2. Cytotoxicity of Chalcones in p53+/+ and p53−/− Cell Lines
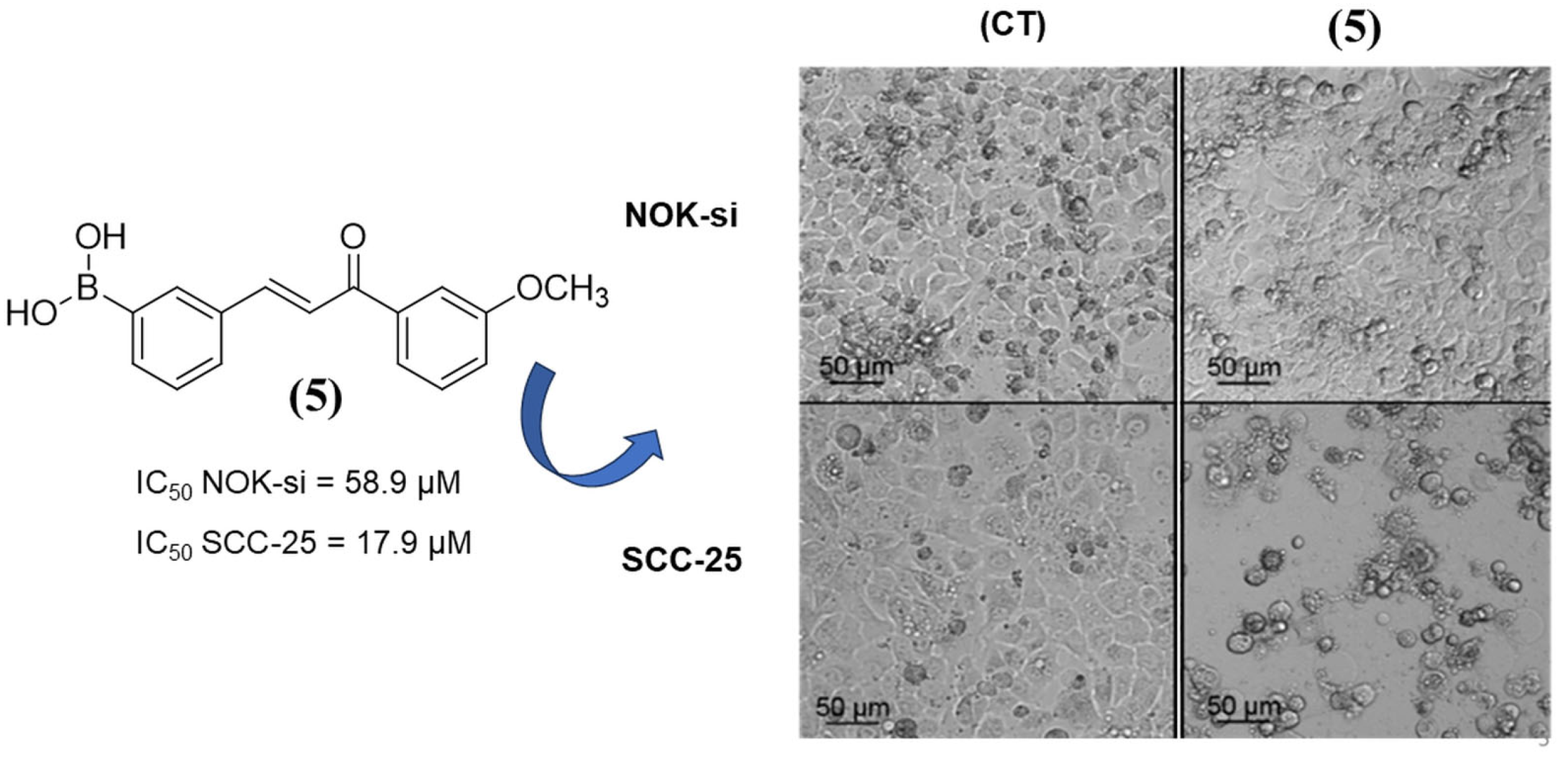
2.2.3. Mechanism of Cell Death Triggered by Chalcone 5
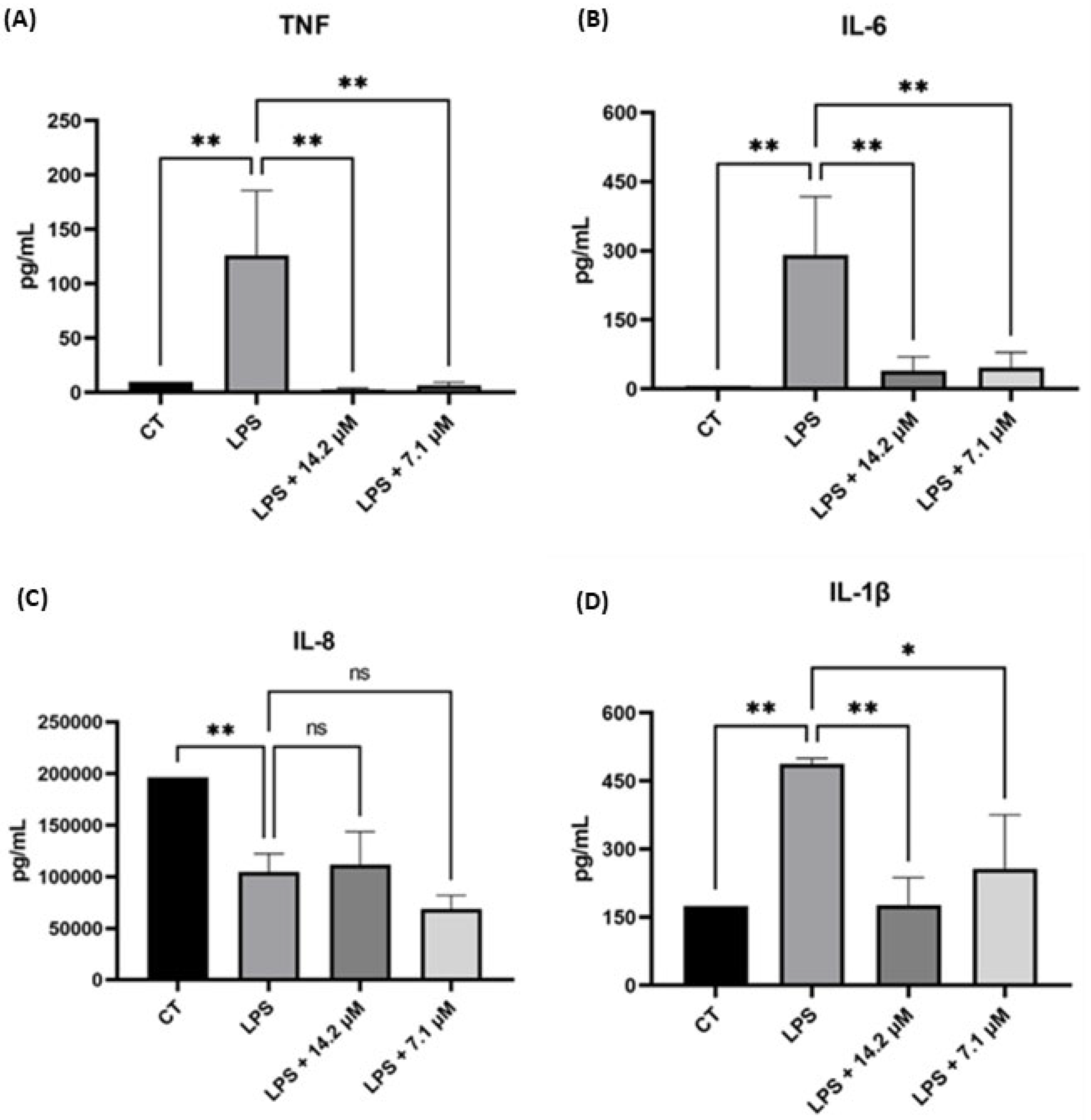
2.2.4. Chalcone 5 Drastically Reduces Levels of Pro-Inflammatory Cytokines: TNF, IL-6, IL-1β
2.2.5. Compound 5 Exhibits a Promising DMPK Profile
3. Discussion
4. Materials and Methods
4.1. General Information
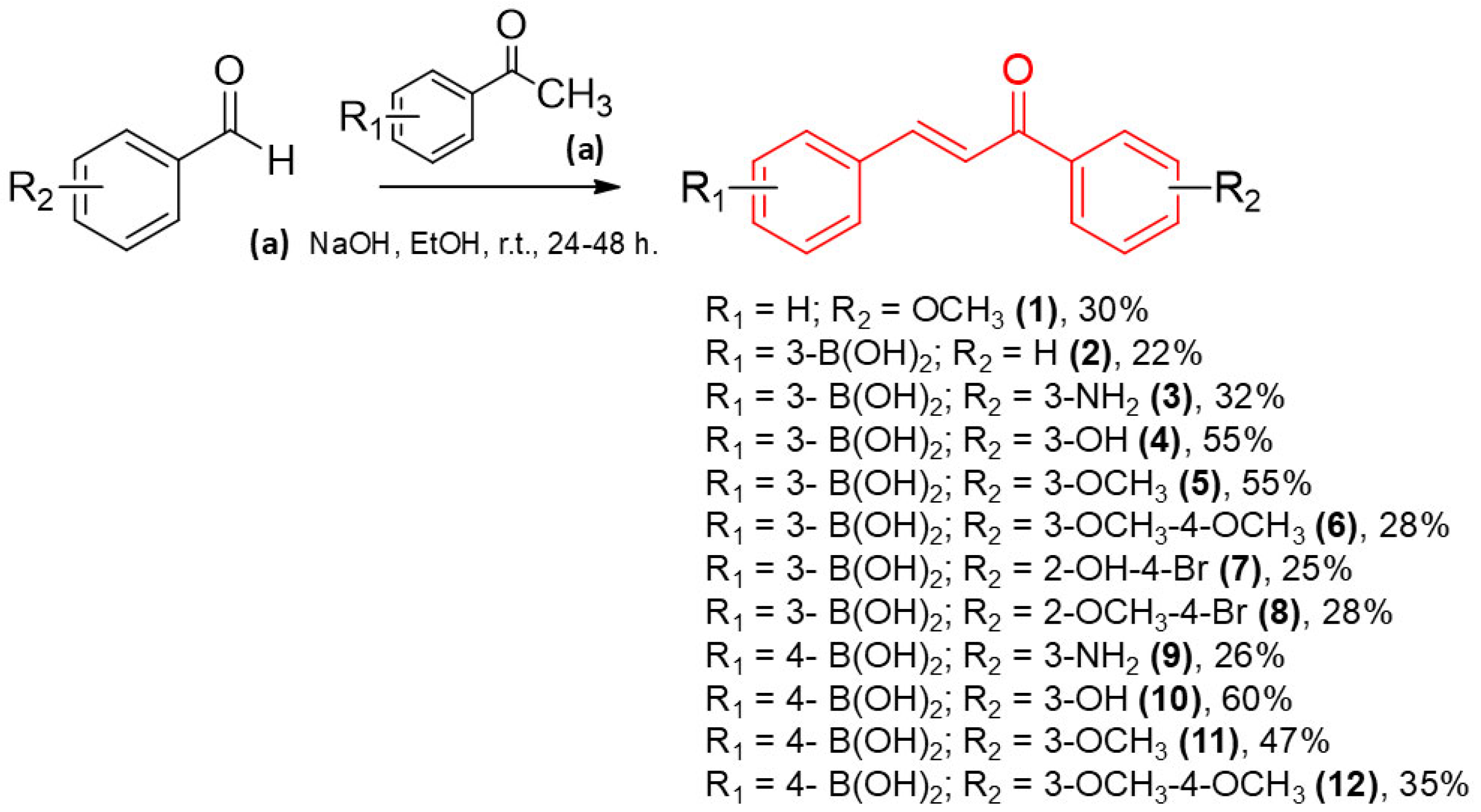
4.2. General Procedure for the Synthesis of Chalcones Derivatives (1–12)
- (E)-3-(3-methoxyphenyl)-1-phenylprop-2-en-1-one (1)
- (E)-(3-(3-oxo-3-phenylprop-1-en-1-yl)phenyl)boronic acid (2)
- (E)-(3-(3-(3-aminophenyl)-3-oxoprop-1-en-1-yl)phenyl)boronic acid (3)
- (E)-(3-(3-(3-hydroxyphenyl)-3-oxoprop-1-en-1-yl)phenyl)boronic acid (4)
- (E)-(3-(3-(3-methoxyphenyl)-3-oxoprop-1-en-1-yl)phenyl)boronic acid (5)
- (E)-(3-(3-(3,4-dimethoxyphenyl)-3-oxoprop-1-en-1-yl)phenyl)boronic acid (6)
- (E)-(3-(3-(2-bromo-4-hydroxyphenyl)-3-oxoprop-1-en-1-yl)phenyl)boronic acid (7)
- (E)-(3-(3-(2-bromo-4-methoxyphenyl)-3-oxoprop-1-en-1-yl)phenyl)boronic acid (8)
- (E)-(4-(3-(3-aminophenyl)-3-oxoprop-1-en-1-yl)phenyl)boronic acid (9)
- (E)-(4-(3-(3-hydroxyphenyl)-3-oxoprop-1-en-1-yl)phenyl)boronic acid (10)
- (E)-(4-(3-(3-methoxyphenyl)-3-oxoprop-1-en-1-yl)phenyl)boronic acid (11)
- (E)-(4-(3-(3,4-dimethoxyphenyl)-3-oxoprop-1-en-1-yl)phenyl)boronic acid (12)
4.3. Determination of Partition Coefficient (Log P)
4.4. Biological Evaluation
4.4.1. Cell Lines
4.4.2. Cytotoxicity Assay on HNC Cell Lines (MTT Protocol)
4.4.3. Cytotoxicity Assay on HCT p53+/+ and HCT p53−/− Cell Lines
4.4.4. Apoptosis/Necrosis Assay
4.4.5. Anti-Inflammatory Assay
4.5. DMPK Assays
4.5.1. Equipment
4.5.2. Determination of Kinetic Solubility
4.5.3. Parallel Artificial Membrane Permeability Assays (PAMPA)
4.5.4. Determination of Distribution Coefficient (eLogD)
4.5.5. Metabolic Stability in Mouse (CD-1) and Human Liver Microsomes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Johnson, D.E.; Burtness, B.; Leemans, C.R.; Lui, V.W.Y.; Bauman, J.E.; Grandis, J.R. Head and Neck Squamous Cell Carcinoma. Nat. Rev. Dis. Primers 2020, 6, 92. [Google Scholar] [CrossRef] [PubMed]
- Wierzbicka, M.; Pietruszewska, W.; Maciejczyk, A.; Markowski, J. Markowski, Trends in Incidence and Mortality of Head and Neck Cancer Subsites Among Elderly Patients: A Population-Based Analysis. Cancers 2025, 17, 548. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Network. Comprehensive Genomic Characterization of Head and Neck Squamous Cell Carcinomas. Nature 2015, 517, 576–582. [Google Scholar] [CrossRef]
- Leemans, C.R.; Braakhuis, B.J.M.; Brakenhoff, R.H. The Molecular Biology of Head and Neck Cancer. Nat. Rev. Cancer 2011, 11, 9–22. [Google Scholar] [CrossRef]
- Nakano, Y.; Kobayashi, W.; Sugai, S.; Kimura, H.; Yagihashil, S. Expression of Tumor Necrosis Factor-α and Interleukin-6 in Oral Squamous Cell Carcinoma. Jpn J. Cancer Res. 1999, 90, 858–866. [Google Scholar] [CrossRef]
- Benito-Ramal, E.; Egido-Moreno, S.; González-Navarro, B.; Jané-Salas, E.; Roselló-Llabrés, X.; López-López, J. Role of Selected Salivary Inflammatory Cytokines in the Diagnosis and Prognosis of Oral Squamous Cell Carcinoma: A Systematic Review and Meta-analysis. Med. Oral Patol. Oral Cir. Bucal 2023, 28, e474–e486. [Google Scholar] [CrossRef] [PubMed]
- Sahebjamee, M.; Eslami, M.; AtarbashiMoghadam, F.; Sarafnejad, A. Salivary Concentration of TNF-α, IL-1α, IL-6, and IL-8 in Oral Squamous Cell Carcinoma. Med. Oral Patol. Oral Cir. Bucal 2008, 13, E292–E295. [Google Scholar] [PubMed]
- Scheff, N.N.; Ye, Y.; Bhattacharya, A.; MacRae, J.; Hickman, D.N.; Sharma, A.K.; Dolan, J.C.; Schmidt, B.L. Tumor Necrosis Factor Alpha Secreted from Oral Squamous Cell Carcinoma Contributes to Cancer Pain and Associated Inflammation. Pain 2017, 158, 2396–2409. [Google Scholar] [CrossRef] [PubMed]
- Goertzen, C.; Mahdi, H.; Laliberte, C.; Meirson, T.; Eymael, D.; Gil-Henn, H.; Magalhaes, M. Oral Inflammation Promotes Oral Squamous Cell Carcinoma Invasion. Oncotarget 2018, 9, 29047–29063. [Google Scholar] [CrossRef]
- De Sousa, L.G.; Ferrarotto, R. Pembrolizumab in the First-Line Treatment of Advanced Head and Neck Cancer. Expert Rev. Anticancer Ther. 2021, 21, 1321–1331. [Google Scholar] [CrossRef] [PubMed]
- Vermorken, J.B.; Mesia, R.; Rivera, F.; Remenar, E.; Kawecki, A.; Rottey, S.; Erfan, J.; Zabolotnyy, D.; Kienzer, H.-R.; Cupissol, D.; et al. Platinum-Based Chemotherapy Plus Cetuximab in Head and Neck Cancer. N. Engl. J. Med. 2008, 359, 1116–1127. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; Li, Y.; Xiong, L.; Wang, W.; Wu, M.; Yuan, T.; Yang, W.; Tian, C.; Miao, Z.; Wang, T.; et al. Small Molecules in Targeted Cancer Therapy: Advances, Challenges, and Future Perspectives. Signal Transduct. Target Ther. 2021, 6, 201. [Google Scholar] [CrossRef] [PubMed]
- Bedard, P.L.; Hyman, D.M.; Davids, M.S.; Siu, L.L. Small Molecules, Big Impact: 20 Years of Targeted Therapy in Oncology. Lancet 2020, 395, 1078–1088. [Google Scholar] [CrossRef] [PubMed]
- Jasim, H.A.; Nahar, L.; Jasim, M.A.; Moore, S.A.; Ritchie, K.J.; Sarker, S.D. Chalcones: Synthetic Chemistry Follows Where Nature Leads. Biomolecules 2021, 11, 1203. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, C.; Zhang, W.; Sheng, C.; Zhang, W.; Xing, C.; Miao, Z. Chalcone: A Privileged Structure in Medicinal Chemistry. Chem. Rev. 2017, 117, 7762–7810. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, Y.; Li, J.; Chen, X.; Fu, X.; Sun, S.; Wu, Q. Chalcone Derivatives: Role in Anticancer Therapy. Biomolecules 2021, 11, 894. [Google Scholar] [CrossRef]
- Rudrapal, M.; Khan, J.; Bin Dukhyil, A.A.; Alarousy, R.M.I.I.; Attah, E.I.; Sharma, T.; Khairnar, S.J.; Bendale, A.R. Chalcone Scaffolds, Bioprecursors of Flavonoids: Chemistry, Bioactivities, and Pharmacokinetics. Molecules 2021, 26, 7177. [Google Scholar] [CrossRef] [PubMed]
- Mahapatra, D.K.; Bharti, S.K.; Asati, V. Asati, Anti-Cancer Chalcones: Structural and Molecular Target Perspectives. Eur. J. Med. Chem. 2015, 98, 69–114. [Google Scholar] [CrossRef]
- Priya, S.; Islam, M.; Kasana, S.; Kurmi, B.D.; Gupta, G.D.; Patel, P. Therapeutic Potential of Chalcone-1,2,3-Triazole Hybrids as Anti-Tumor Agents: A Systematic Review and SAR Studies. Future Med. Chem. 2025, 17, 449–465. [Google Scholar] [CrossRef]
- Bamaraki, I.; Altintop, M.D.; Arslan, R.; Altınok, F.A.; Özdemir, A.; Dallali, I.; Hasan, A.; Türkmen, N.B. Design, Synthesis, and In Vivo Evaluation of a New Series of Indole-Chalcone Hybrids as Analgesic and Anti-Inflammatory Agents. ACS Omega 2024, 9, 12175–12183. [Google Scholar] [CrossRef] [PubMed]
- El-Atawy, M.A.; Hanna, D.H.; Bashal, A.H.; Ahmed, H.A.; Alshammari, E.M.; Hamed, E.A.; Aljohani, A.R.; Omar, A.Z. Synthesis, Characterization, Antioxidant, and Anticancer Activity Against Colon Cancer Cells of Some Cinnamaldehyde-Based Chalcone Derivatives. Biomolecules 2024, 14, 216. [Google Scholar] [CrossRef] [PubMed]
- Prates, J.L.B.; Pavan, A.R.; dos Santos, J.L. Boron in Medicinal and Organic Chemistry. Curr. Org. Chem. 2021, 25, 1853–1867. [Google Scholar] [CrossRef]
- Prates, J.L.B.; Silva, S.D.M.; Medina-Alarcón, K.P.; Santos, K.S.D.; Belizario, J.A.; Lopes, J.R.; Marin-Dett, F.H.; Campos, D.L.; Campos, D.L.; Fusco-Almeida, A.M.; et al. Synthesis and Evaluation of Boron-Containing Heterocyclic Compounds with Antimicrobial and Anticancer Activities. Molecules 2025, 30, 1117. [Google Scholar] [CrossRef] [PubMed]
- Stoll, R.; Renner, C.; Hansen, S.; Palme, S.; Klein, C.; Belling, A.; Zeslawski, W.; Kamionka, M.; Rehm, T.; Mühlhahn, P.; et al. Chalcone Derivatives Antagonize Interactions Between the Human Oncoprotein MDM2 and p53. Biochemistry 2000, 40, 336–344. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.K.; Hager, E.; Pettit, C.; Gurulingappa, H.; Davidson, N.E.; Khan, S.R. Design, Synthesis, and Evaluation of Novel Boronic-Chalcone Derivatives as Antitumor Agents. J. Med. Chem. 2003, 46, 2813–2815. [Google Scholar] [CrossRef] [PubMed]
- Achanta, G.; Modzelewska, A.; Feng, L.; Khan, S.R.; Huang, P. A Boronic-Chalcone Derivative Exhibits Potent Anticancer Activity Through Inhibition of the Proteasome. Mol. Pharmacol. 2006, 70, 426–433. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.; Wang, K.; Edler, M.C.; Hamel, E.; Mooberry, S.L.; Paige, M.A.; Brown, M.L. A Boronic Acid Chalcone Analog of Combretastatin A-4 as a Potent Anti-Proliferation Agent. Bioorg. Med. Chem. 2010, 18, 971–977. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Wang, W.; Ou, H.; Ning, J.; Zhou, Y.; Ke, J.; Hou, A.; Chen, L.; Li, P.; Ma, Y.; et al. Identification of Chalcone Analogues as Anti-Inflammatory Agents Through the Regulation of NF-κB and JNK Activation. RSC Med. Chem. 2024, 15, 2002–2017. [Google Scholar] [CrossRef] [PubMed]
- Jabbar, Z.; Irfan, H.M.; Alamgeer; Ullah, A.; Mahmoud, M.H.; Batiha, G.E.; Ahsan, H.; Khan, A.; Kashtoh, H. Trans-Chalcone (1–3-Diphenyl-2-Propen-1-One) as a Therapeutic Candidate in Joint Inflammation via Reduction of TNF, IL-1β, IL-6, and IL-17 in Rodents: An In Vivo Study by RT-PCR and ELISA Analysis. ACS Omega 2024, 9, 22123–22135. [Google Scholar] [CrossRef]
- Ibrahim, N.S.; Sayed, H.A.; Sharaky, M.; Diab, H.M.; Elwahy, A.H.M.; Abdelhamid, I.A. Synthesis, Cytotoxicity, Anti-Inflammatory, Anti-Metastatic and Anti-Oxidant Activities of Novel Chalcones Incorporating 2-Phenoxy-N-Arylacetamide and Thiophene Moieties: Induction of Apoptosis in MCF7 and HEP2 Cells. Naunyn Schmiedebergs Arch. Pharmacol. 2024, 397, 10091–10107. [Google Scholar] [CrossRef] [PubMed]
- DeFrancesco, H.; Dudley, J.; Coca, A. Boron Chemistry: An Overview. In Boron Reagents in Synthesis; Coca, A., Ed.; American Chemical Society: Washington, DC, USA, 2016; Volume 1236, pp. 1–25. [Google Scholar]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef] [PubMed]
- Obach, R.S. Prediction of Human Clearance of Twenty-Nine Drugs from Hepatic Microsomal Intrinsic Clearance Data: An Examination of In Vitro Half-Life Approach and Nonspecific Binding to Microsomes. Drug Metab. Dispos. 1999, 27, 1350–1359. [Google Scholar] [CrossRef]
- OECD. Test No. 117: Partition Coefficient (n-Octanol/Water), HPLC Method. In OECD Guidelines for the Testing of Chemicals, Section 1; OECD Publishing: Paris, France, 2022. [Google Scholar]
- Arbeláez, M.I.M.; de Paula e Silva, A.C.A.; Navegante, G.; Valente, V.; Barbugli, P.A.; Vergani, C.E. Proto-Oncogenes and Cell Cycle Gene Expression in Normal and Neoplastic Oral Epithelial Cells Stimulated with Soluble Factors from Single and Dual Biofilms of Candida albicans and Staphylococcus aureus. Front. Cell Infect. Microbiol. 2021, 11, 62704. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, F.S.; Paula e Silva, A.C.A.; Barbugli, P.A.; Cinca, N.; Dosta, S.; Cano, I.G.; Guilemany, J.M.; Vergani, C.E.; Benedetti, A.V. Anti-Biofilm Activity and In Vitro Biocompatibility of Copper Surface Prepared by Cold Gas Spray. Surf. Coat. Technol. 2021, 411, 126981. [Google Scholar] [CrossRef]
- Baxter, E.W.; Graham, A.E.; Re, N.A.; Carr, I.M.; Robinson, J.I.; Mackie, S.L.; Morgan, A.W. Standardized Protocols for Differentiation of THP-1 Cells to Macrophages with Distinct M(IFNγ+LPS), M(IL-4) and M(IL-10) Phenotypes. J. Immunol. Methods 2020, 478, 112721. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | SCC-25 IC50 (µM) | NOK-si IC50 (µM) | Detroit IC50 (µM) | SI * | eLog P *** |
|---|---|---|---|---|---|
| (1) | 24.9 ± 1.09 | 101.5 ± 0.17 | – ** | 4.08 | 3.73 |
| (2) | 133.7 ± 12.29 | 133.1 ± 3.25 | – | 1.00 | 2.68 |
| (3) | 45.2 ± 13.71 | 36.2 ± 10.69 | – | 1.88 | 1.49 |
| (4) | 9.8 ± 1.00 | 18.5 ± 4.43 | 31.6 ± 3.13 | 1.88 | 1.81 |
| (5) | 17.9 ± 0.35 | 58.9 ± 10.63 | 33.1 ± 5.10 | 3.29 | 2.92 |
| (6) | 27.2 ± 3.84 | 20.8 ± 2.17 | – | 0.77 | 2.30 |
| (7) | 41.1 ± 4.62 | 54.0 ± 10.98 | – | 1.31 | 3.46 |
| (8) | 33.4 ± 1.25 | 27.4 ± 3.05 | – | 0.82 | 4.68 |
| (9) | 108.3 ± 22.46 | 74.6 ± 14.97 | – | 0.69 | 1.18 |
| (10) | 43.8 ± 4.43 | 25.7 ± 11.56 | – | 0.59 | 1.64 |
| (11) | 65.9 ± 10.98 | 96.2 ± 21.26 | – | 1.46 | 2.81 |
| (12) | 38.4 ± 1.63 | 29.5 ± 6.08 | – | 0.77 | 2.16 |
| 5-FU | - | 1800 ± 27.67 | - | - | - |
| Compounds | HCT p53+/+ IC50 (µM) | HCT p53−/− IC50 (µM) |
|---|---|---|
| (1) | 6.83 ± 1.15 | 7.56 ± 0.40 |
| (2) | 4.4 ± 0.53 | 3.63 ± 0.47 |
| (4) | 2.33 ± 0.20 | 2.16 ± 0.25 |
| (5) | 2.5 ± 0.17 | 2.16 ± 0.31 |
| (7) | 3.63 ± 0.40 | 4.2 ± 0.35 |
| (8) | 2.06 ± 0.45 | 1.70 ± 0.72 |
| (11) | 3.96 ± 0.55 | 3.8 ± 0.26 |
| LogD7.4 | Kinect Solubility (KS) pH = 7.4 * (µg/mL) | PAMPA (Mean Pe ** 10−6 cm/s) | Human Liver Microsome (HLM) | Mouse Liver Microsome (MsLM) | ||
|---|---|---|---|---|---|---|
| T1/2 (min.) | Clint(mic) (µg/min/mg) | T1/2 (min.) | Clint(mic) (µg/min/mg) | |||
| 3.92 | >39.65 | 17.50 | 48.81 | 56.80 | 77.02 | 36.00 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lopes, J.R.; Marin-Dett, F.H.; Silva, R.A.M.; Chelucci, R.C.; Saraiva, L.; Sousa, M.E.; Ferreira, L.L.G.; Andricopulo, A.D.; Barbugli, P.A.; Dos Santos, J.L. Design and Synthesis of Boronic Chalcones with Dual Anticancer and Anti-Inflammatory Activity. Molecules 2025, 30, 3032. https://doi.org/10.3390/molecules30143032
Lopes JR, Marin-Dett FH, Silva RAM, Chelucci RC, Saraiva L, Sousa ME, Ferreira LLG, Andricopulo AD, Barbugli PA, Dos Santos JL. Design and Synthesis of Boronic Chalcones with Dual Anticancer and Anti-Inflammatory Activity. Molecules. 2025; 30(14):3032. https://doi.org/10.3390/molecules30143032
Chicago/Turabian StyleLopes, Juliana Romano, Freddy Humberto Marin-Dett, Rita Alexandra Machado Silva, Rafael Consolin Chelucci, Lucília Saraiva, Maria Emília Sousa, Leonardo Luiz Gomes Ferreira, Adriano Defini Andricopulo, Paula Aboud Barbugli, and Jean Leandro Dos Santos. 2025. "Design and Synthesis of Boronic Chalcones with Dual Anticancer and Anti-Inflammatory Activity" Molecules 30, no. 14: 3032. https://doi.org/10.3390/molecules30143032
APA StyleLopes, J. R., Marin-Dett, F. H., Silva, R. A. M., Chelucci, R. C., Saraiva, L., Sousa, M. E., Ferreira, L. L. G., Andricopulo, A. D., Barbugli, P. A., & Dos Santos, J. L. (2025). Design and Synthesis of Boronic Chalcones with Dual Anticancer and Anti-Inflammatory Activity. Molecules, 30(14), 3032. https://doi.org/10.3390/molecules30143032