
Extended Antimicrobial Profile of Chromone–Butenafine Hybrids

 ,
,  , and
, and 
Abstract
1. Introduction
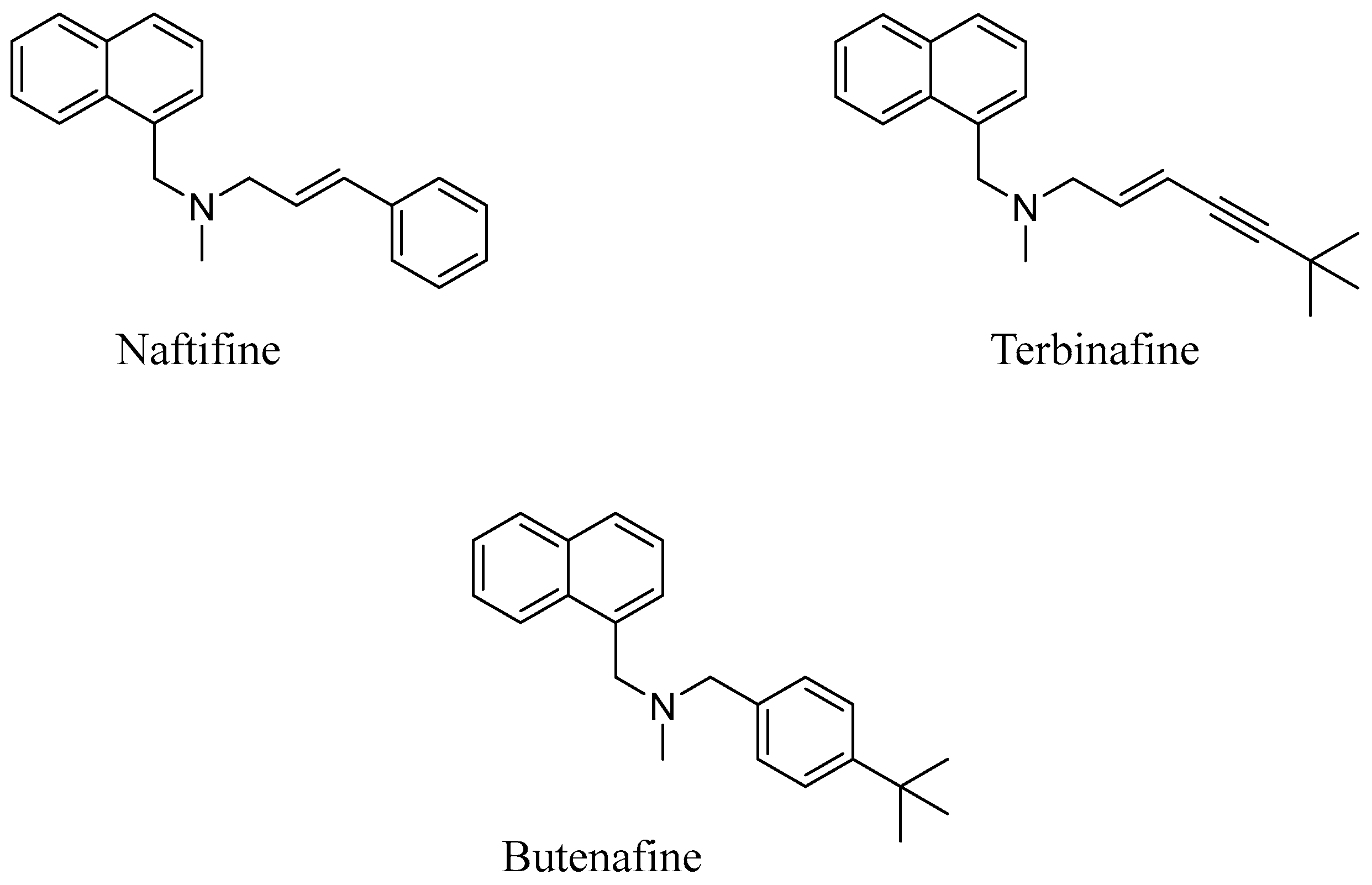
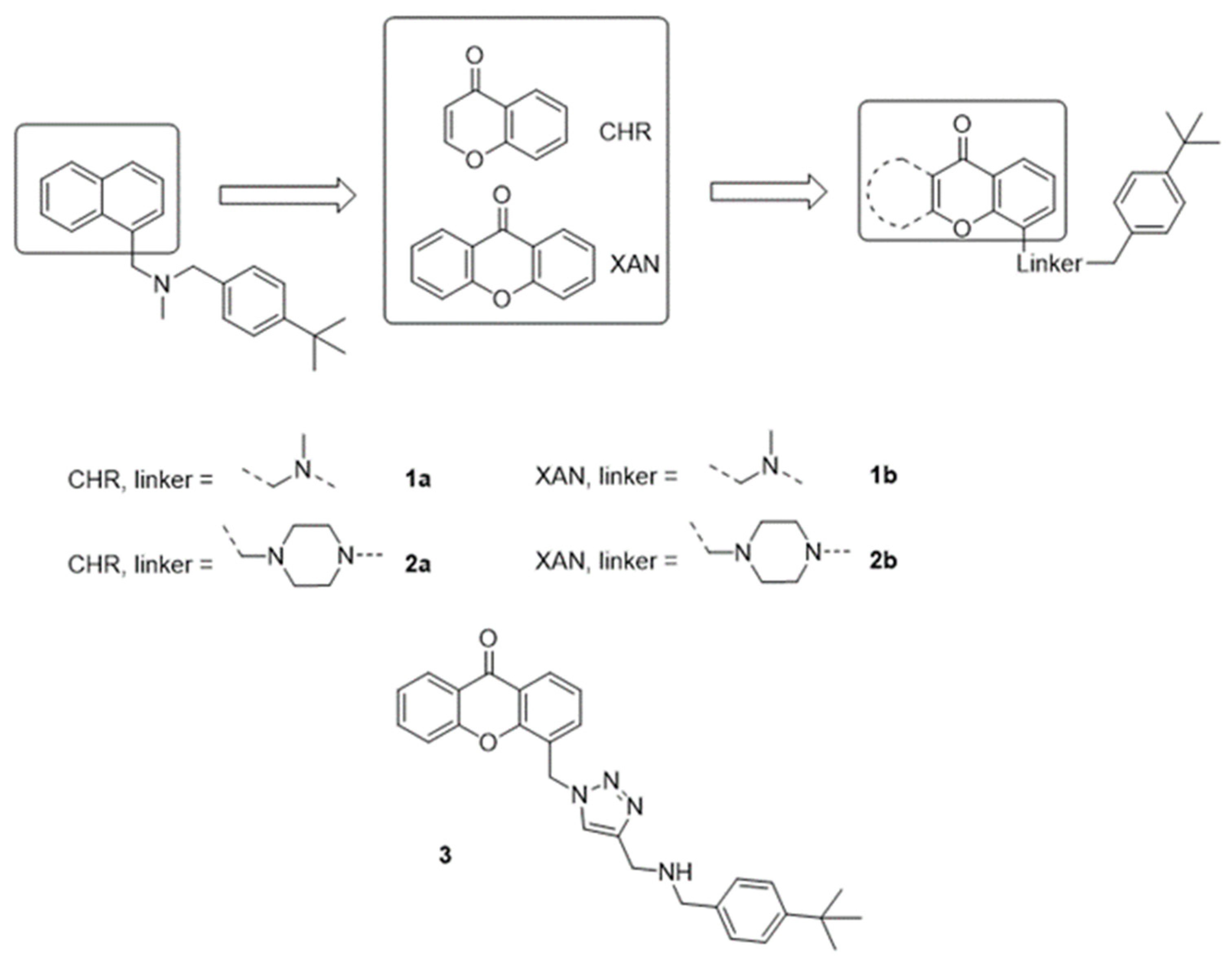
2. Design
3. Results and Discussion
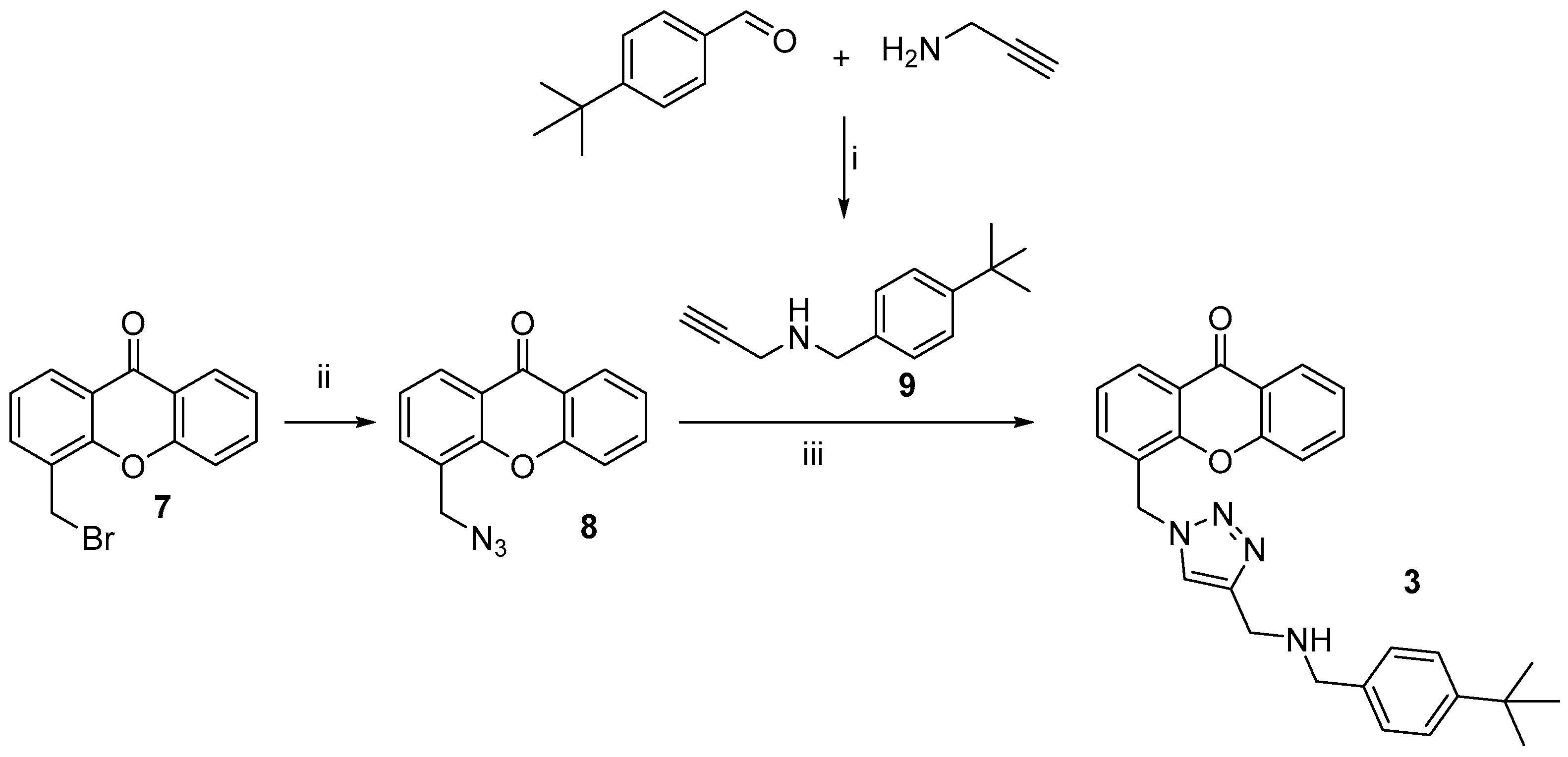
3.1. Chemistry

3.2. Biological Evaluations
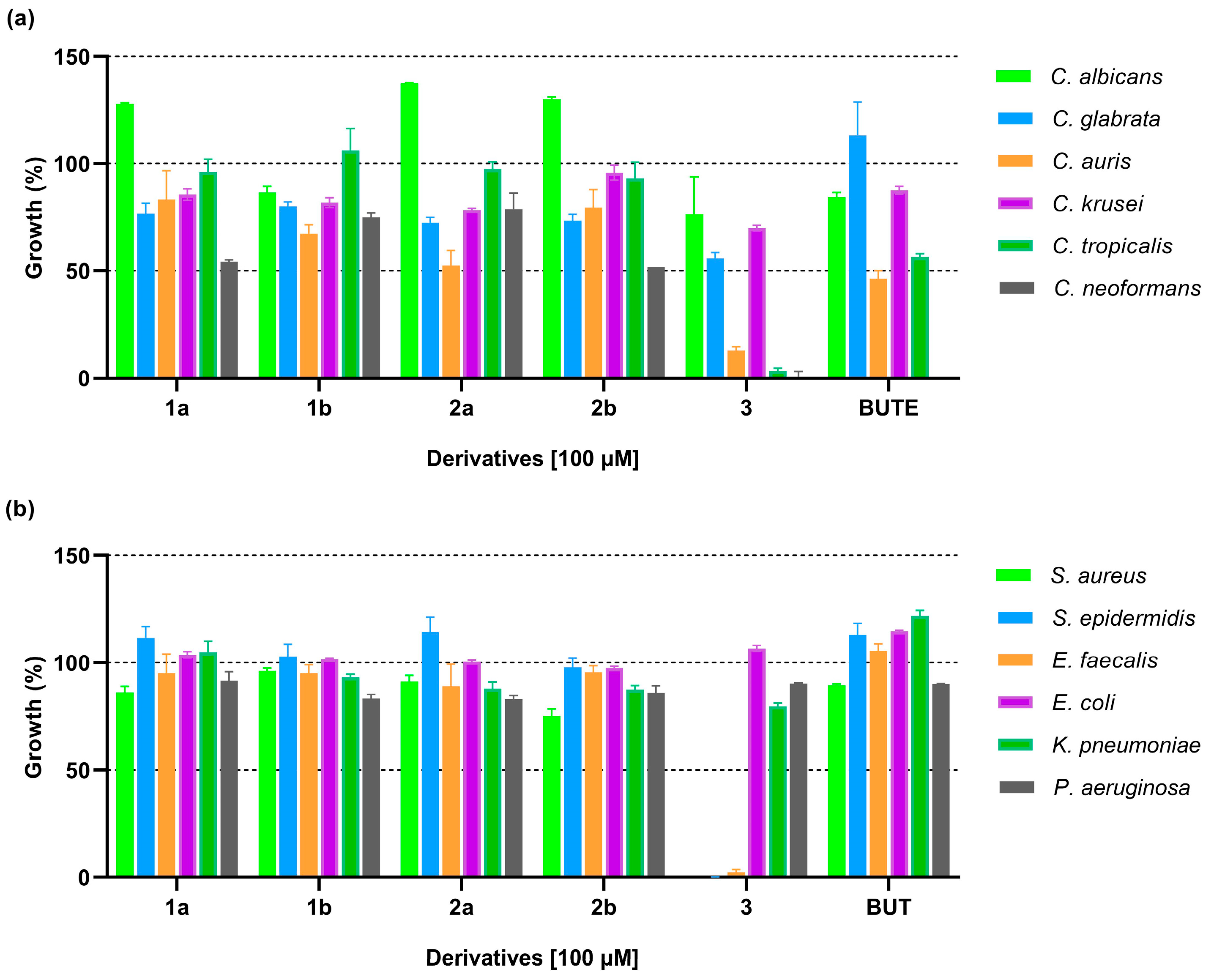
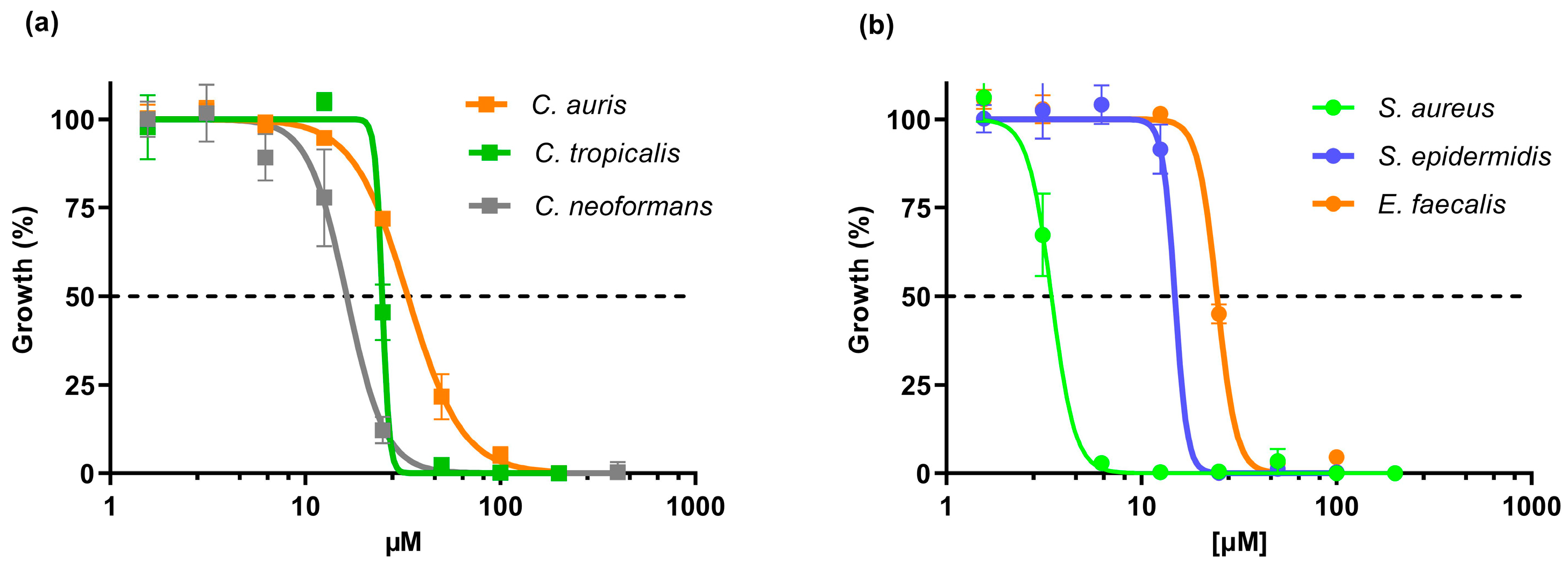
3.2.1. In Vitro Antimicrobial Activity
3.2.2. Selectivity of Action
3.2.3. Combined Effect with Derivative 3 and Colistin
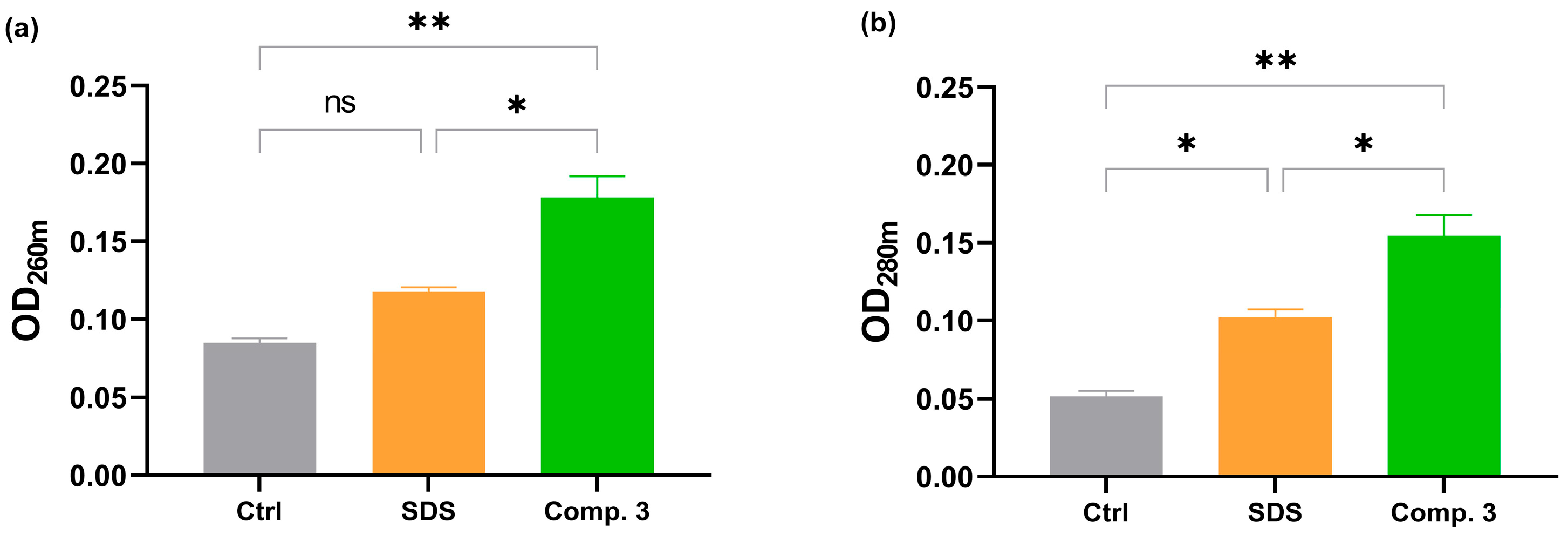
3.2.4. Leakage of Intracellular Contents
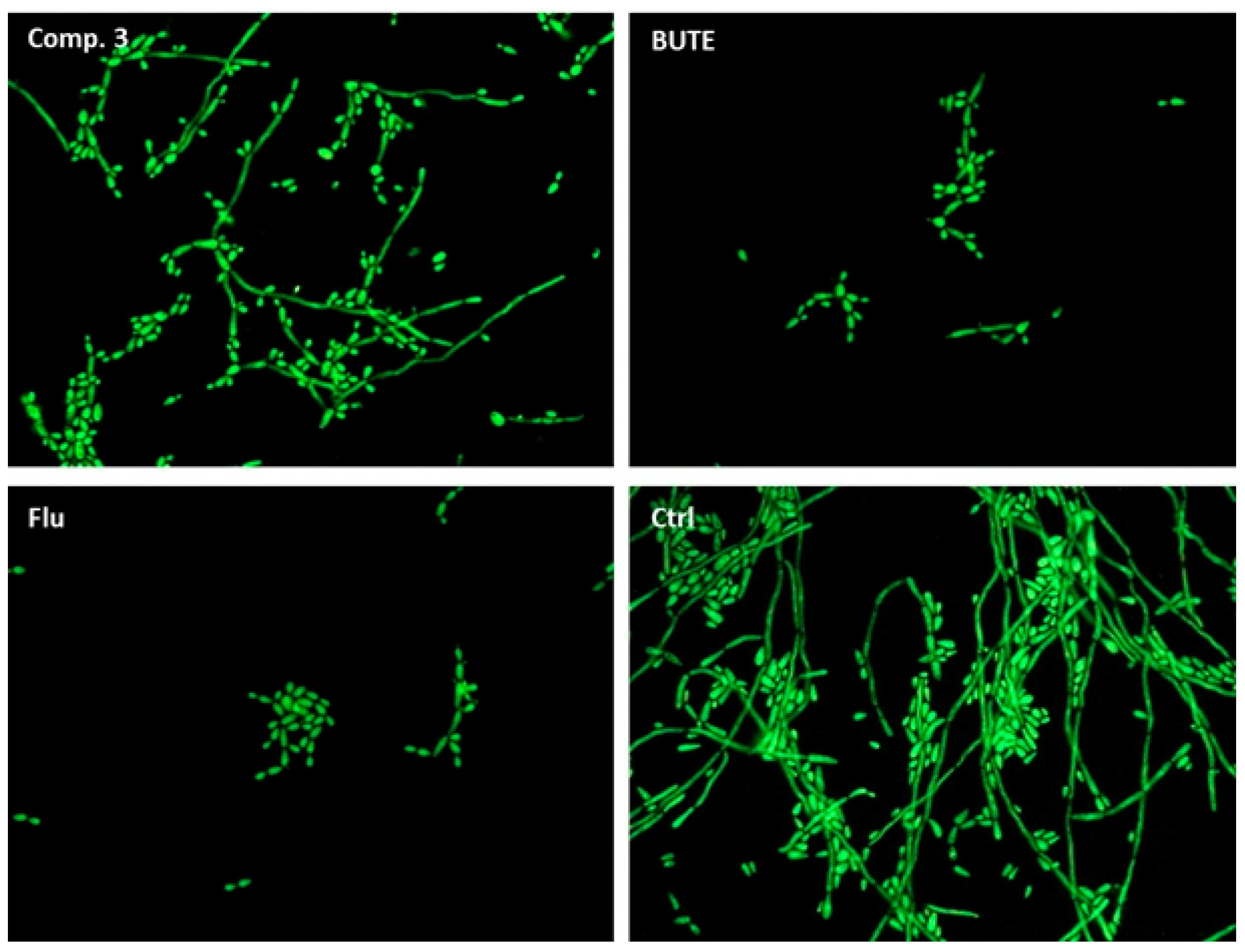
3.3. Inhibition of Yeast-to-Hyphae Morphological Transition
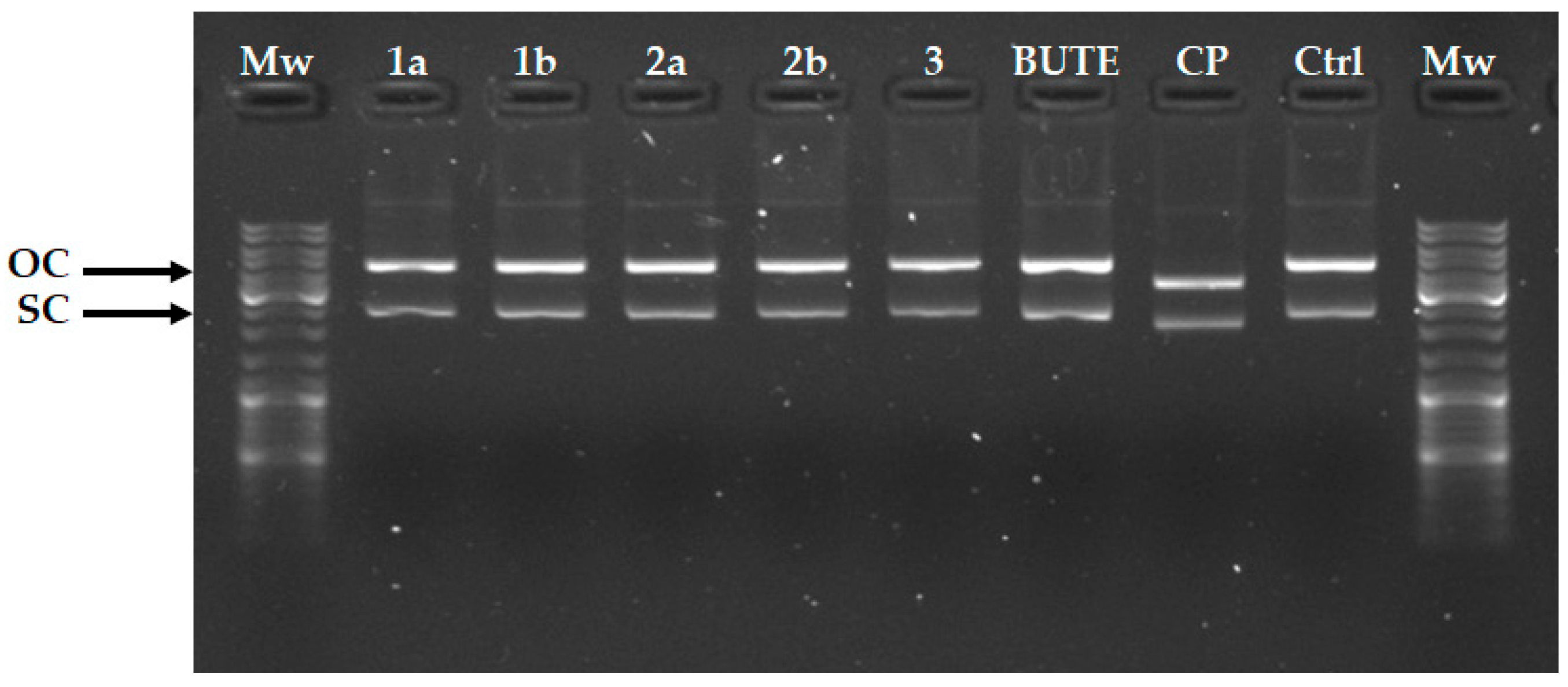
3.4. In Vitro DNA Binding Interactions
4. Materials and Methods
4.1. Chemistry
4.2. Compounds and Reference Drugs
4.3. Biological Evaluation
4.3.1. Microbial Strains and Growth Conditions
4.3.2. MIC and IC50 Determination
4.3.3. Cell Viability and Proliferation Assay
4.3.4. Hemolytic Activity Assay
4.4. In Vitro DNA Cleavage/Mobility Shift Assays
4.5. Analysis of the Yeast-to-Hyphae Transition
4.6. Leakage of Intracellular Contents
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| IFIs | Invasive fungal infections |
| hRBCs | Human red blood cells |
| MIC | Minimum inhibitory concentration |
| MHC | Minimum hemolytic concentration |
| TI | Therapeutic index |
| OM | Outer membrane |
References
- Reddy, G.K.K.; Padmavathi, A.R.; Nancharaiah, Y.V. Fungal infections: Pathogenesis, antifungals and alternate treatment approaches. Curr. Res. Microb. Sci. 2022, 3, 100137. [Google Scholar] [CrossRef]
- Sanguinetti, M.; Posteraro, B.; Beigelman-Aubry, C.; Lamoth, F.; Dunet, V.; Slavin, M.; Richardson, M.D. Diagnosis and treatment of invasive fungal infections: Looking ahead. J. Antimicrob. Chemother. 2019, 74, ii27–ii37. [Google Scholar] [CrossRef] [PubMed]
- Revie, N.M.; Iyer, K.R.; Robbins, N.; Cowen, L.E. Antifungal drug resistance: Evolution, mechanisms and impact. Curr. Opin. Microbiol. 2018, 45, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Hasim, S.; Coleman, J.J. Targeting the Fungal Cell Wall: Current Therapies and Implications for Development of Alternative Antifungal Agents. Future Med. Chem. 2019, 11, 869–883. [Google Scholar] [CrossRef] [PubMed]
- Ibe, C.; Munro, C.A. Fungal cell wall: An underexploited target for antifungal therapies. PLoS Pathog. 2021, 17, e1009470. [Google Scholar] [CrossRef]
- Sant, D.G.; Tupe, S.G.; Ramana, C.V.; Deshpande, M.V. Fungal cell membrane-promising drug target for antifungal therapy. J. Appl. Microbiol. 2016, 121, 1498–1510. [Google Scholar] [CrossRef]
- Halat, D.H.; Younes, S.; Mourad, N.; Rahal, M. Allylamines, Benzylamines, and Fungal Cell Permeability: A Review of Mechanistic Effects and Usefulness against Fungal Pathogens. Membranes 2022, 12, 1171. [Google Scholar] [CrossRef]
- Zhang, W.; Sunami, K.; Liu, S.; Zhuang, Z.; Sakihama, Y.; Zhou, D.-Y.; Suzuki, T.; Murai, Y.; Hashimoto, M.; Hashidoko, Y. Accumulation of squalene in filamentous fungi Trichoderma virens PS1-7 in the presence of butenafine hydrochloride, squalene epoxidase inhibitor: Biosynthesis of 13C-enriched squalene. Biosci. Biotechnol. Biochem. 2023, 87, 1129–1138. [Google Scholar] [CrossRef]
- Mingeot-Leclercq, M.-P.; Gallet, X.; Flore, C.; Van Bambeke, F.; Peuvot, J.; Brasseur, R. Experimental and Conformational Analyses of Interactions between Butenafine and Lipids. Antimicrob. Agents Chemother. 2001, 45, 3347–3354. [Google Scholar] [CrossRef]
- Pinheiro, P.d.S.M.; Franco, L.S.; Montagnoli, T.L.; Fraga, C.A.M. Fraga, Molecular hybridization: A powerful tool for multitarget drug discovery. Expert Opin. Drug Discov. 2024, 19, 451–470. [Google Scholar] [CrossRef]
- Ivasiv, V.; Albertini, C.; Gonçalves, A.E.; Rossi, M.; Bolognesi, M.L. Molecular Hybridization as a Tool for Designing Multitarget Drug Candidates for Complex Diseases. Curr. Top. Med. Chem. 2019, 19, 1694–1711. [Google Scholar] [CrossRef] [PubMed]
- Duarte, C.; Barreiro, E.; Fraga, C. Privileged Structures: A Useful Concept for the Rational Design of New Lead Drug Candidates. Mini-Rev. Med. Chem. 2007, 7, 1108–1119. [Google Scholar] [CrossRef] [PubMed]
- Mohsin, N.U.A.; Irfan, M.; Hassan, S.U.; Saleem, U. Current Strategies in Development of New Chromone Derivatives with Diversified Pharmacological Activities: A Review. Pharm. Chem. J. 2020, 54, 241–257. [Google Scholar] [CrossRef] [PubMed]
- Pinto, M.M.M.; Sousa, M.E.; Nascimento, M.S.J. Xanthone Derivatives: New Insights in Biological Activities. Curr. Med. Chem. 2005, 12, 2517–2538. [Google Scholar] [CrossRef]
- Salmoiraghi, I.; Rossi, M.; Valenti, P.; Da Re, P. Allylamine Type Xanthone Antimycotics. Arch. Pharm. 1998, 331, 225–227. [Google Scholar] [CrossRef]
- Shaquiquzzaman, M.; Verma, G.; Marella, A.; Akhter, M.; Akhtar, W.; Khan, M.F.; Tasneem, S.; Alam, M.M. Piperazine scaffold: A remarkable tool in generation of diverse pharmacological agents. Eur. J. Med. Chem. 2015, 102, 487–529. [Google Scholar] [CrossRef]
- Jha, A.; Kumar, A. Piperazine reveals as a cell wall targeting antifungal agent against Candida albicans. Gene Rep. 2024, 37, 102044. [Google Scholar] [CrossRef]
- Tian, G.; Song, Q.; Liu, Z.; Guo, J.; Cao, S.; Long, S. Recent advances in 1,2,3- and 1,2,4-triazole hybrids as antimicrobials and their SAR: A critical review. Eur. J. Med. Chem. 2023, 259, 115603. [Google Scholar] [CrossRef]
- Marzi, M.; Farjam, M.; Kazeminejad, Z.; Shiroudi, A.; Kouhpayeh, A.; Zarenezhad, E. A Recent Overview of 1,2,3-Triazole-Containing Hybrids as Novel Antifungal Agents: Focusing on Synthesis, Mechanism of Action, and Structure-Activity Relationship (SAR). J. Chem. 2022, 2022, 1–50. [Google Scholar] [CrossRef]
- Chathangad, S.N.; Vijayan, V.N.; George, J.A.; Sadhukhan, S. Mitigating Antimicrobial Resistance through Strategic Design of Multimodal Antibacterial Agents Based on 1,2,3-Triazole with Click Chemistry. ACS Bio. Med. Chem. Au 2025. [Google Scholar] [CrossRef]
- Dai, T.; Li, Q.; Zhang, X.; Yang, C. Substituent-Oriented Synthesis of Substituted Pyrazoles/Chromeno [3,2-c]pyrazoles via Sequential Reactions of Chromones/3-Chlorochromones and Tosylhydrazones. J. Org. Chem. 2019, 84, 5913–5921. [Google Scholar] [CrossRef] [PubMed]
- Pickert, M.; Frahm, W. Substituted Xanthones as Antimycobacterial Agents, Part 1: Synthesis and Assignment of1H/13C NMR Chemical Shifts. Arch. Pharm. 1998, 331, 177–192. [Google Scholar] [CrossRef]
- Gobbi, S.; Zimmer, C.; Belluti, F.; Rampa, A.; Hartmann, R.W.; Recanatini, M.; Bisi, A. Novel highly potent and selective nonsteroidal aromatase inhibitors: Synthesis, biological evaluation and structure-activity relationships investigation. J. Med. Chem. 2010, 53, 5347–5351. [Google Scholar] [CrossRef]
- Ma, R.; Gu, Y.; Wang, Y.-E.; Fei, R.; Xiong, D.; Mao, J. One-Pot Synthesis of Indolin-3-ones Mediated by LiN(SiMe3)2/CsF. Org. Lett. 2024, 26, 5082–5086. [Google Scholar] [CrossRef]
- Chen, L.; Chen, H.; Chen, P.; Zhang, W.; Wu, C.; Sun, C.; Luo, W.; Zheng, L.; Liu, Z.; Liang, G. Development of 2-amino-4-phenylthiazole analogues to disrupt myeloid differentiation factor 88 and prevent inflammatory responses in acute lung injury. Eur. J. Med. Chem. 2019, 161, 22–38. [Google Scholar] [CrossRef] [PubMed]
- Morigi, R.; Esposito, D.; Calvaresi, M.; Marforio, T.D.; Gentilomi, G.A.; Bonvicini, F.; Locatelli, A. Isatin Bis-Imidathiazole Hybrids Identified as FtsZ Inhibitors with On-Target Activity Against Staphylococcus aureus. Antibiotics 2024, 13, 992. [Google Scholar] [CrossRef] [PubMed]
- Bonvicini, F.; Locatelli, A.; Morigi, R.; Leoni, A.; Gentilomi, G.A. Isatin Bis-Indole and Bis-Imidazothiazole Hybrids: Synthesis and Antimicrobial Activity. Molecules 2022, 27, 5781. [Google Scholar] [CrossRef]
- CLSI Standard M07; Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically. 12th ed. Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2024.
- Lockhart, S.R.; Etienne, K.A.; Vallabhaneni, S.; Farooqi, J.; Chowdhary, A.; Govender, N.P.; Colombo, A.L.; Calvo, B.; Cuomo, C.A.; Desjardins, C.A.; et al. Simultaneous Emergence of Multidrug-Resistant Candida auris on 3 Continents Confirmed by Whole-Genome Sequencing and Epidemiological Analyses. Clin. Infect. Dis. 2017, 64, 134–140. [Google Scholar] [CrossRef]
- Kneale, M.; Bartholomew, J.S.; Davies, E.; Denning, D.W. Global access to antifungal therapy and its variable cost. J. Antimicrob. Chemother. 2016, 71, 3599–3606. [Google Scholar] [CrossRef]
- Iwatani, W.; Arika, T.; Yamaguchi, H. Two mechanisms of butenafine action in Candida albicans. Antimicrob. Agents Chemother. 1993, 37, 785–788. [Google Scholar] [CrossRef]
- Zhou, Z.; Chen, K.; He, L. Efficient and Recyclable Cobalt(II)/Ionic Liquid Catalytic System for CO2 Conversion to Prepare 2-Oxazolinones at Atmospheric Pressure. Chin. J. Chem. 2019, 37, 1223–1228. [Google Scholar] [CrossRef]
- Bonvicini, F.; Manet, I.; Belluti, F.; Gobbi, S.; Rampa, A.; Gentilomi, G.A.; Bisi, A. Targeting the Bacterial Membrane with a New Polycyclic Privileged Structure: A Powerful Tool To Face Staphylococcus aureus Infections. ACS Infect. Dis. 2019, 5, 1524–1534. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | IC50 1 | SI 2 | MIC 3 | TI 4 |
|---|---|---|---|---|
| C. tropicalis | 24.8 | 1.3 | 50 | >2 |
| C. auris | 33.6 | 1.0 | 100 | >1 |
| C. neoformans | 16.3 | 2.0 | 25 | >4 |
| S. aureus | 3.5 | 9.2 | 6.25 | >16 |
| S. epidermidis | 14.8 | 2.2 | 25 | >4 |
| E. faecalis | 24.4 | 1.3 | 50 | >2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bonvicini, F.; Menegaldo, L.; Orioli, R.; Belluti, F.; Gentilomi, G.A.; Gobbi, S.; Bisi, A. Extended Antimicrobial Profile of Chromone–Butenafine Hybrids. Molecules 2025, 30, 2973. https://doi.org/10.3390/molecules30142973
Bonvicini F, Menegaldo L, Orioli R, Belluti F, Gentilomi GA, Gobbi S, Bisi A. Extended Antimicrobial Profile of Chromone–Butenafine Hybrids. Molecules. 2025; 30(14):2973. https://doi.org/10.3390/molecules30142973
Chicago/Turabian StyleBonvicini, Francesca, Lisa Menegaldo, Rebecca Orioli, Federica Belluti, Giovanna Angela Gentilomi, Silvia Gobbi, and Alessandra Bisi. 2025. "Extended Antimicrobial Profile of Chromone–Butenafine Hybrids" Molecules 30, no. 14: 2973. https://doi.org/10.3390/molecules30142973
APA StyleBonvicini, F., Menegaldo, L., Orioli, R., Belluti, F., Gentilomi, G. A., Gobbi, S., & Bisi, A. (2025). Extended Antimicrobial Profile of Chromone–Butenafine Hybrids. Molecules, 30(14), 2973. https://doi.org/10.3390/molecules30142973