
Dual Inhibitors of Acetylcholinesterase and Monoamine Oxidase-B for the Treatment of Alzheimer’s Disease



Abstract

1. Introduction
1.1. Alzheimer’s Disease
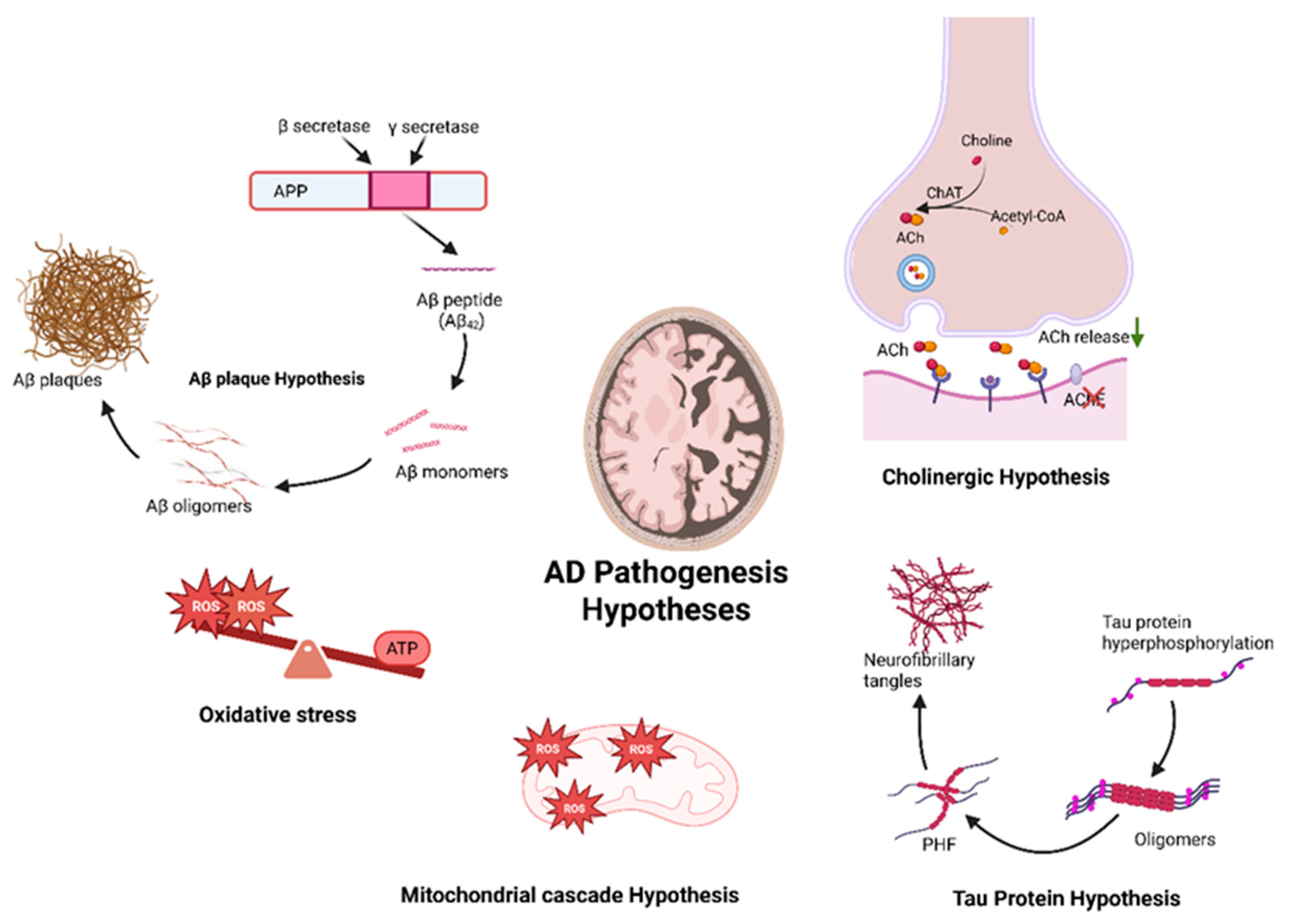
1.2. Proposed Pathomechanism of AD
1.2.1. The Cholinergic Hypothesis
1.2.2. The Amyloid Hypothesis
1.2.3. The Tau (τ) Protein Hypothesis
1.2.4. The Mitochondrial Cascade Hypothesis
1.2.5. Other Theories
2. Treatment of AD
2.1. Non-Pharmacological Treatment
2.2. Pharmacological Treatment
2.2.1. Acetylcholinesterase Inhibitors
2.2.2. Memantine
2.2.3. New Approaches to the Treatment of AD

| Approach Name | Description | Advantages | Disadvantages |
|---|---|---|---|
| Enzyme inhibitors | Targets enzymes like AChE to increase acetylcholine levels in the brain | Temporarily alleviates cognitive symptoms | Limited efficacy; does not modify disease progression |
| NMDA Receptor Antagonists | Antagonizes NMDA | Temporarily improves dementia symptoms | Side effects; limited long-term benefits |
| Gene Therapy | Modifies genes to address underlying causes of Alzheimer’s disease | Targets disease at a genetic level | Complex; potential ethical concerns |
| Lipid Nanoparticles | Utilizes nanoparticles for drug delivery, particularly for crossing the blood–brain barrier | Enhances drug delivery to the brain | Technical challenges; potential toxicity |
| Dendrimer-Based Therapy | Uses dendrimers for targeted drug delivery | Precise drug delivery; reduced side effects | Limited research; potential toxicity |
| Monoclonal Antibody Treatment Lecanemab Donanemab Aducanumab | Aims to clear Aβ fibrils/plaques | Disease-modifying effects Slows down cognitive decline in early AD | ARIA risk; high cost; controversial efficacy |
2.3. Multi-Target Drug Design Strategy
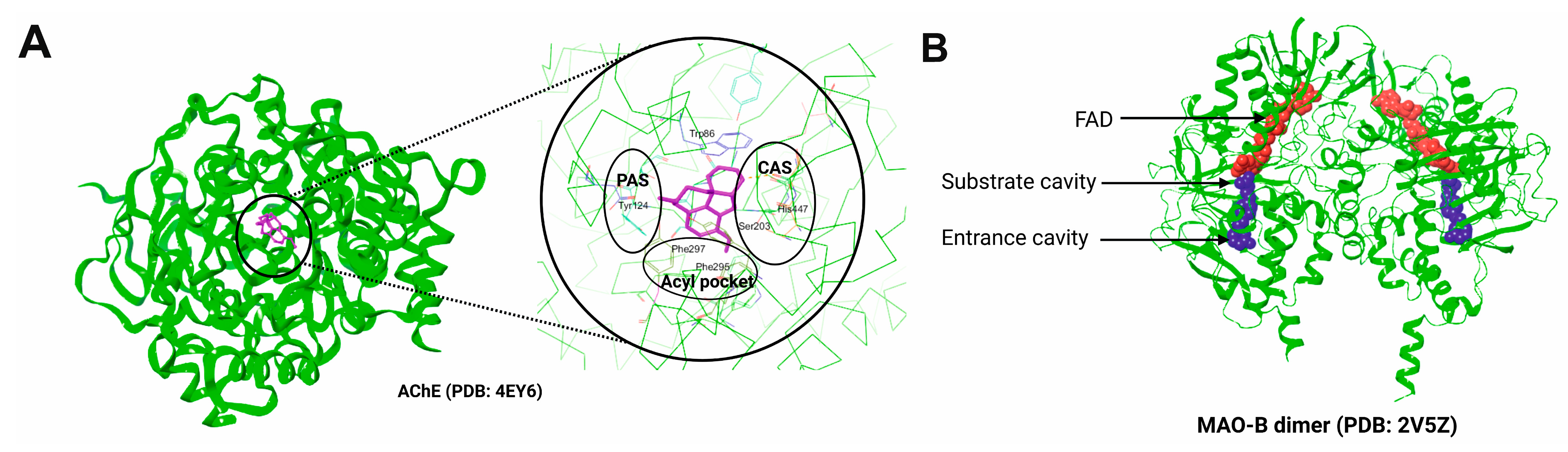
3. AChE and MAO-B as Selected Molecular Targets
3.1. Structural Aspects of AChE and MAO-B
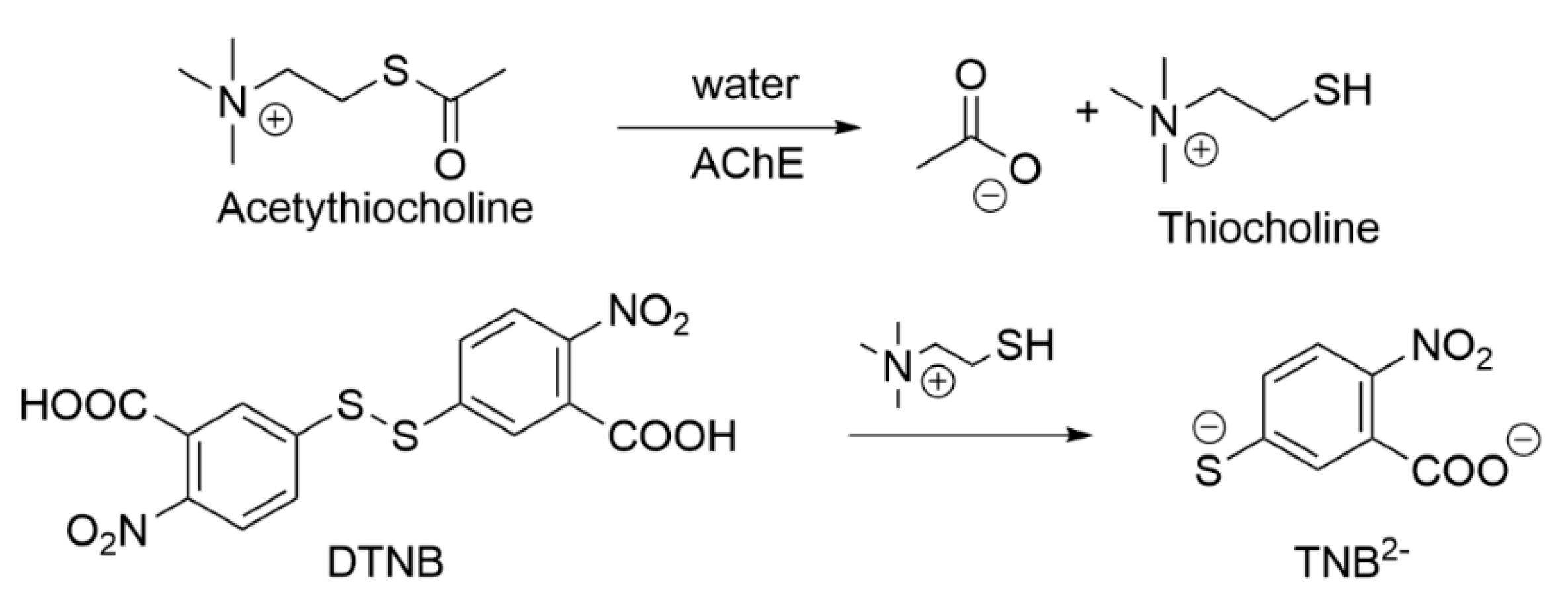
3.2. Chemical Methods for Testing AChE and MAO Activity
3.2.1. Ellman’s Method
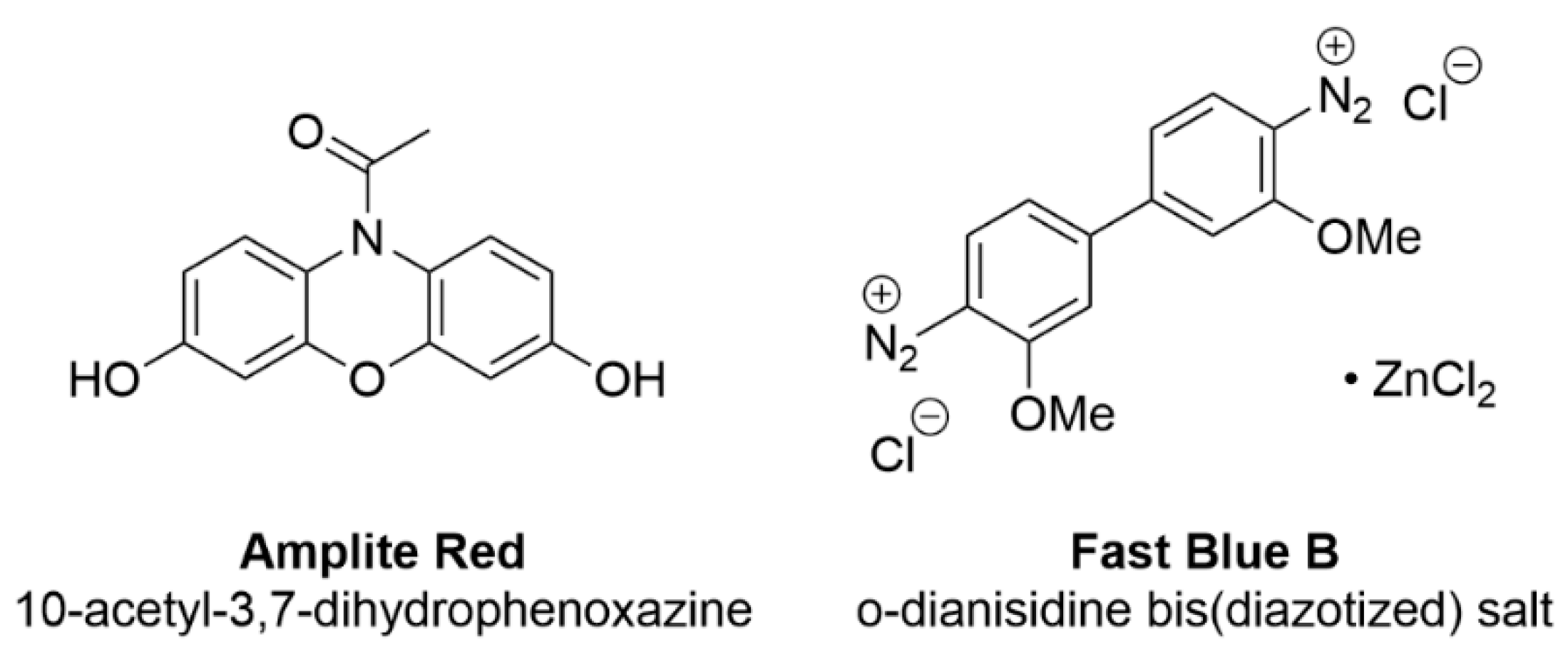
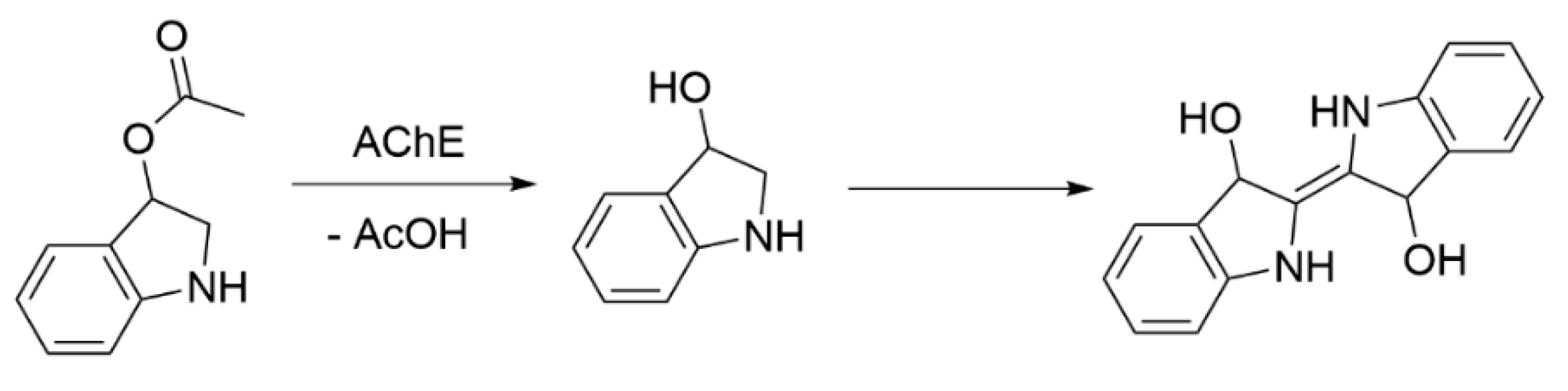
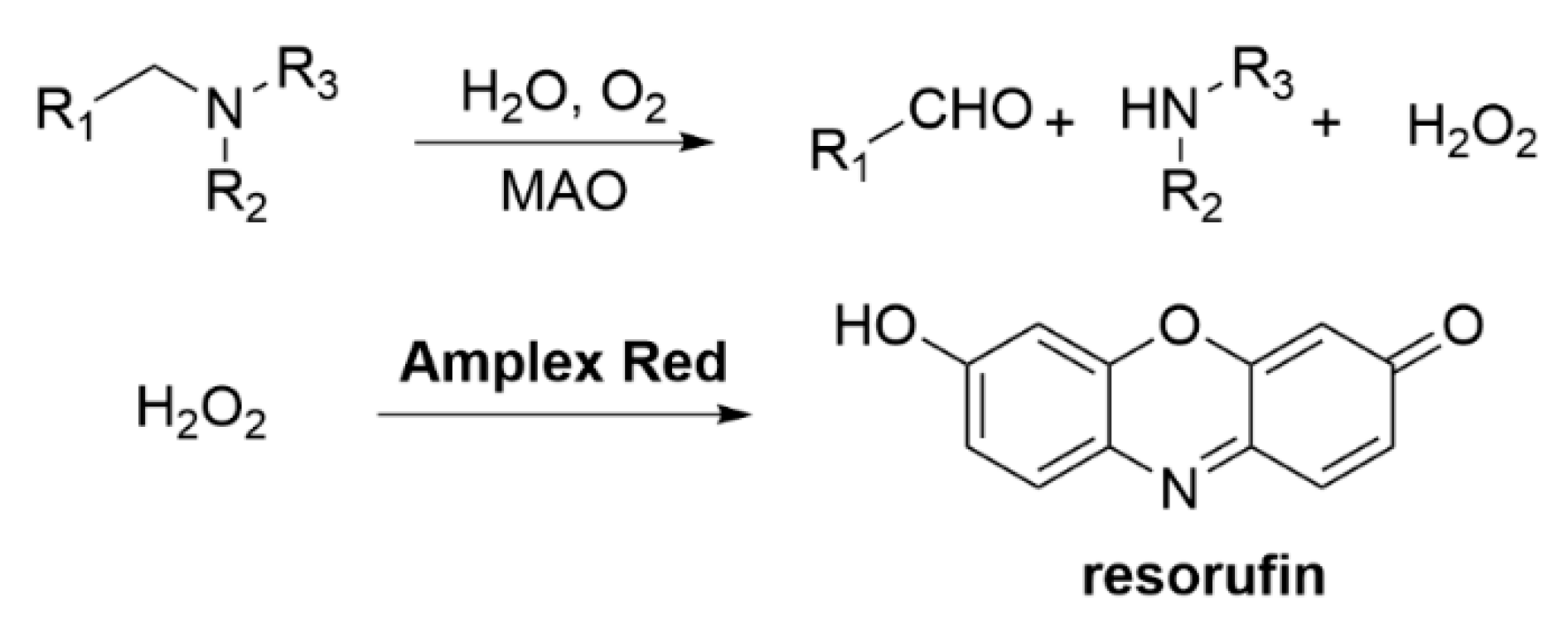
3.2.2. Other Methods for Testing AChE Activity
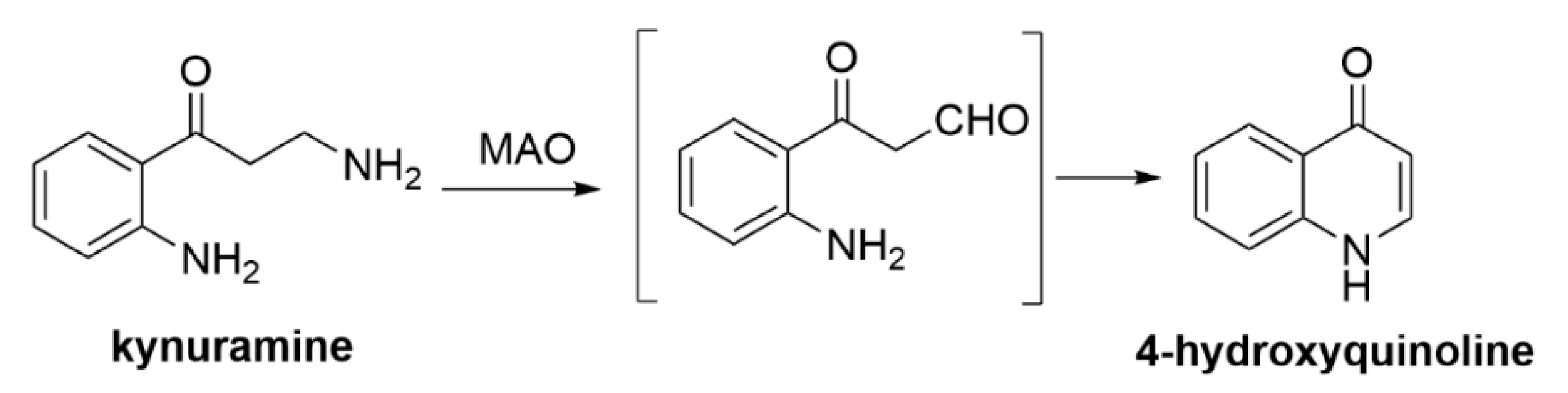
3.2.3. MAO-B Inhibition Assays
4. Overview of Dual AChE and MAO-B Modulators
- Modified structures of known active substances (rasagiline, rivastigmine, selegiline, donepezil, tacrine);
- Carbamate derivatives of N-propargylaminoindans and N-propargylphenethylamines;
- Chalcone-based inhibitors;
- Coumarin-based inhibitors;
- Chromone-based inhibitors;
- Benzo-5-membered ring-based inhibitors;
- Imine and hydrazone inhibitors;
- 3,4-dihydropyrimidin-2(1H)-one inhibitors;
- Pyridoxine-based inhibitors;
- 1-(2H)-phthalazinone-based inhibitors;
- Diclofenac derivatives.
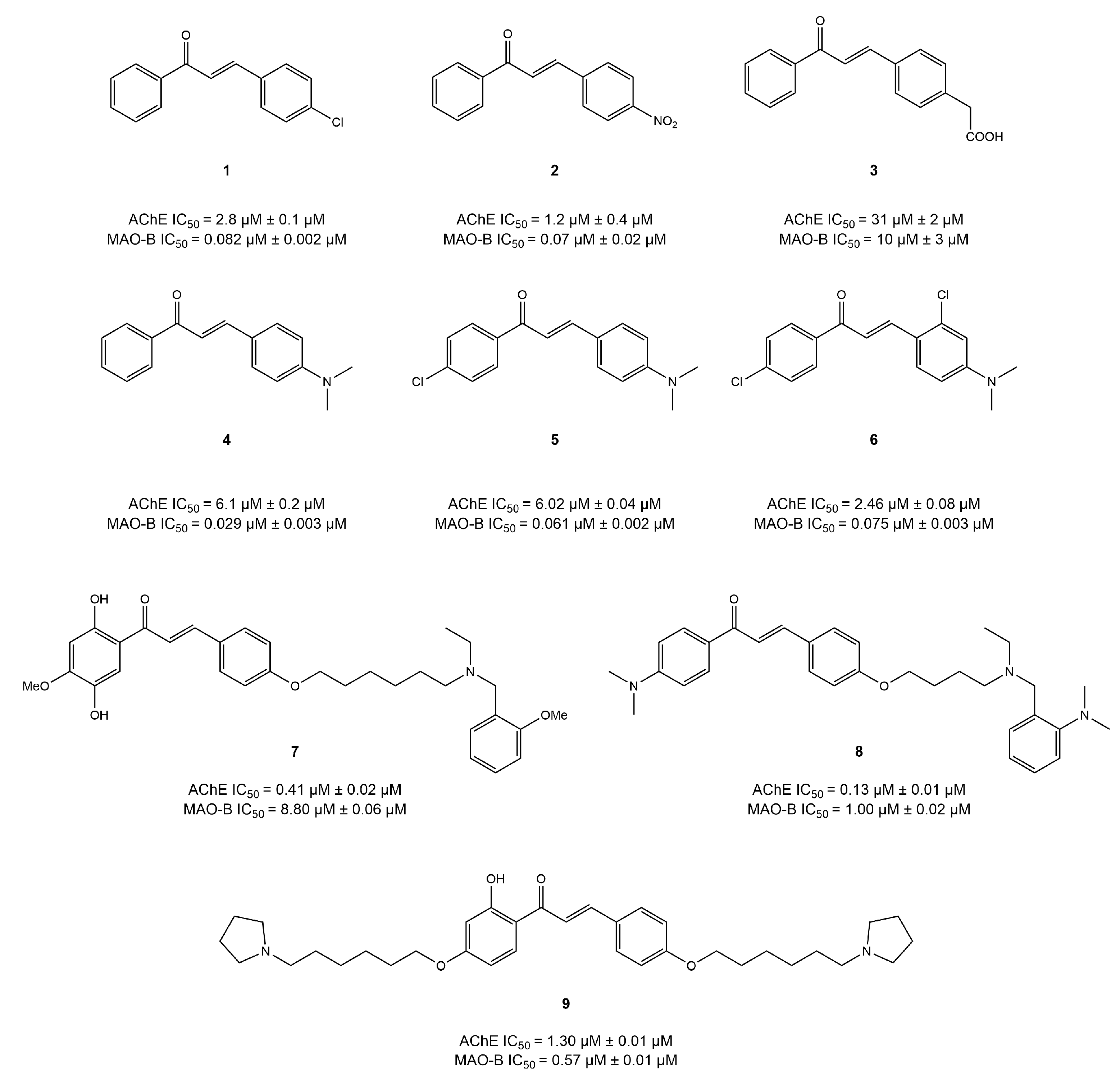
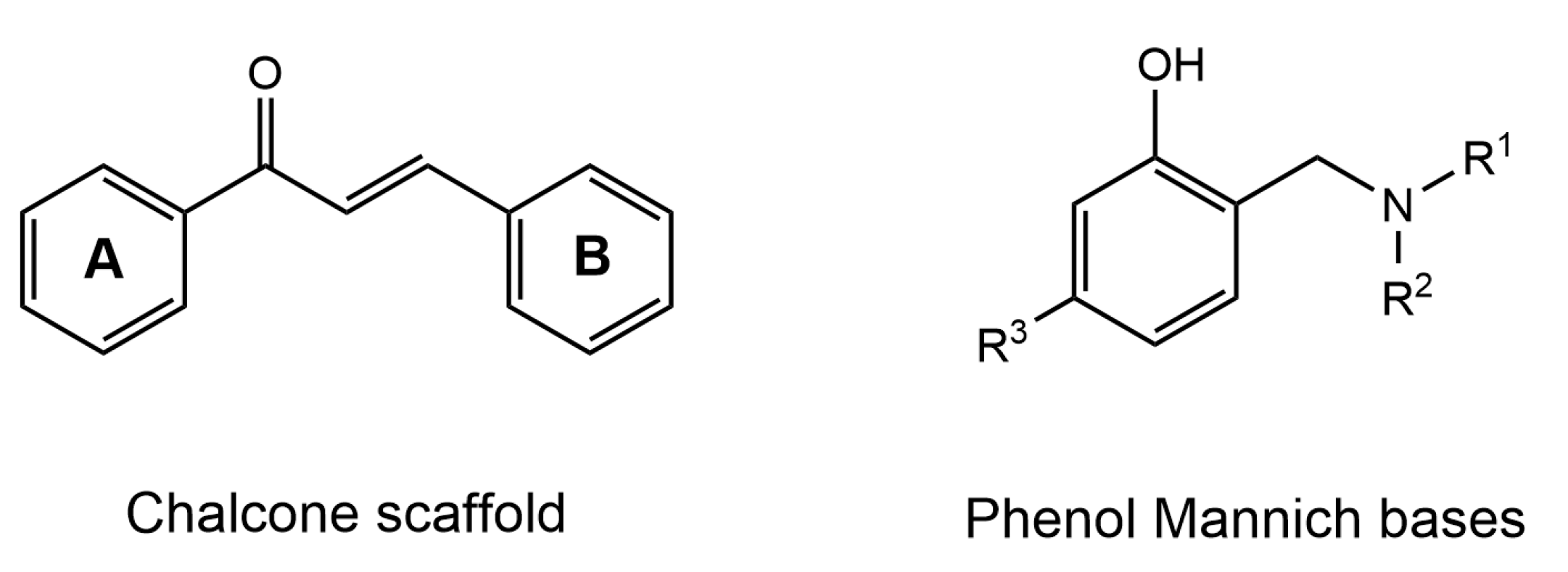
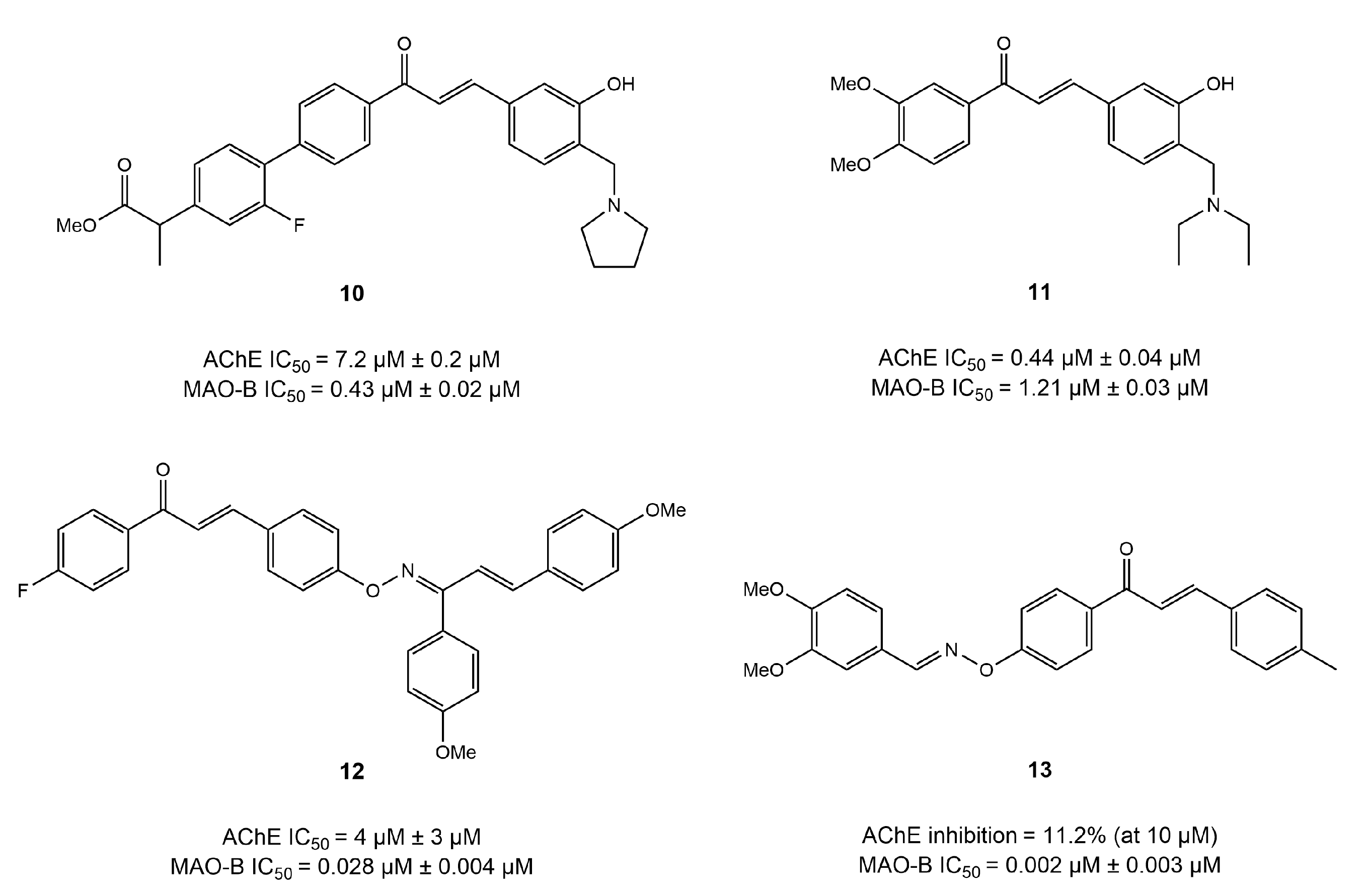
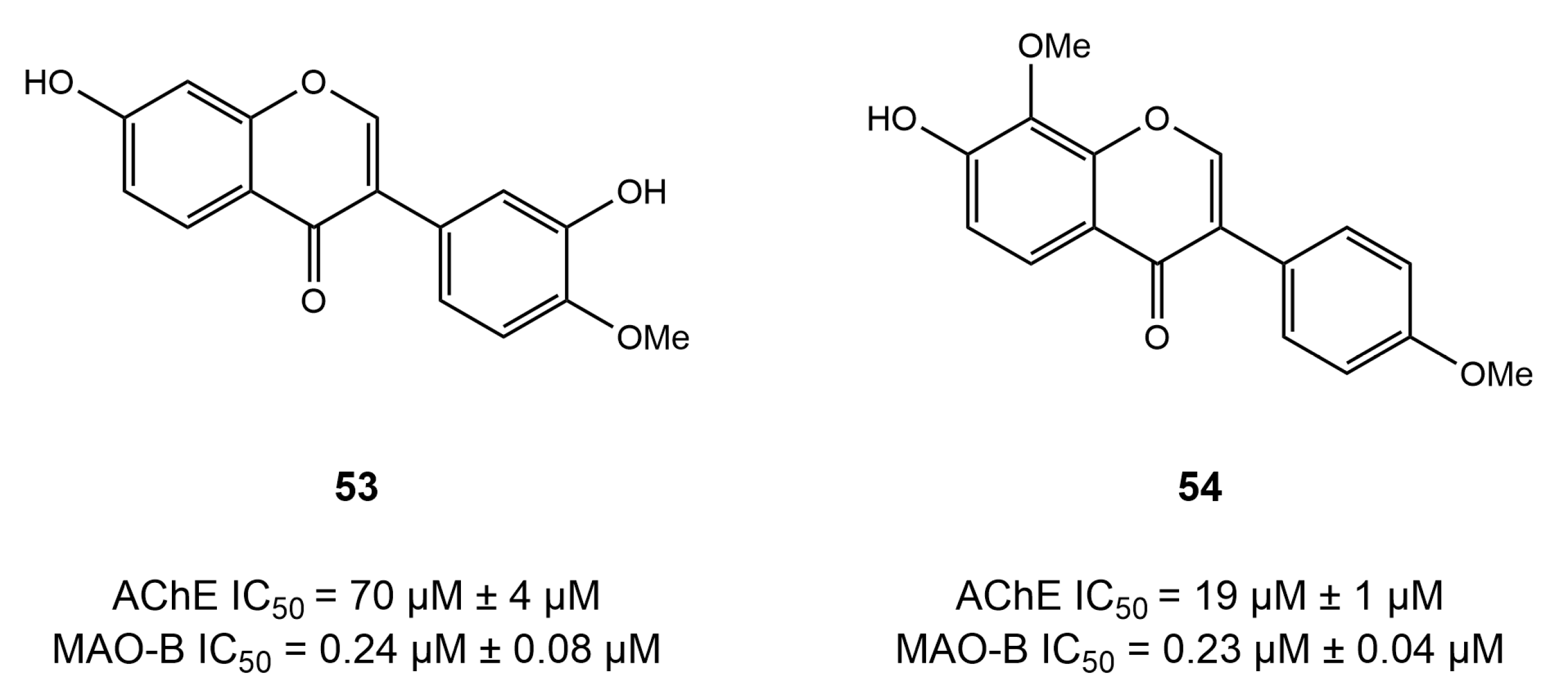
4.1. Chalcone Derivatives
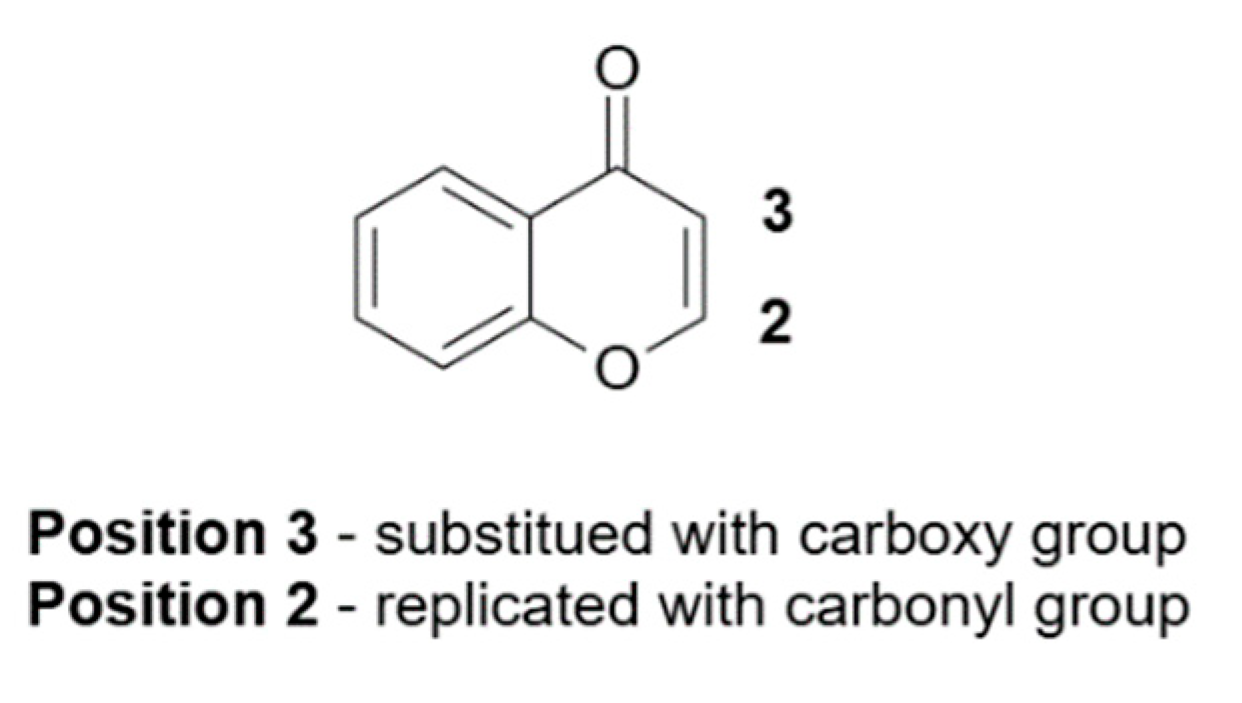
4.2. Coumarin Derivatives
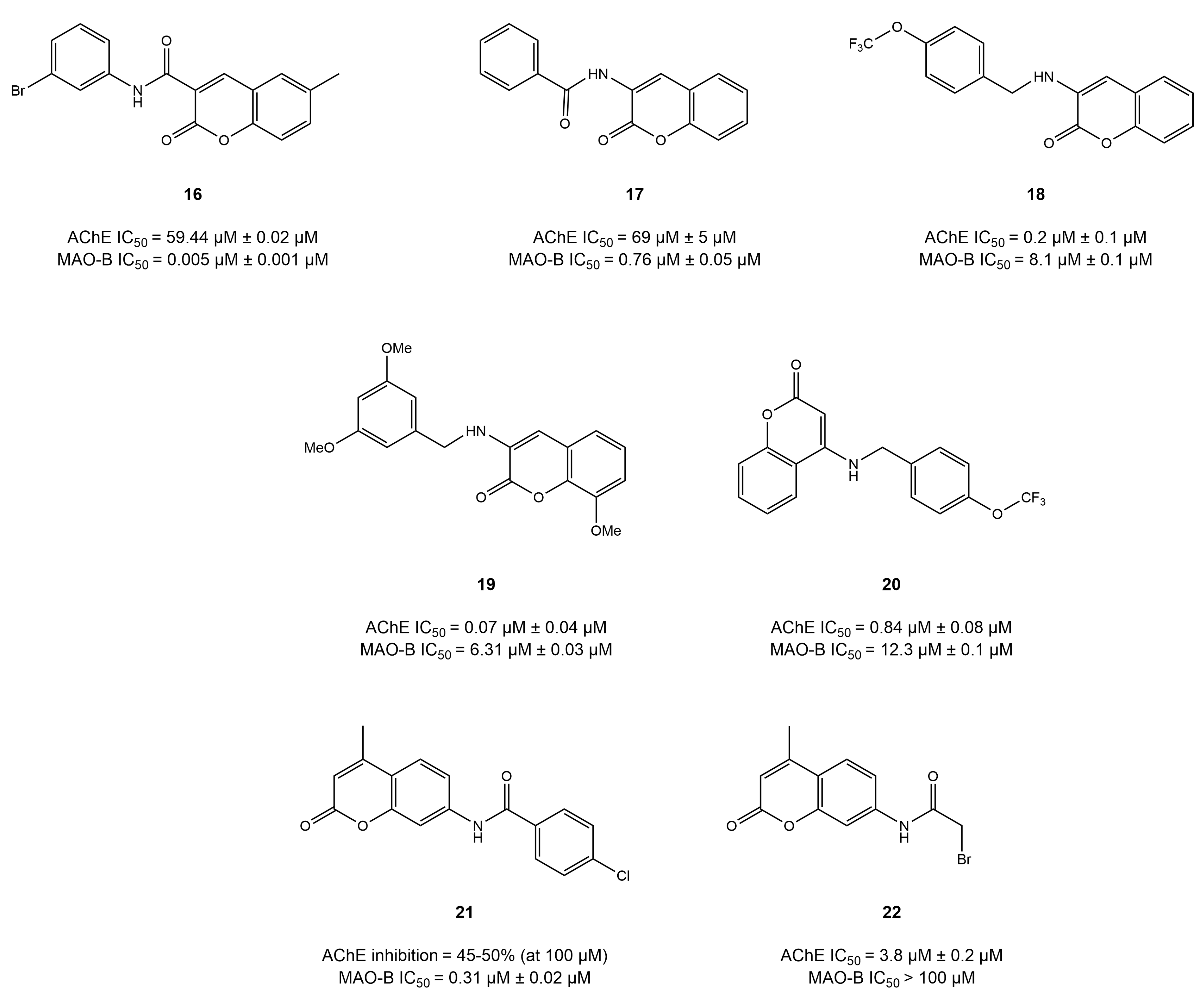
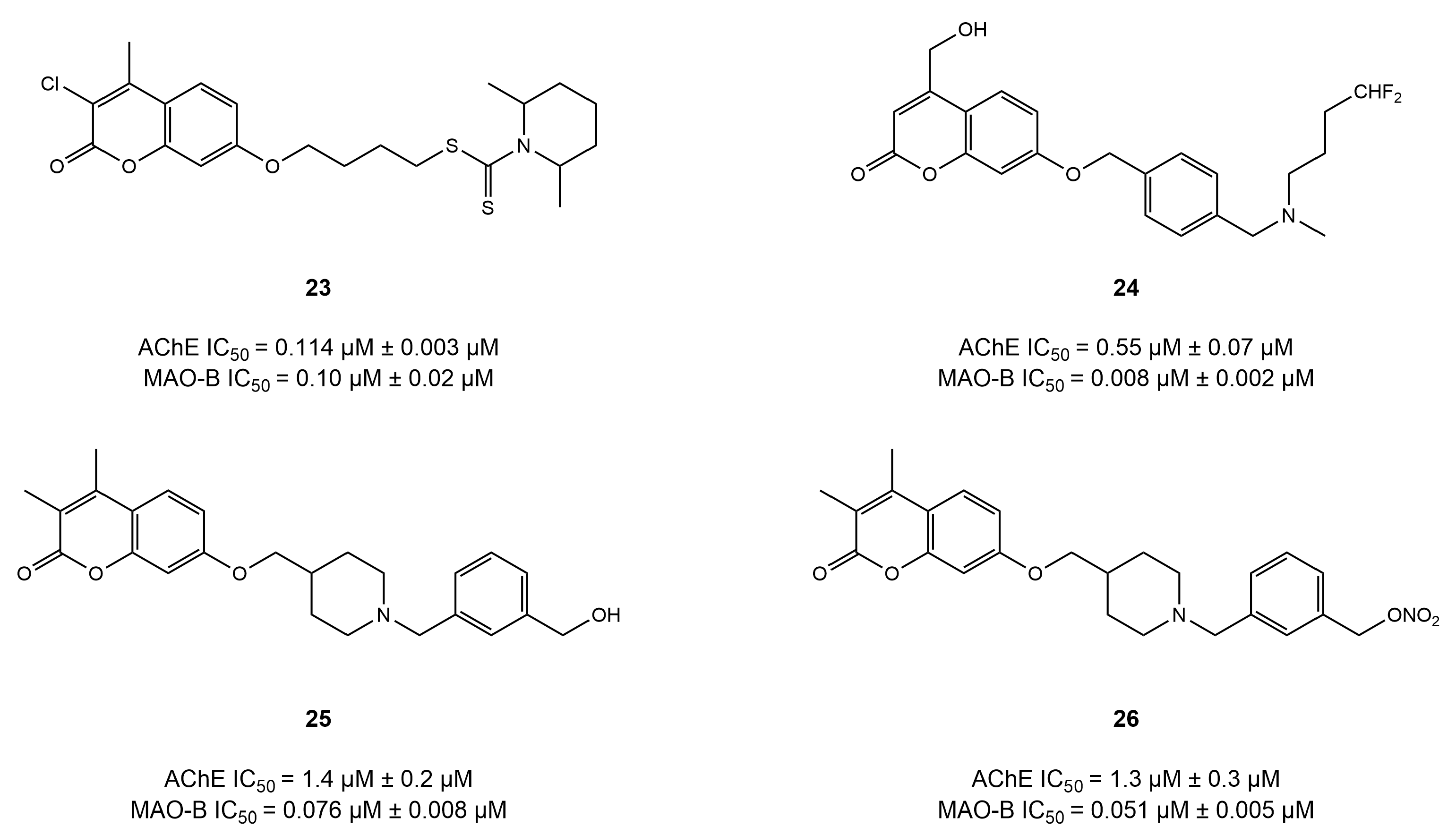
4.3. Chromone Derivatives
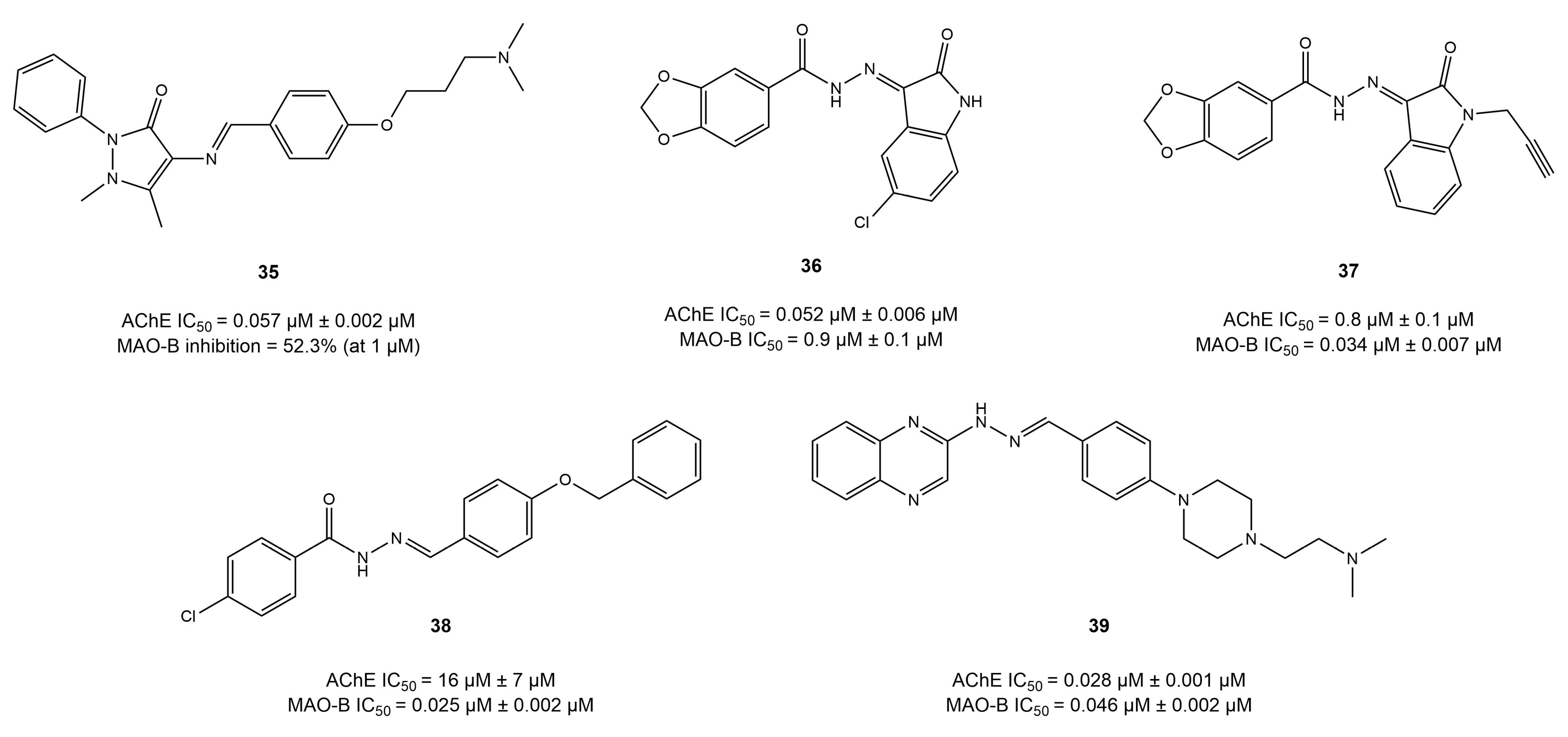
4.4. Imine and Hydrazone Derivatives
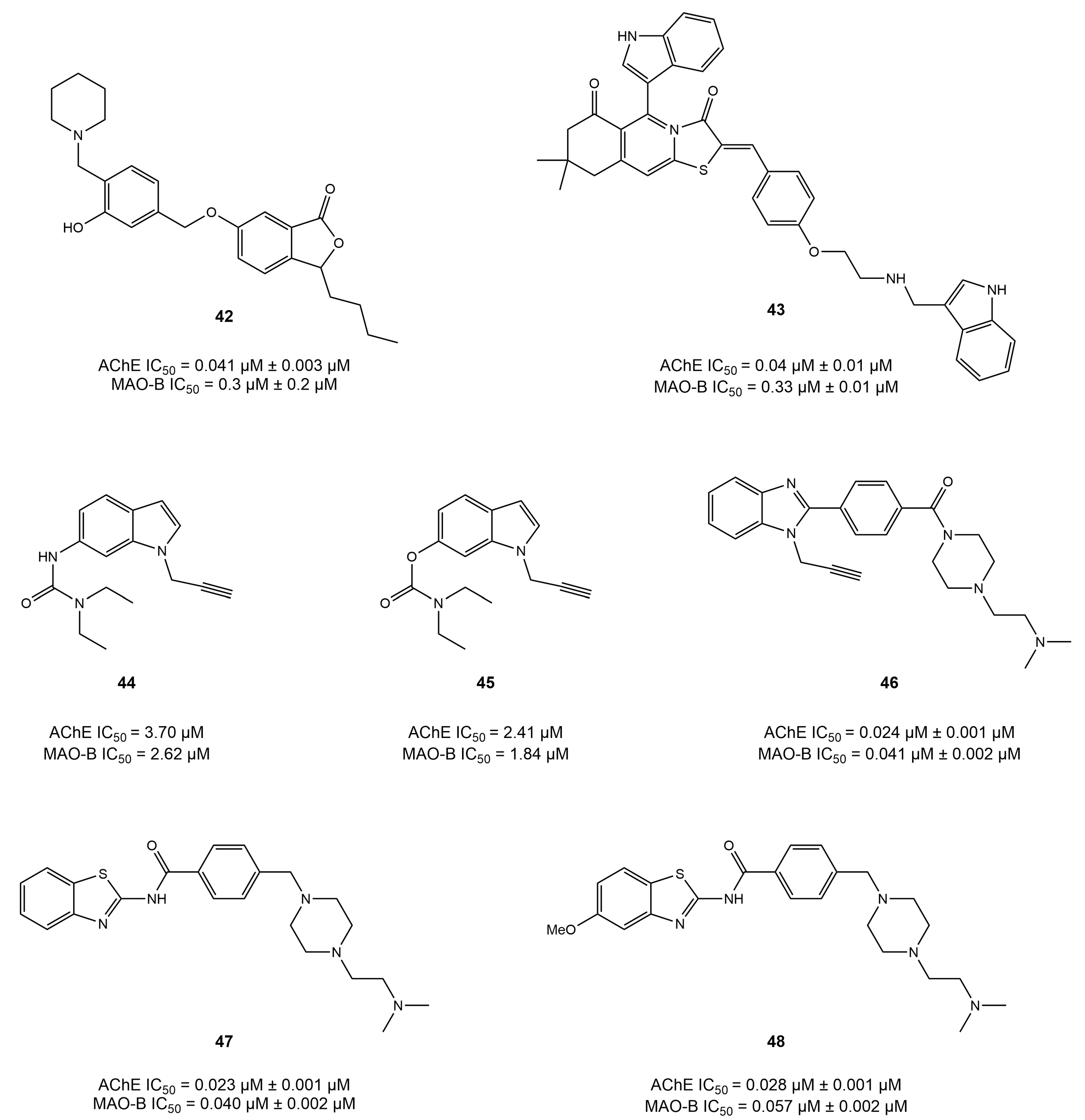
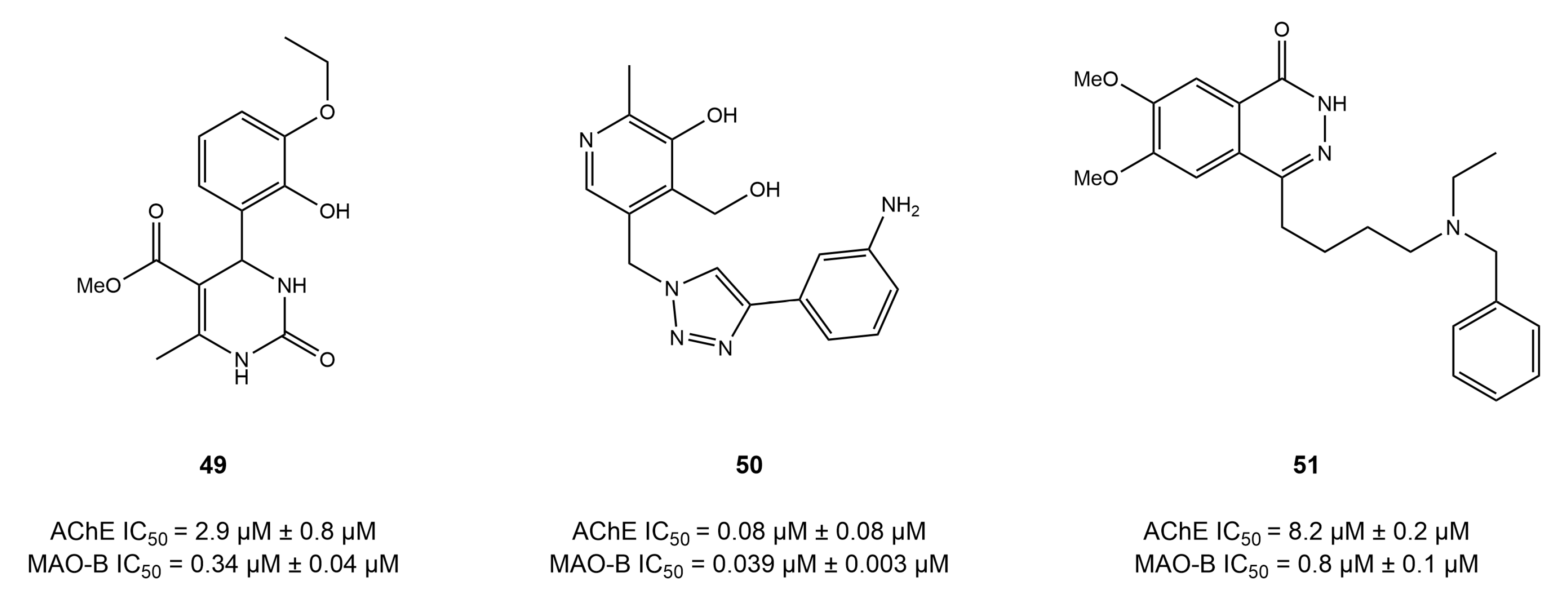
4.5. Benzo Five-Membered Ring-Based Dual Inhibitors
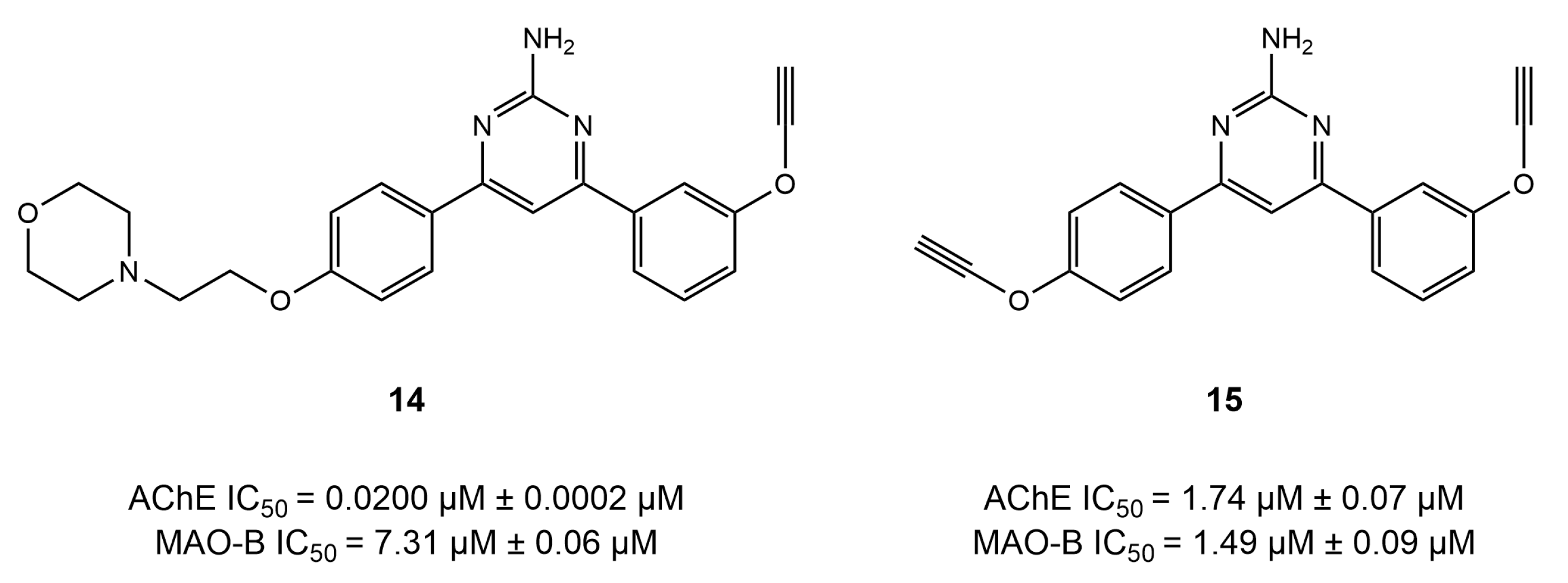
4.6. Diverse Scaffold-Based Dual Inhibitors
5. Computational Methods to Design Dual Inhibitors
5.1. Virtual Screening (VS)
5.2. Other Molecular Modeling Methods
6. Recent Advances in the Development of Inhibitors for the Treatment of AD
7. Conclusions and Prospect
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Declaration of Generative AI and AI-Assisted Technologies in the Writing Process
Abbreviations
| AD | Alzheimer’s disease |
| CNS | Central nervous system |
| AChE | Acetylcholine esterase |
| EOAD | Early-onset Alzheimer’s disease |
| LOAD | Late-onset Alzheimer’s disease |
| ChAT | Choline acetyl transferase |
| VAChT | Vesicular acetyl choline transporter |
| NMDA | N-methyl-D-aspartate |
| BACE1 | Beta-site APP |
| RAGE | Receptor for advanced glycation end products |
| ROS | Reactive oxygen species |
| PHFs | Paired H=helical filaments |
| MAO-B | Monoamine oxidase B |
| APP | Amyloid precursor protein |
| Aβ | Amyloid-β |
| ARIA | Amyloid-related imaging abnormalities |
| NFT | Neurofibrillary tangles |
| PAS | Peripheral anionic site |
| BBB | Blood–brain barrier |
| CAS | Catalytic active site |
| SAR | Structure–activity relationship |
| VS | Virtual screening |
References
- Scheltens, P.; De Strooper, B.; Kivipelto, M.; Holstege, H.; Chételat, G.; Teunissen, C.E.; Cummings, J.; van der Flier, W.M. Alzheimer’s Disease. Lancet 2021, 397, 1577–1590. [Google Scholar] [CrossRef] [PubMed]
- DeTure, M.A.; Dickson, D.W. The Neuropathological Diagnosis of Alzheimer’s Disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef] [PubMed]
- Andrade-Guerrero, J.; Santiago-Balmaseda, A.; Jeronimo-Aguilar, P.; Vargas-Rodríguez, I.; Cadena-Suárez, A.R.; Sánchez-Garibay, C.; Pozo-Molina, G.; Méndez-Catalá, C.F.; Cardenas-Aguayo, M.-C.; Diaz-Cintra, S. Alzheimer’s Disease: An Updated Overview of Its Genetics. Int. J. Mol. Sci. 2023, 24, 3754. [Google Scholar] [CrossRef] [PubMed]
- Akyol, M.A.; Söylemez, B.A.; Küçükgüçlü, Ö. Effects of Early-and Late-Onset Alzheimer’s Disease on Caregiver Burden. Turk. J. Fam. Med. Prim. Care 2023, 17, 79–86. [Google Scholar] [CrossRef]
- Patel, A.O.; Caldwell, A.B.; Ramachandran, S.; Subramaniam, S. Endotype Characterization Reveals Mechanistic Differences Across Brain Regions in Sporadic Alzheimer’s Disease. J. Alzheimer’s Dis. Rep. 2023, 7, 957–972. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.; Mao, C.; Hu, X.; Zhang, S.; Yang, Z.; Hu, Z.; Sun, H.; Fan, Y.; Dong, Y.; Yang, J.; et al. New Insights Into the Pathogenesis of Alzheimer’s Disease. Front. Neurol. 2020, 10, 1312. [Google Scholar] [CrossRef] [PubMed]
- Deletoile, J.; Adeli, H. Graph Theory and Brain Connectivity in Alzheimer’s Disease. Neuroscientist 2017, 23, 616–626. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Singh, A.; Ekavali. A Review on Alzheimer’s Disease Pathophysiology and Its Management: An Update. Pharmacol. Rep. 2015, 67, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Sanabria-Castro, A.; Alvarado-Echeverría, I.; Monge-Bonilla, C. Molecular Pathogenesis of Alzheimer’s Disease: An Update. Ann. Neurosci. 2017, 24, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Breijyeh, Z.; Karaman, R. Comprehensive Review on Alzheimer’s Disease: Causes and Treatment. Molecules 2020, 25, 5789. [Google Scholar] [CrossRef] [PubMed]
- de Castro, A.A.; Soares, F.V.; Pereira, A.F.; Polisel, D.A.; Caetano, M.S.; Leal, D.H.S.; da Cunha, E.F.F.; Nepovimova, E.; Kuca, K.; Ramalho, T.C. Non-Conventional Compounds with Potential Therapeutic Effects against Alzheimer’s Disease. Exp. Rev. Neurother. 2019, 19, 375–395. [Google Scholar] [CrossRef] [PubMed]
- Beck, J.; Lenart, B.; Kintner, D.B.; Sun, D. Na-K-Cl Cotransporter Contributes to Glutamate-Mediated Excitotoxicity. J. Neurosci. 2003, 23, 5061–5068. [Google Scholar] [CrossRef] [PubMed]
- Friedman, L.K. Calcium: A Role for Neuroproduction and Sustained Adaptation. Mol. Interv. 2006, 6, 315–329. [Google Scholar] [CrossRef] [PubMed]
- Girling, K.D.; Wang, Y.T. Neuroprotective Strategies for NMDAR-Mediated Excitotoxicity in Huntington’s Disease. Front. Biol. 2016, 11, 439–458. [Google Scholar] [CrossRef]
- Amorim, J.A.; Coppotelli, G.; Rolo, A.P.; Palmeira, C.M.; Ross, J.M.; Sinclair, D.A. Mitochondrial and Metabolic Dysfunction in Ageing and Age-Related Diseases. Nat. Rev. Endocrinol. 2022, 18, 243–258. [Google Scholar] [CrossRef] [PubMed]
- Paroni, G.; Bisceglia, P.; Seripa, D. Understanding the Amyloid Hypothesis in Alzheimer’s Disease. J. Alzheimer’s Dis. 2019, 68, 493–510. [Google Scholar] [CrossRef] [PubMed]
- Puzzo, D.; Privitera, L.; Leznik, E.; Fà, M.; Staniszewski, A.; Palmeri, A.; Arancio, O. Picomolar Amyloid-β Positively Modulates Synaptic Plasticity and Memory in Hippocampus. J. Neurosci. 2008, 28, 14537–14545. [Google Scholar] [CrossRef] [PubMed]
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s disease. Eur. J. Neurol. 2018, 25, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.D.; Bierhaus, A.; Nawroth, P.P.; Stern, D.M. RAGE and Alzheimer’s Disease: A Progression Factor for Amyloid-β-Induced Cellular Perturbation? J. Alzheimer’s Dis. 2009, 16, 833–843. [Google Scholar] [CrossRef] [PubMed]
- Jellinger, K.A. Neuropathological Subtypes of Alzheimer’s Disease. Acta Neuropathol. 2012, 123, 153–154. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.-Y.; Li, W.; Benedetti, N.J.; Lee, D.H.S. A7 Nicotinic Acetylcholine Receptors Mediate β-Amyloid Peptide-Induced Tau Protein Phosphorylation. J. Biol. Chem. 2003, 278, 31547–31553. [Google Scholar] [CrossRef] [PubMed]
- Götz, J.; Chen, F.; van Dorpe, J.; Nitsch, R.M. Formation of Neurofibrillary Tangles in P301L Tau Transgenic Mice Induced by Aβ42 Fibrils. Science 2001, 293, 1491–1495. [Google Scholar] [CrossRef] [PubMed]
- Stoothoff, W.H.; Johnson, G.V.W. Tau Phosphorylation: Physiological and Pathological Consequences. Biochim. Biophys. Acta 2005, 1739, 280–297. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, R.H.; Burns, J.M.; Khan, S.M. The Alzheimer’s Disease Mitochondrial Cascade Hypothesis. J. Alzheimer’s Dis. 2010, 20, S265–S279. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.X.; Yan, S. Du Amyloid-β-Induced Mitochondrial Dysfunction. J. Alzheimer’s Dis. 2007, 12, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Pavlov, P.F.; Wiehager, B.; Sakai, J.; Frykman, S.; Behbahani, H.; Winblad, B.; Ankarcrona, M. Mitochondrial γ-secretase Participates in the Metabolism of Mitochondria-associated Amyloid Precursor Protein. FASEB J. 2011, 25, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Bonda, D.J.; Lee, H.; Blair, J.A.; Zhu, X.; Perry, G.; Smith, M.A. Role of Metal Dyshomeostasis in Alzheimer Disease. Metallomics 2011, 3, 267–270. [Google Scholar] [CrossRef] [PubMed]
- Greenough, M.A.; Camakaris, J.; Bush, A.I. Metal Dyshomeostasis and Oxidative Stress in Alzheimer’s Disease. Neurochem. Int. 2013, 62, 540–555. [Google Scholar] [CrossRef] [PubMed]
- Adlard, P.A.; Parncutt, J.M.; Finkelstein, D.I.; Bush, A.I. Cognitive Loss in Zinc Transporter-3 Knock-Out Mice: A Phenocopy for the Synaptic and Memory Deficits of Alzheimer’s Disease? J. Neurosci. 2010, 30, 1631–1636. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-Y.; Cole, T.B.; Palmiter, R.D.; Suh, S.W.; Koh, J.-Y. Contribution by Synaptic Zinc to the Gender-Disparate Plaque Formation in Human Swedish Mutant APP Transgenic Mice. Proc. Natl. Acad. Sci. USA 2002, 99, 7705–7710. [Google Scholar] [CrossRef] [PubMed]
- Makhaeva, G.F.; Lushchekina, S.V.; Boltneva, N.P.; Sokolov, V.B.; Grigoriev, V.V.; Serebryakova, O.G.; Vikhareva, E.A.; Aksinenko, A.Y.; Barreto, G.E.; Aliev, G.; et al. Conjugates of γ-Carbolines and Phenothiazine as New Selective Inhibitors of Butyrylcholinesterase and Blockers of NMDA Receptors for Alzheimer Disease. Sci. Rep. 2015, 5, 13164. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.T.; Beal, M.F. Mitochondrial Dysfunction and Oxidative Stress in Neurodegenerative Diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Kern, A.; Mavrikaki, M.; Ullrich, C.; Albarran-Zeckler, R.; Brantley, A.F.; Smith, R.G. Hippocampal Dopamine/DRD1 Signaling Dependent on the Ghrelin Receptor. Cell 2015, 163, 1176–1190. [Google Scholar] [CrossRef] [PubMed]
- Carniglia, L.; Ramírez, D.; Durand, D.; Saba, J.; Turati, J.; Caruso, C.; Scimonelli, T.N.; Lasaga, M. Neuropeptides and Microglial Activation in Inflammation, Pain, and Neurodegenerative Diseases. Med. Inflamm. 2017, 2017, 5048616. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Guo, L.; Sui, S.; Driskill, C.; Phensy, A.; Wang, Q.; Gauba, E.; Zigman, J.M.; Swerdlow, R.H.; Kroener, S.; et al. Disrupted Hippocampal Growth Hormone Secretagogue Receptor 1α Interaction with Dopamine Receptor D1 Plays a Role in Alzheimer′s Disease. Sci. Transl. Med. 2019, 11, eaav6278. [Google Scholar] [CrossRef] [PubMed]
- Kaushal, V.; Dye, R.; Pakavathkumar, P.; Foveau, B.; Flores, J.; Hyman, B.; Ghetti, B.; Koller, B.H.; LeBlanc, A.C. Neuronal NLRP1 Inflammasome Activation of Caspase-1 Coordinately Regulates Inflammatory Interleukin-1-Beta Production and Axonal Degeneration-Associated Caspase-6 Activation. Cell Death Diff. 2015, 22, 1676–1686. [Google Scholar] [CrossRef] [PubMed]
- Knopman, D.S.; Amieva, H.; Petersen, R.C.; Chételat, G.; Holtzman, D.M.; Hyman, B.T.; Nixon, R.A.; Jones, D.T. Alzheimer Disease. Nat. Rev. Dis. Prim. 2021, 7, 33. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R., Jr.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimer’s Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef] [PubMed]
- Solomon, A.; Turunen, H.; Ngandu, T.; Peltonen, M.; Levälahti, E.; Helisalmi, S.; Antikainen, R.; Bäckman, L.; Hänninen, T.; Jula, A.; et al. Effect of the Apolipoprotein E Genotype on Cognitive Change During a Multidomain Lifestyle Intervention. JAMA Neurol. 2018, 75, 462. [Google Scholar] [CrossRef] [PubMed]
- Ngandu, T.; Lehtisalo, J.; Solomon, A.; Levälahti, E.; Ahtiluoto, S.; Antikainen, R.; Bäckman, L.; Hänninen, T.; Jula, A.; Laatikainen, T.; et al. A 2 Year Multidomain Intervention of Diet, Exercise, Cognitive Training, and Vascular Risk Monitoring versus Control to Prevent Cognitive Decline in at-Risk Elderly People (FINGER): A Randomised Controlled Trial. Lancet 2015, 385, 2255–2263. [Google Scholar] [CrossRef] [PubMed]
- Andrieu, S.; Guyonnet, S.; Coley, N.; Cantet, C.; Bonnefoy, M.; Bordes, S.; Bories, L.; Cufi, M.-N.; Dantoine, T.; Dartigues, J.-F.; et al. Effect of Long-Term Omega 3 Polyunsaturated Fatty Acid Supplementation with or without Multidomain Intervention on Cognitive Function in Elderly Adults with Memory Complaints (MAPT): A Randomised, Placebo-Controlled Trial. Lancet Neurol. 2017, 16, 377–389. [Google Scholar] [CrossRef] [PubMed]
- van Charante, E.P.M.; Richard, E.; Eurelings, L.S.; van Dalen, J.-W.; Ligthart, S.A.; van Bussel, E.F.; Hoevenaar-Blom, M.P.; Vermeulen, M.; van Gool, W.A. Effectiveness of a 6-Year Multidomain Vascular Care Intervention to Prevent Dementia (preDIVA): A Cluster-Randomised Controlled Trial. Lancet 2016, 388, 797–805. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.L.; Morstorf, T.; Zhong, K. Alzheimer’s Disease Drug-Development Pipeline: Few Candidates, Frequent Failures. Alzheimer’s Res. Ther. 2014, 6, 37. [Google Scholar] [CrossRef] [PubMed]
- Colgin, L.L.; Moser, E.I. Gamma Oscillations in the Hippocampus. Physiology 2010, 25, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Goutagny, R.; Gu, N.; Cavanagh, C.; Jackson, J.; Chabot, J.-G.; Quirion, R.; Krantic, S.; Williams, S. Alterations in Hippocampal Network Oscillations and Theta-Gamma Coupling Arise before Aβ Overproduction in a Mouse Model of Alzheimer’s Disease. Eur. J. Neurosci. 2013, 37, 1896–1902. [Google Scholar] [CrossRef] [PubMed]
- Mably, A.J.; Gereke, B.J.; Jones, D.T.; Colgin, L.L. Impairments in Spatial Representations and Rhythmic Coordination of Place Cells in the 3xTg Mouse Model of Alzheimer’s Disease. Hippocampus 2017, 27, 378–392. [Google Scholar] [CrossRef] [PubMed]
- Booth, C.A.; Witton, J.; Nowacki, J.; Tsaneva-Atanasova, K.; Jones, M.W.; Randall, A.D.; Brown, J.T. Altered Intrinsic Pyramidal Neuron Properties and Pathway-Specific Synaptic Dysfunction Underlie Aberrant Hippocampal Network Function in a Mouse Model of Tauopathy. J. Neurosci. 2016, 36, 350–363. [Google Scholar] [CrossRef] [PubMed]
- Farlow, M.R.; Miller, M.L.; Pejovic, V. Treatment Options in Alzheimer’s Disease: Maximizing Benefit, Managing Expectations. Dement. Geriatr. Cogn. Disord. 2008, 25, 408–422. [Google Scholar] [CrossRef] [PubMed]
- Masters, C.L.; Bateman, R.; Blennow, K.; Rowe, C.C.; Sperling, R.A.; Cummings, J.L. Alzheimer’s Disease. Nat. Rev. Dis. Primers 2015, 1, 15056. [Google Scholar] [CrossRef] [PubMed]
- Schilström, B.; Ivanov, V.B.; Wiker, C.; Svensson, T.H. Galantamine Enhances Dopaminergic Neurotransmission In Vivo Via Allosteric Potentiation of Nicotinic Acetylcholine Receptors. Neuropsychopharmacology 2007, 32, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, D.G.; Francis, P.T.; Schwam, E.; Payne-Parrish, J. Cholinesterase Inhibitors Used in the Treatment of Alzheimer’s Disease. Drugs Aging 2004, 21, 453–478. [Google Scholar] [CrossRef] [PubMed]
- Johannsen, P. Long-Term Cholinesterase Inhibitor Treatment of Alzheimer’s Disease. CNS Drugs 2004, 18, 757–768. [Google Scholar] [CrossRef] [PubMed]
- Winblad, B.; Black, S.E.; Homma, A.; Schwam, E.M.; Moline, M.; Xu, Y.; Perdomo, C.A.; Swartz, J.; Albert, K. Donepezil Treatment in Severe Alzheimer’s Disease: A Pooled Analysis of Three Clinical Trials. Curr. Med. Res. Opin. 2009, 25, 2577–2587. [Google Scholar] [CrossRef] [PubMed]
- Castellani, R.J.; Rolston, R.K.; Smith, M.A. Alzheimer Disease. Disease-a-Month 2010, 56, 484–546. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.L. Alzheimer’s Disease. N. Eng. J. Med. 2004, 351, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, N.; Cappell, J.; Eryavec, G.M.; Lanctôt, K.L. Changes in Nursing Burden Following Memantine for Agitation and Aggression in Long-Term Care Residents with Moderate to Severe Alzheimerʼs Disease. CNS Drugs 2011, 25, 425–433. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, R.; Hofer, E.; Bouwman, F.H.; Buerger, K.; Cordonnier, C.; Fladby, T.; Galimberti, D.; Georges, J.; Heneka, M.T.; Hort, J.; et al. EFNS-ENS/EAN Guideline on Concomitant Use of Cholinesterase Inhibitors and Memantine in Moderate to Severe Alzheimer’s Disease. Eur. J. Neurol. 2015, 22, 889–898. [Google Scholar] [CrossRef] [PubMed]
- Kishi, T.; Matsunaga, S.; Oya, K.; Nomura, I.; Ikuta, T.; Iwata, N. Memantine for Alzheimer’s Disease: An Updated Systematic Review and Meta-Analysis. J. Alzheimer’s Dis. 2017, 60, 401–425. [Google Scholar] [CrossRef] [PubMed]
- Nowak, D.; Słupski, W.; Rutkowska, M. Nowe Strategie Terapeutyczne Choroby Alzheimera. Postep. Hig. Med. Dosw. 2021, 75, 474–490. [Google Scholar] [CrossRef]
- Adlard, P.A.; Cherny, R.A.; Finkelstein, D.I.; Gautier, E.; Robb, E.; Cortes, M.; Volitakis, I.; Liu, X.; Smith, J.P.; Perez, K.; et al. Rapid Restoration of Cognition in Alzheimer’s Transgenic Mice with 8-Hydroxy Quinoline Analogs Is Associated with Decreased Interstitial Aβ. Neuron 2008, 59, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Prachayasittikul, V.; Prachayasittikul, S.; Ruchirawat, S.; Prachayasittikul, V. 8-Hydroxyquinolines: A Review of Their Metal Chelating Properties and Medicinal Applications. Drug. Des. Devel. Ther. 2013, 7, 1157–1178. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, Y.; Wang, J.; Xia, Y.; Zhang, J.; Chen, L. Recent Advances in Alzheimer’s Disease: Mechanisms, Clinical Trials and New Drug Development Strategies. Sig. Transduct. Target. Ther. 2024, 9, 211. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.Y.; Howard, R. Can We Learn Lessons from the FDA’s Approval of Aducanumab? Nat. Rev. Neurol. 2021, 17, 715–722. [Google Scholar] [CrossRef] [PubMed]
- Donanemab in Early Symptomatic Alzheimer Disease: The TRAILBLAZER-ALZ 2 Randomized Clinical Trial—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/37459141/ (accessed on 28 October 2024).
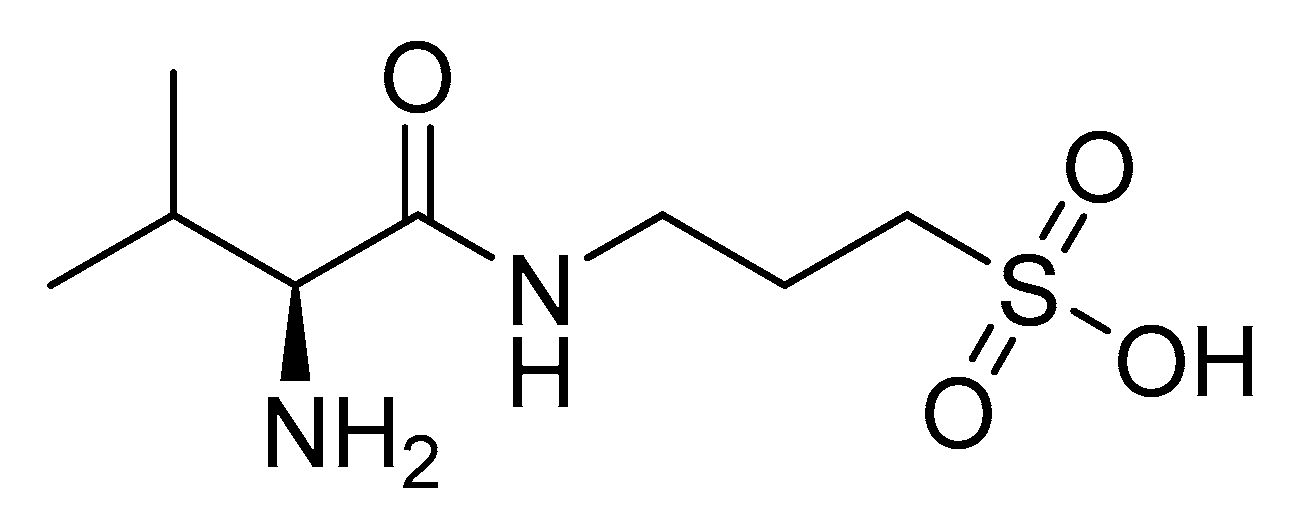
- Alzheon Topline Results from Pivotal APOLLOE4 Phase 3 Trial of Oral Valiltramiprosate/ALZ-801 in Patients with Early Alzheimer’s Disease Carrying Two Copies of APOE4 Gene. Alzheon|Preserving Future Memories 2025. Available online: https://alzheon.com/topline-results-from-pivotal-apolloe4-phase-3-trial-of-oral-valiltramiprosate-alz-801-in-patients-with-early-alzheimers-disease-carrying-two-copies-of-apoe4-gene/ (accessed on 29 October 2024).
- Jastrzębski, M.K.; Wójcik, P.; Stępnicki, P.; Kaczor, A.A. Effects of Small Molecules on Neurogenesis: Neuronal Prolifera-2 Tion and Differentiation. Acta Pharm. Sin. B 2024, 14, 20–37. [Google Scholar] [CrossRef] [PubMed]
- Dave, B.P.; Shah, Y.B.; Maheshwari, K.G.; Mansuri, K.A.; Prajapati, B.S.; Postwala, H.I.; Chorawala, M.R. Pathophysiological Aspects and Therapeutic Armamentarium of Alzheimer’s Disease: Recent Trends and Future Development. Cell. Mol. Neurobiol. 2023, 43, 3847–3884. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Kumar, V.; Anand, P.; Kumar, V.; Dwivedi, A.R.; Kumar, V. Advancements in the Development of Multi-Target Directed Ligands for the Treatment of Alzheimer’s Disease. Bioorg. Med. Chem. 2022, 61, 116742. [Google Scholar] [CrossRef] [PubMed]
- Marco-Contelles, J.; Unzeta, M.; Bolea, I.; Esteban, G.; Ramsay, R.R.; Romero, A.; Martínez-Murillo, R.; Carreiras, M.C.; Ismaili, L. ASS234, As a New Multi-Target Directed Propargylamine for Alzheimer’s Disease Therapy. Front. Neurosci. 2016, 10, 294. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, R.; Chakrabarti, S.S.; Kaur, U.; Parashar, G.; Banerjee, A.; Rawal, R.K. Multi-Target Directed Ligands (MTDLs): Promising Coumarin Hybrids for Alzheimer’s Disease. Curr. Alzheimer Res. 2021, 18, 802–830. [Google Scholar] [CrossRef] [PubMed]
- Zou, D.; Liu, R.; Lv, Y.; Guo, J.; Zhang, C.; Xie, Y. Latest Advances in Dual Inhibitors of Acetylcholinesterase and Monoamine Oxidase B against Alzheimer’s Disease. J. Enzyme Inhib. Med. Chem. 2023, 38, 2270781. [Google Scholar] [CrossRef] [PubMed]
- Vecchio, I.; Sorrentino, L.; Paoletti, A.; Marra, R.; Arbitrio, M. The State of the Art on Acetylcholinesterase Inhibitors in the Treatment of Alzheimer’s Disease. J. Centr. Nerv. Sys. Dis. 2021, 13. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Lv, Y.; Bai, R.; Xie, Y. Structural Exploration of Multifunctional Monoamine Oxidase B Inhibitors as Potential Drug Candidates against Alzheimer’s Disease. Bioorg. Chem. 2021, 114, 105070. [Google Scholar] [CrossRef] [PubMed]
- Pourshojaei, Y.; Abiri, A.; Eskandari, K.; Haghighijoo, Z.; Edraki, N.; Asadipour, A. Phenoxyethyl Piperidine/Morpholine Derivatives as PAS and CAS Inhibitors of Cholinesterases: Insights for Future Drug Design. Sci. Rep. 2019, 9, 19855. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Saha, A.; Roy, K. In Silico Modeling for Dual Inhibition of Acetylcholinesterase (AChE) and Butyrylcholinesterase (BuChE) Enzymes in Alzheimer’s Disease. Comp. Biol. Chem. 2020, 88, 107355. [Google Scholar] [CrossRef] [PubMed]
- Cheung, J.; Rudolph, M.J.; Burshteyn, F.; Cassidy, M.S.; Gary, E.N.; Love, J.; Franklin, M.C.; Height, J.J. Structures of Human Acetylcholinesterase in Complex with Pharmacologically Important Ligands. J. Med. Chem. 2012, 55, 10282–10286. [Google Scholar] [CrossRef] [PubMed]
- Allgardsson, A.; Berg, L.; Akfur, C.; Hörnberg, A.; Worek, F.; Linusson, A.; Ekström, F.J. Structure of a Prereaction Complex between the Nerve Agent Sarin, Its Biological Target Acetylcholinesterase, and the Antidote HI-6. Proc. Natl. Acad. Sci. USA 2016, 113, 5514–5519. [Google Scholar] [CrossRef] [PubMed]
- Gerlits, O.; Fajer, M.; Cheng, X.; Blumenthal, D.K.; Radić, Z.; Kovalevsky, A. Structural and Dynamic Effects of Paraoxon Binding to Human Acetylcholinesterase by X-Ray Crystallography and Inelastic Neutron Scattering. Structure 2022, 30, 1538–1549.e3. [Google Scholar] [CrossRef] [PubMed]
- Dileep, K.V.; Ihara, K.; Mishima-Tsumagari, C.; Kukimoto-Niino, M.; Yonemochi, M.; Hanada, K.; Shirouzu, M.; Zhang, K.Y.J. Crystal Structure of Human Acetylcholinesterase in Complex with Tacrine: Implications for Drug Discovery. Int. J. Biol. Macromol. 2022, 210, 172–181. [Google Scholar] [CrossRef] [PubMed]
- Binda, C.; Wang, J.; Pisani, L.; Caccia, C.; Carotti, A.; Salvati, P.; Edmondson, D.E.; Mattevi, A. Structures of Human Monoamine Oxidase B Complexes with Selective Noncovalent Inhibitors: Safinamide and Coumarin Analogs. J. Med. Chem. 2007, 50, 5848–5852. [Google Scholar] [CrossRef] [PubMed]
- Reis, J.; Manzella, N.; Cagide, F.; Mialet-Perez, J.; Uriarte, E.; Parini, A.; Borges, F.; Binda, C. Tight-Binding Inhibition of Human Monoamine Oxidase B by Chromone Analogs: A Kinetic, Crystallographic, and Biological Analysis. J. Med. Chem. 2018, 61, 4203–4212. [Google Scholar] [CrossRef] [PubMed]
- Osmaniye, D.; Evren, A.E.; Sağlık, B.N.; Levent, S.; Özkay, Y.; Kaplancıklı, Z.A. Design, Synthesis, Biological Activity, Molecular Docking, and Molecular Dynamics of Novel Benzimidazole Derivatives as Potential AChE/MAO-B Dual Inhibitors. Arch. Pharm. 2022, 355, 2100450. [Google Scholar] [CrossRef] [PubMed]
- Ellman, G.L.; Courtney, K.D.; Andres, V.; Feather-Stone, R.M. A New and Rapid Colorimetric Determination of Acetylcholinesterase Activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Atatreh, N.; Al Rawashdah, S.; Al Neyadi, S.S.; Abuhamdah, S.M.; Ghattas, M.A. Discovery of New Butyrylcholinesterase Inhibitors via Structure-Based Virtual Screening. J. Enzyme Inhib. Med. Chem. 2019, 34, 1373–1379. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Li, A.J.; Zhao, J.; Santillo, M.F.; Xia, M. Acetylcholinesterase Inhibition Assays for High-Throughput Screening. Methods Mol. Biol. 2022, 2474, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Ali-Shtayeh, M.S.; Jamous, R.M.; Zaitoun, S.Y.A.; Qasem, I.B. In-Vitro Screening of Acetylcholinesterase Inhibitory Activity of Extracts from Palestinian Indigenous Flora in Relation to the Treatment of Alzheimer’s Disease. Funct. Foods Health Dis. 2014, 4, 381–400. [Google Scholar] [CrossRef]
- Pohanka, M.; Hrabinova, M.; Kuca, K.; Simonato, J.-P. Assessment of Acetylcholinesterase Activity Using Indoxylacetate and Comparison with the Standard Ellman’s Method. Int. J. Mol. Sci. 2011, 12, 2631–2640. [Google Scholar] [CrossRef] [PubMed]
- Hestrin, S. The Reaction of Acetylcholine and Other Carboxylic Acid Derivatives with Hydroxylamine, and Its Analytical Application. J. Biol. Chem. 1949, 180, 249–261. [Google Scholar] [CrossRef] [PubMed]
- Pilz, W. Acetylcholinesterase. In Methods of Enzymatic Analysis; Bergmeyer, H.-U., Ed.; Academic Press: Cambridge, MA, USA, 1965; pp. 765–770. ISBN 978-0-12-395630-9. [Google Scholar]
- Frawley, J.P.; Hagan, E.C.; Fitzhugh, O.G. A Comparative Pharmacological and Toxicological Study of Organic Phosphate-Anticholinesterase Compounds. J. Pharmacol. Exp. Ther. 1952, 105, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Hamada, H.; Gerk, P.M. Selectivity of Dietary Phenolics for Inhibition of Human Monoamine Oxidases A and B. BioMed Res. Int. 2019, 2019, 8361858. [Google Scholar] [CrossRef] [PubMed]
- Lammertsma, A.A.; Bench, C.J.; Price, G.W.; Cremer, J.E.; Luthra, S.K.; Turton, D.; Wood, N.D.; Frackowiak, R.S.J. Measurement of Cerebral Monoamine Oxidase B Activity Using L-[11C]Deprenyl and Dynamic Positron Emission Tomography. J. Cereb. Blood Flow Metab. 1991, 11, 545–556. [Google Scholar] [CrossRef] [PubMed]
- Kumlien, E.; Bergström, M.; Lilja, A.; Andersson, J.; Szekeres, V.; Westerberg, C.E.; Westerberg, G.; Antoni, G.; Långström, B. Positron Emission Tomography with [11C]Deuterium-Deprenyl in Temporal Lobe Epilepsy. Epilepsia 1995, 36, 712–721. [Google Scholar] [CrossRef] [PubMed]
- Guang, H.-M.; Du, G.-H. High-Throughput Screening for Monoamine Oxidase-A and Monoamine Oxidase-B Inhibitors Using One-Step Fluorescence Assay. Acta Pharmacol. Sin. 2006, 27, 760–766. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.-R.; Huang, J.-B.; Yang, S.-L.; Hong, F.-F. Role of Cholinergic Signaling in Alzheimer’s Disease. Molecules 2022, 27, 1816. [Google Scholar] [CrossRef] [PubMed]
- Naoi, M.; Maruyama, W.; Shamoto-Nagai, M. Neuroprotective Function of Rasagiline and Selegiline, Inhibitors of Type B Monoamine Oxidase, and Role of Monoamine Oxidases in Synucleinopathies. Int. J. Mol. Sci. 2022, 23, 11059. [Google Scholar] [CrossRef] [PubMed]
- Mathew, B.; Parambi, D.G.T.; Mathew, G.E.; Uddin, M.S.; Inasu, S.T.; Kim, H.; Marathakam, A.; Unnikrishnan, M.K.; Carradori, S. Emerging Therapeutic Potentials of Dual-acting MAO and AChE Inhibitors in Alzheimer’s and Parkinson’s Diseases. Arch. Pharm. 2019, 352, 1900177. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.M.; Kang, M.-G.; Hong, A.; Park, J.-E.; Kim, S.H.; Lee, J.P.; Baek, S.C.; Park, D.; Nam, S.-J.; Cho, M.-L.; et al. Potent and Selective Inhibition of Human Monoamine Oxidase-B by 4-Dimethylaminochalcone and Selected Chalcone Derivatives. Int. J. Biol. Macromol. 2019, 137, 426–432. [Google Scholar] [CrossRef] [PubMed]
- Jeong, G.S.; Kaipakasseri, S.; Lee, S.R.; Marraiki, N.; Batiha, G.E.-S.; Dev, S.; Palakkathondi, A.; Kavully, F.S.; Gambacorta, N.; Nicolotti, O.; et al. Selected 1,3-Benzodioxine-Containing Chalcones as Multipotent Oxidase and Acetylcholinesterase Inhibitors. ChemMedChem 2020, 15, 2257–2263. [Google Scholar] [CrossRef] [PubMed]
- Sang, Z.; Song, Q.; Cao, Z.; Deng, Y.; Zhang, L. Design, Synthesis, and Evaluation of Chalcone-Vitamin E-Donepezil Hybrids as Multi-Target-Directed Ligands for the Treatment of Alzheimer’s Disease. J. Enzyme Inhib. Med. Chem. 2022, 37, 69–85. [Google Scholar] [CrossRef] [PubMed]
- Sang, Z.; Song, Q.; Cao, Z.; Deng, Y.; Tan, Z.; Zhang, L. Design, Synthesis and Evaluation of Novel Dimethylamino Chalcone-O-Alkylamines Derivatives as Potential Multifunctional Agents against Alzheimer’s Disease. Eur. J. Med. Chem. 2021, 216, 113310. [Google Scholar] [CrossRef] [PubMed]
- Bai, P.; Wang, K.; Zhang, P.; Shi, J.; Cheng, X.; Zhang, Q.; Zheng, C.; Cheng, Y.; Yang, J.; Lu, X.; et al. Development of Chalcone-O-Alkylamine Derivatives as Multifunctional Agents against Alzheimer’s Disease. Eur. J. Med. Chem. 2019, 183, 111737. [Google Scholar] [CrossRef] [PubMed]
- Tian, C.; Qiang, X.; Song, Q.; Cao, Z.; Ye, C.; He, Y.; Deng, Y.; Zhang, L. Flurbiprofen-Chalcone Hybrid Mannich Base Derivatives as Balanced Multifunctional Agents against Alzheimer’s Disease: Design, Synthesis and Biological Evaluation. Bioorg. Chem. 2020, 94, 103477. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Song, Q.; Cao, Z.; Li, Y.; Tian, C.; Yang, Z.; Zhang, H.; Deng, Y. Design, Synthesis and Evaluation of Chalcone Mannich Base Derivatives as Multifunctional Agents for the Potential Treatment of Alzheimer’s Disease. Bioorg. Chem. 2019, 87, 395–408. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.M.; Rangarajan, T.M.; Chaudhary, R.; Singh, R.P.; Singh, M.; Singh, R.P.; Tondo, A.R.; Gambacorta, N.; Nicolotti, O.; Mathew, B.; et al. Novel Class of Chalcone Oxime Ethers as Potent Monoamine Oxidase-B and Acetylcholinesterase Inhibitors. Molecules 2020, 25, 2356. [Google Scholar] [CrossRef] [PubMed]
- Kumar, B.; Dwivedi, A.R.; Sarkar, B.; Gupta, S.K.; Krishnamurthy, S.; Mantha, A.K.; Parkash, J.; Kumar, V. 4,6-Diphenylpyrimidine Derivatives as Dual Inhibitors of Monoamine Oxidase and Acetylcholinesterase for the Treatment of Alzheimer’s Disease. ACS Chem. Neurosci. 2019, 10, 252–265. [Google Scholar] [CrossRef] [PubMed]
- Kamel, N.N.; Aly, H.F.; Fouad, G.I.; El-Karim, S.S.A.; Anwar, M.M.; Syam, Y.M.; Elseginy, S.A.; Ahmed, K.A.; Booles, H.F.; Shalaby, M.B. Anti-Alzheimer Activity of New Coumarin-Based Derivatives Targeting Acetylcholinesterase Inhibition. RSC Adv. 2023, 13, 18496–18510. [Google Scholar] [CrossRef] [PubMed]
- Viña, D.; Matos, M.J.; Yáñez, M.; Santana, L.; Uriarte, E. 3-Substituted Coumarins as Dual Inhibitors of AChE and MAO for the Treatment of Alzheimer’s Disease. Med. Chem. Commun. 2012, 3, 213–218. [Google Scholar] [CrossRef]
- Zhang, Q.; Hao, C.; Miao, Y.; Yun, Y.; Sun, X.; Pan, Y.; Sun, J.; Wang, X. Design and Synthesis of Benzyl Aminocoumarin and Its Anti-Alzheimer’s Activity. New J. Chem. 2021, 45, 17287–17300. [Google Scholar] [CrossRef]
- Rodríguez-Enríquez, F.; Viña, D.; Uriarte, E.; Laguna, R.; Matos, M.J. 7-Amidocoumarins as Multitarget Agents against Neurodegenerative Diseases: Substitution Pattern Modulation. ChemMedChem 2021, 16, 179–186. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Liu, J.; Lan, J.-S.; Ding, J.; Sun, Y.; Fang, Y.; Jiang, N.; Yang, Z.; Sun, L.; Jin, Y.; et al. Coumarin-Dithiocarbamate Hybrids as Novel Multitarget AChE and MAO-B Inhibitors against Alzheimer’s Disease: Design, Synthesis and Biological Evaluation. Bioorg. Chem. 2018, 81, 512–528. [Google Scholar] [CrossRef] [PubMed]
- Rullo, M.; Cipolloni, M.; Catto, M.; Colliva, C.; Miniero, D.V.; Latronico, T.; de Candia, M.; Benicchi, T.; Linusson, A.; Giacchè, N.; et al. Probing Fluorinated Motifs onto Dual AChE-MAO B Inhibitors: Rational Design, Synthesis, Biological Evaluation, and Early-ADME Studies. J. Med. Chem. 2022, 65, 3962–3977. [Google Scholar] [CrossRef] [PubMed]
- Pisani, L.; Iacobazzi, R.M.; Catto, M.; Rullo, M.; Farina, R.; Denora, N.; Cellamare, S.; Altomare, C.D. Investigating Alkyl Nitrates as Nitric Oxide Releasing Precursors of Multitarget Acetylcholinesterase-Monoamine Oxidase B Inhibitors. Eur. J. Med. Chem. 2019, 161, 292–309. [Google Scholar] [CrossRef] [PubMed]
- Carradori, S.; Silvestri, R. New Frontiers in Selective Human MAO-B Inhibitors. J. Med. Chem. 2015, 58, 6717–6732. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.-B.; Yin, F.-C.; Huang, M.; Jiang, N.; Lan, J.-S.; Kong, L.-Y. Chromone and Donepezil Hybrids as New Multipotent Cholinesterase and Monoamine Oxidase Inhibitors for the Potential Treatment of Alzheimer’s Disease. RSC Med. Chem. 2020, 11, 225. [Google Scholar] [CrossRef] [PubMed]
- Estrada-Valencia, M.; Herrera-Arozamena, C.; Pérez, C.; Viña, D.; Morales-García, J.A.; Pérez-Castillo, A.; Ramos, E.; Romero, A.; Laurini, E.; Pricl, S.; et al. New Flavonoid—N,N-Dibenzyl(N-Methyl)Amine Hybrids: Multi-Target-Directed Agents for Alzheimer’s Disease Endowed with Neurogenic Properties. J. Enzyme Inhib. Med. Chem. 2019, 34, 712. [Google Scholar] [CrossRef] [PubMed]
- Reis, J.; Cagide, F.; Valencia, M.E.; Teixeira, J.; Bagetta, D.; Pérez, C.; Uriarte, E.; Oliveira, P.J.; Ortuso, F.; Alcaro, S.; et al. Multi-Target-Directed Ligands for Alzheimer’s Disease: Discovery of Chromone-Based Monoamine Oxidase/Cholinesterase Inhibitors. Eur. J. Med. Chem. 2018, 158, 781–800. [Google Scholar] [CrossRef] [PubMed]
- Łażewska, D.; Bajda, M.; Kaleta, M.; Zaręba, P.; Doroz-Płonka, A.; Siwek, A.; Alachkar, A.; Mogilski, S.; Saad, A.; Kuder, K.; et al. Rational Design of New Multitarget Histamine H3 Receptor Ligands as Potential Candidates for Treatment of Alzheimer’s Disease. Eur. J. Med. Chem. 2020, 207, 112743. [Google Scholar] [CrossRef] [PubMed]
- Campora, M.; Canale, C.; Gatta, E.; Tasso, B.; Laurini, E.; Relini, A.; Pricl, S.; Catto, M.; Tonelli, M. Multitarget Biological Profiling of New Naphthoquinone and Anthraquinone-Based Derivatives for the Treatment of Alzheimer’s Disease. ACS Chem. Neurosci. 2021, 12, 447–461. [Google Scholar] [CrossRef] [PubMed]
- Çevik, U.A.; Osmaniye, D.; Sağlik, B.N.; Çavuşoğlu, B.K.; Levent, S.; Karaduman, A.B.; Ilgin, S.; Karaburun, A.Ç.; Özkay, Y.; Kaplancikli, Z.A.; et al. Multifunctional Quinoxaline-Hydrazone Derivatives with Acetylcholinesterase and Monoamine Oxidases Inhibitory Activities as Potential Agents against Alzheimer’s Disease. Med. Chem. Res. 2020, 29, 1000–1011. [Google Scholar] [CrossRef]
- Tok, F.; Koçyiğit-Kaymakçıoğlu, B.; Sağlık, B.N.; Levent, S.; Özkay, Y.; Kaplancıklı, Z.A. Synthesis and Biological Evaluation of New Pyrazolone Schiff Bases as Monoamine Oxidase and Cholinesterase Inhibitors. Bioorg. Chem. 2019, 84, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Vishnu, M.S.; Pavankumar, V.; Kumar, S.; Raja, A.S. Experimental and Computational Evaluation of Piperonylic Acid Derived Hydrazones Bearing Isatin Moieties as Dual Inhibitors of Cholinesterases and Monoamine Oxidases. ChemMedChem 2019, 14, 1359–1376. [Google Scholar] [CrossRef] [PubMed]
- Palakkathondi, A.; Oh, J.M.; Dev, S.; Rangarajan, T.M.; Kaipakasseri, S.; Kavully, F.S.; Gambacorta, N.; Nicolotti, O.; Kim, H.; Mathew, B. (Hetero-)(Arylidene)Arylhydrazides as Multitarget-Directed Monoamine Oxidase Inhibitors. ACS Comb. Sci. 2020, 22, 592–599. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Zhang, J.; Wang, H.; Mao, F.; Bao, K.; Liu, W.; Zhu, J.; Li, X.; Zhang, H.; Li, J. Rational Design of Novel Selective Dual-Target Inhibitors of Acetylcholinesterase and Monoamine Oxidase B as Potential Anti-Alzheimer’s Disease Agents. ACS Chem. Neurosci. 2019, 10, 482–496. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Xiao, G.; Li, Y.; Liu, H.; Song, Q.; Zhang, X.; Yang, Z.; Zheng, Y.; Tan, Z.; Deng, Y. Multifunctional 5,6-Dimethoxybenzo[d]Isothiazol-3(2H)-One-N-Alkylbenzylamine Derivatives with Acetylcholinesterase, Monoamine Oxidases and β-Amyloid Aggregation Inhibitory Activities as Potential Agents against Alzheimer’s Disease. Bioorg. Med. Chem. 2018, 26, 1885–1895. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Shi, Y.; Zhang, X.; Yu, G.; Li, J.; Cong, S.; Deng, Y. Discovery of Novel 3-Butyl-6-Benzyloxyphthalide Mannich Base Derivatives as Multifunctional Agents against Alzheimer’s Disease. Bioorg. Med. Chem. 2022, 58, 116660. [Google Scholar] [CrossRef] [PubMed]
- Nadeem, M.S.; Khan, J.A.; Kazmi, I.; Rashid, U. Design, Synthesis, and Bioevaluation of Indole Core Containing 2-Arylidine Derivatives of Thiazolopyrimidine as Multitarget Inhibitors of Cholinesterases and Monoamine Oxidase A/B for the Treatment of Alzheimer Disease. ACS Omega 2022, 7, 9369. [Google Scholar] [CrossRef] [PubMed]
- Denya, I.; Malan, S.F.; Enogieru, A.B.; Omoruyi, S.I.; Ekpo, O.E.; Kapp, E.; Zindo, F.T.; Joubert, J. Design, Synthesis and Evaluation of Indole Derivatives as Multifunctional Agents against Alzheimer’s Disease. MedChemComm 2018, 9, 357. [Google Scholar] [CrossRef] [PubMed]
- Karaca, Ş.; Osmaniye, D.; Sağlık, B.N.; Levent, S.; Ilgın, S.; Özkay, Y.; Karaburun, A.Ç.; Kaplancıklı, Z.A.; Gundogdu-Karaburun, N. Synthesis of Novel Benzothiazole Derivatives and Investigation of Their Enzyme Inhibitory Effects against Alzheimer’s Disease. RSC Adv. 2022, 12, 23626. [Google Scholar] [CrossRef] [PubMed]
- Saleem Khan, M.; Asif Nawaz, M.; Jalil, S.; Rashid, F.; Hameed, A.; Asari, A.; Mohamad, H.; Ur Rehman, A.; Iftikhar, M.; Iqbal, J.; et al. Deep Eutectic Solvent Mediated Synthesis of 3,4-Dihydropyrimidin-2(1H)-Ones and Evaluation of Biological Activities Targeting Neurodegenerative Disorders. Bioorg. Chem. 2022, 118, 105457. [Google Scholar] [CrossRef] [PubMed]
- Jia, Z.; Wen, H.; Huang, S.; Luo, Y.; Gao, J.; Wang, R.; Wan, K.; Xue, W. “Click” Assembly of Novel Dual Inhibitors of AChE and MAO-B from Pyridoxine Derivatives for the Treatment of Alzheimer’s Disease. Heterocycl. Commun. 2022, 28, 18–25. [Google Scholar] [CrossRef]
- Youdim, M.B.H. Site-Activated Multi Target Iron Chelators with Acetylcholinesterase (AChE) and Monoamine Oxidase (MAO) Inhibitory Activities for Alzheimer’s Disease Therapy. J. Neural Transm. 2022, 129, 715–721. [Google Scholar] [CrossRef] [PubMed]
- Nadeem, M.S.; Khan, J.A.; Rashid, U. Fluoxetine and Sertraline Based Multitarget Inhibitors of Cholinesterases and Monoamine Oxidase-A/B for the Treatment of Alzheimer’s Disease: Synthesis, Pharmacology and Molecular Modeling Studies. Int. J. Biol. Macromol. 2021, 193, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Gogineni, V.; Nael, M.A.; Chaurasiya, N.D.; Elokely, K.M.; McCurdy, C.R.; Rimoldi, J.M.; Cutler, S.J.; Tekwani, B.L.; León, F. Computationally Assisted Lead Optimization of Novel Potent and Selective MAO-B Inhibitors. Biomedicines 2021, 9, 1304. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Jia, W.; Wang, J.; Yang, Z.; Liu, Y.; Huang, D.; Mei, X.; Xiong, X.; Shi, J.; Tang, Y. Design, Synthesis, and Biological Evaluation of Novel Donepezil-Tacrine Hybrids as Multi-Functional Agents with Low Neurotoxicity against Alzheimer’s Disease. Bioorg. Chem. 2024, 143, 107010. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Yi, C.; Chen, K.; Wang, T.; Deng, K.; Jin, C.; Hao, G. Enhancing Monoamine Oxidase B Inhibitory Activity via Chiral Fluorination: Structure-Activity Relationship, Biological Evaluation, and Molecular Docking Study. Eur. J. Med. Chem. 2022, 228, 114025. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Zhou, S.; Nao, J. Kaempferol as a Therapeutic Agent in Alzheimer’s Disease: Evidence from Preclinical Studies. Ageing Res. Rev. 2023, 87, 101910. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Ayyannan, S.R. Identification of New Small Molecule Monoamine Oxidase-B Inhibitors through Pharmacophore-Based Virtual Screening, Molecular Docking and Molecular Dynamics Simulation Studies. J. Biomol. Struct. Dyn. 2023, 41, 6789–6810. [Google Scholar] [CrossRef] [PubMed]
- Khan, B.A.; Hamdani, S.S.; Jalil, S.; Ejaz, S.A.; Iqbal, J.; Shawky, A.M.; Alqahtani, A.M.; Gabr, G.A.; Ibrahim, M.A.; Sidhom, P.A. Synthesis and Evaluation of Novel S-Alkyl Phthalimide-and S-Benzyl-Oxadiazole-Quinoline Hybrids as Inhibitors of Monoamine Oxidase and Acetylcholinesterase. Pharmaceuticals 2022, 16, 11. [Google Scholar] [CrossRef] [PubMed]
- Salman, M.M.; Al-Obaidi, Z.; Kitchen, P.; Loreto, A.; Bill, R.M.; Wade-Martins, R. Advances in Applying Computer-Aided Drug Design for Neurodegenerative Diseases. Int. J. Mol. Sci. 2021, 22, 4688. [Google Scholar] [CrossRef] [PubMed]
- James, J.P.; Sasidharan, P.; Mandal, S.P.; Dixit, S.R. Virtual Screening of Alkaloids and Flavonoids as Acetylcholinesterase and MAO-B Inhibitors by Molecular Docking and Dynamic Simulation Studies. Polycycl. Aromat. Compd. 2023, 43, 5453–5477. [Google Scholar] [CrossRef]
- Mateev, E.; Kondeva-Burdina, M.; Georgieva, M.; Zlatkov, A. Repurposing of FDA-Approved Drugs as Dual-Acting MAO-B and AChE Inhibitors against Alzheimer’s Disease: An in Silico and in Vitro Study. J. Mol. Graphics Model. 2023, 122, 108471. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Sun, Y.; Guo, Y.; Wang, Z.; Huang, L.; Li, X. Dual Functional Cholinesterase and MAO Inhibitors for the Treatment of Alzheimer’s Disease: Synthesis, Pharmacological Analysis and Molecular Modeling of Homoisoflavonoid Derivatives. J. Enzyme Inhib. Med. Chem. 2016, 31, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.M.; Jang, H.-J.; Kim, W.J.; Kang, M.-G.; Baek, S.C.; Lee, J.P.; Park, D.; Oh, S.-R.; Kim, H. Calycosin and 8-O-Methylretusin Isolated from Maackia Amurensis as Potent and Selective Reversible Inhibitors of Human Monoamine Oxidase-B. Int. J. Biol. Macromol. 2020, 151, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Hou, Q.; Li, Y. Dual Inhibition of AChE and MAO-B in Alzheimer’s Disease: Machine Learning Approaches and Model Interpretations. Mol. Divers. 2024, 29, 3113–3130. [Google Scholar] [CrossRef] [PubMed]
- Bajad, N.G.; Singh, R.B.; Gajendra, T.A.; Gutti, G.; Kumar, A.; Krishnamurthy, S.; Singh, S.K. Development of Multi-Targetable Chalcone Derivatives Bearing N-Aryl Piperazine Moiety for the Treatment of Alzheimer’s Disease. Bioorg. Chem. 2024, 143, 107082. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, T.; Zhan, F.; Cheng, F.; Lu, L.; Zhang, B.; Li, J.; Hu, Z.; Zhou, S.; Jia, Y.; et al. Design, Synthesis, and Biological Evaluation of Novel Chromanone Derivatives as Multifunctional Agents for the Treatment of Alzheimer’s Disease. ACS Chem. Neurosci. 2022, 13, 3488–3501. [Google Scholar] [CrossRef] [PubMed]
- Viayna, E.; Coquelle, N.; Cieslikiewicz-Bouet, M.; Cisternas, P.; Oliva, C.A.; Sánchez-López, E.; Ettcheto, M.; Bartolini, M.; De Simone, A.; Ricchini, M.; et al. Discovery of a Potent Dual Inhibitor of Acetylcholinesterase and Butyrylcholinesterase with Antioxidant Activity That Alleviates Alzheimer-like Pathology in Old APP/PS1 Mice. J. Med. Chem. 2021, 64, 812–839. [Google Scholar] [CrossRef] [PubMed]
- Fronza, M.G.; Alves, D.; Praticò, D.; Savegnago, L. The Neurobiology and Therapeutic Potential of Multi-Targeting β-Secretase, Glycogen Synthase Kinase 3β and Acetylcholinesterase in Alzheimer’s Disease. Ageing Res. Rev. 2023, 90, 102033. [Google Scholar] [CrossRef] [PubMed]
- Stern, N.; Gacs, A.; Tátrai, E.; Flachner, B.; Hajdú, I.; Dobi, K.; Bágyi, I.; Dormán, G.; Lőrincz, Z.; Cseh, S.; et al. Dual Inhibitors of AChE and BACE-1 for Reducing Aβ in Alzheimer’s Disease: From In Silico to In Vivo. Int. J. Mol. Sci. 2022, 23, 13098. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer’s Disease Drug Development Pipeline: 2023-Cummings-2023-Alzheimer’s & Dementia: Translational Research & Clinical Interventions—Wiley Online Library. Available online: https://alz-journals.onlinelibrary.wiley.com/doi/full/10.1002/trc2.12385 (accessed on 29 October 2024).
- Sodium Oligomannate: First Approval—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/32020555/ (accessed on 29 October 2024).







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



| Molecular Signaling Pathways | Therapeutic Targets | Gene Function |
|---|---|---|
| Amyloid Cascade Signaling | Aβ, APP, BACE1, PSEN1, PSEN2, ApoE | Amyloid precursor protein (APP)—synthesis of Aβ peptides, implicated in plaque formation; BACE1—β-secretase enzyme; PSEN1 and—components of the γ-secretase complex; ApoE—apolipoprotein E involved in Aβ clearance and metabolism |
| τ and Neurofibrillary Tangles | τ protein, kinases | τ protein—microtubule stabilization; kinases—regulate τ phosphorylation in normal conditions; aberrant phosphorylation leads to neurofibrillary tangle formation |
| Genetic Mutation | APP, PSEN1, PSEN2, ApoE4 | Variations in APP, PSEN1, PSEN2, and ApoE4 associated with increased risk of Alzheimer’s disease |
| Neurotransmitter Signaling | NMDA receptor, AChE | NMDA receptor—critical for synaptic plasticity; AChE—enzyme involved in acetylcholine breakdown; glutamatergic and GABAergic receptors—essential for neurotransmission |
| Mitochondrial Dysfunction | Mitochondrial respiratory enzymes, mitochondrial ROS | Enzymes involved in mitochondrial respiration and ROS production |
| Oxidative Stress | Reactive oxygen species, mitochondrial dysfunction | ROS—reactive oxygen species; mitochondrial dysfunction—impaired energy production and oxidative stress |
| Neuroinflammatory Signaling | Proinflammatory elements such as chemokines | Various cytokines, chemokines, and reactive species implicated in neuroinflammation |
| Ubiquitin–Proteasomal System | Ubiquitin activating enzyme (E1), ubiquitin conjugating enzyme (E2), UCHL1, 26S proteasome | Components of the ubiquitin–proteasome system involved in protein degradation |
| Protein Misfolding and Molecular Chaperones | Heat shock proteins (HSPs), HSP60 and HSP70 | Molecular chaperones (HSPs)—assist in protein folding and preventing misfolding |
| Insulin Signaling | Insulin receptors, insulin degrading enzymes, Akt | Insulin-related proteins involved in glucose metabolism and signaling pathways |
| Excitotoxicity | Glutamate, NMDA receptors | Glutamate receptors—involved in excitatory neurotransmission |
| Neurotrophic Factors Signaling | Brain-derived neurotrophic factor, nerve growth factor, neurotrophin-3 (NT-3), neurotrophin-4/5, Trk receptors | Neurotrophic factors—promote neuronal survival and growth |
| Wnt/βCatenin Signaling | βcatenin, GSK3β | β-catenin—key component in Wnt signaling; GSK-3β—glycogen synthase kinase 3β |
| Leptin Signaling | Leptin, leptin receptor (ObR) | Leptin—hormone involved in energy regulation; ObR—Leptin receptor |
| Approach Name | Description | Advantages | Disadvantages |
|---|---|---|---|
| Virtual Screening | In silico screening of large compound libraries to identify potential candidates | Accelerated lead identification | Dependent on accurate databases and models |
| Molecular Docking | Predicts the preferred orientation of one molecule to a second when bound to form a stable complex | Efficient exploration of ligand–receptor interactions; rapid evaluation of binding affinities | Accuracy depends on force-field parameters and model parameters |
| Molecular Dynamics | Simulates the motion of atoms and molecules over time, allowing exploration of dynamic behavior | Provides insights into dynamic behavior of biomolecular systems; allows for assessment of structural stability | Computationally intensive; may require significant resources |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Asim, A.; Jastrzębski, M.K.; Kaczor, A.A. Dual Inhibitors of Acetylcholinesterase and Monoamine Oxidase-B for the Treatment of Alzheimer’s Disease. Molecules 2025, 30, 2975. https://doi.org/10.3390/molecules30142975
Asim A, Jastrzębski MK, Kaczor AA. Dual Inhibitors of Acetylcholinesterase and Monoamine Oxidase-B for the Treatment of Alzheimer’s Disease. Molecules. 2025; 30(14):2975. https://doi.org/10.3390/molecules30142975
Chicago/Turabian StyleAsim, Ayesha, Michał K. Jastrzębski, and Agnieszka A. Kaczor. 2025. "Dual Inhibitors of Acetylcholinesterase and Monoamine Oxidase-B for the Treatment of Alzheimer’s Disease" Molecules 30, no. 14: 2975. https://doi.org/10.3390/molecules30142975
APA StyleAsim, A., Jastrzębski, M. K., & Kaczor, A. A. (2025). Dual Inhibitors of Acetylcholinesterase and Monoamine Oxidase-B for the Treatment of Alzheimer’s Disease. Molecules, 30(14), 2975. https://doi.org/10.3390/molecules30142975