
Pyrrolopyrimidines: Design, Synthesis and Antitumor Properties of Novel Tricyclic Pyrrolo [2,3-d]pyrimidine Derivatives

 , ,
, ,  ,
,  and
and
Abstract

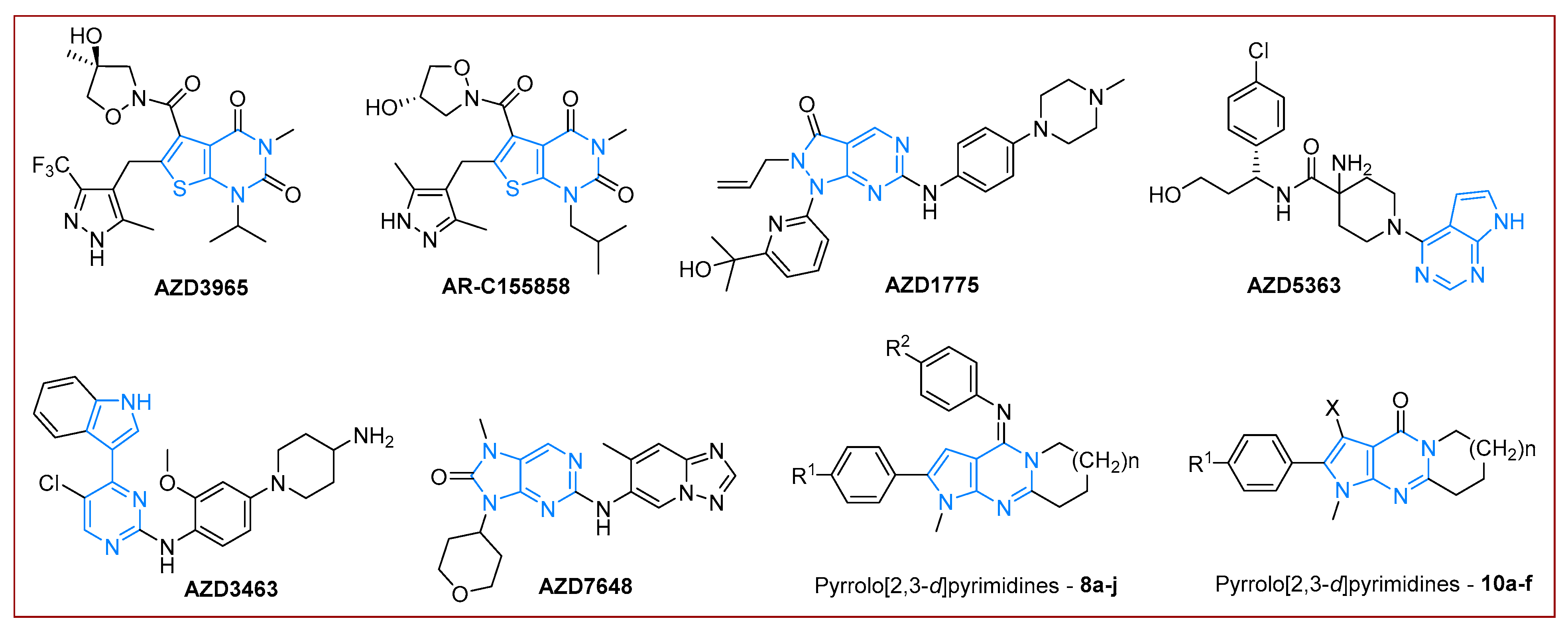
1. Introduction
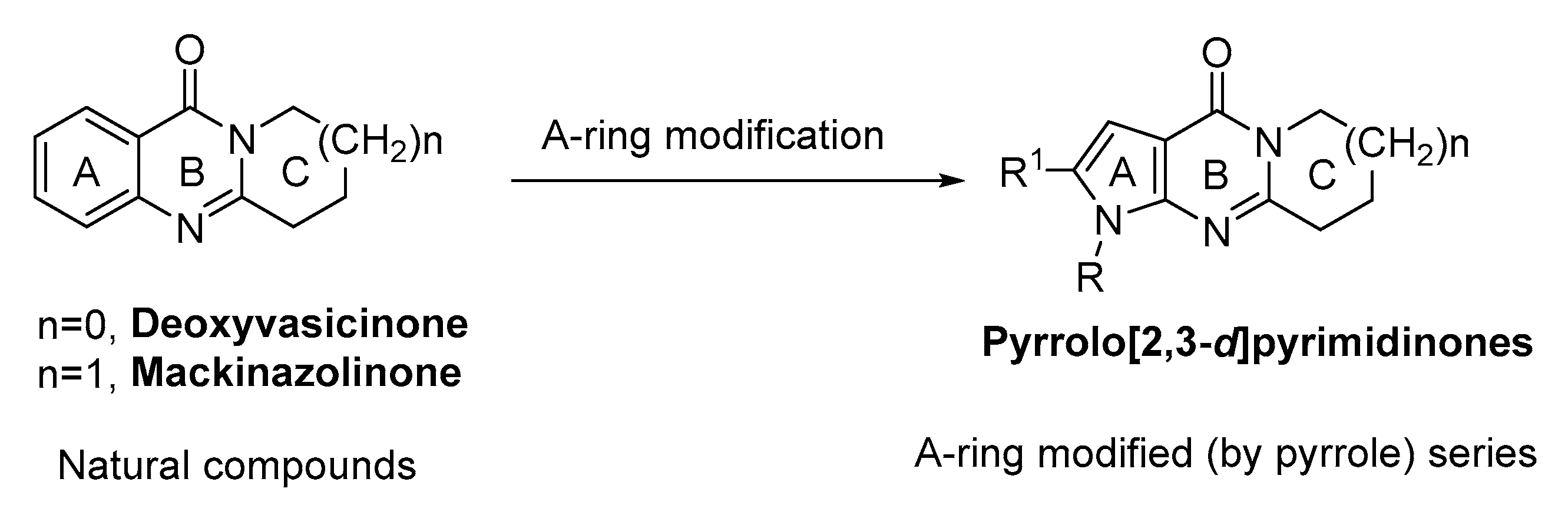
2. Results and Discussion
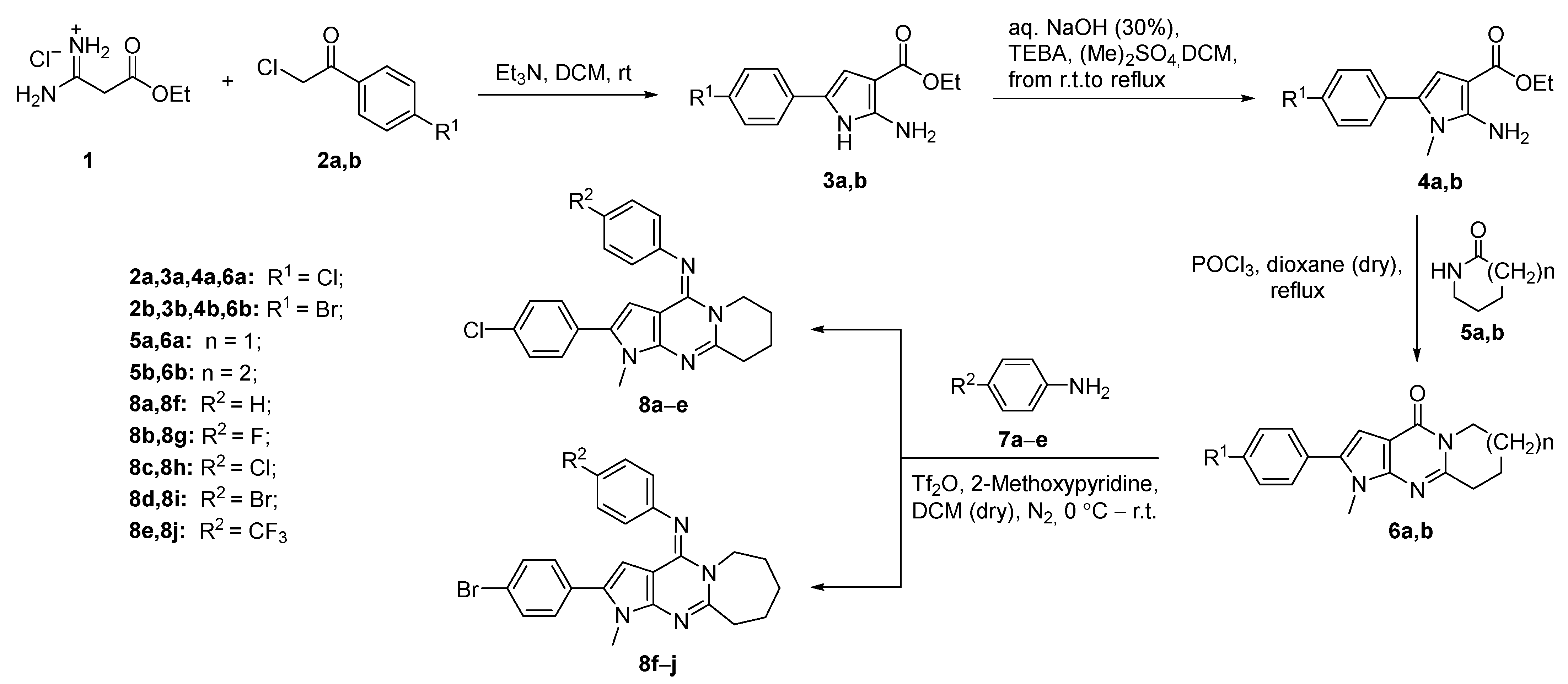
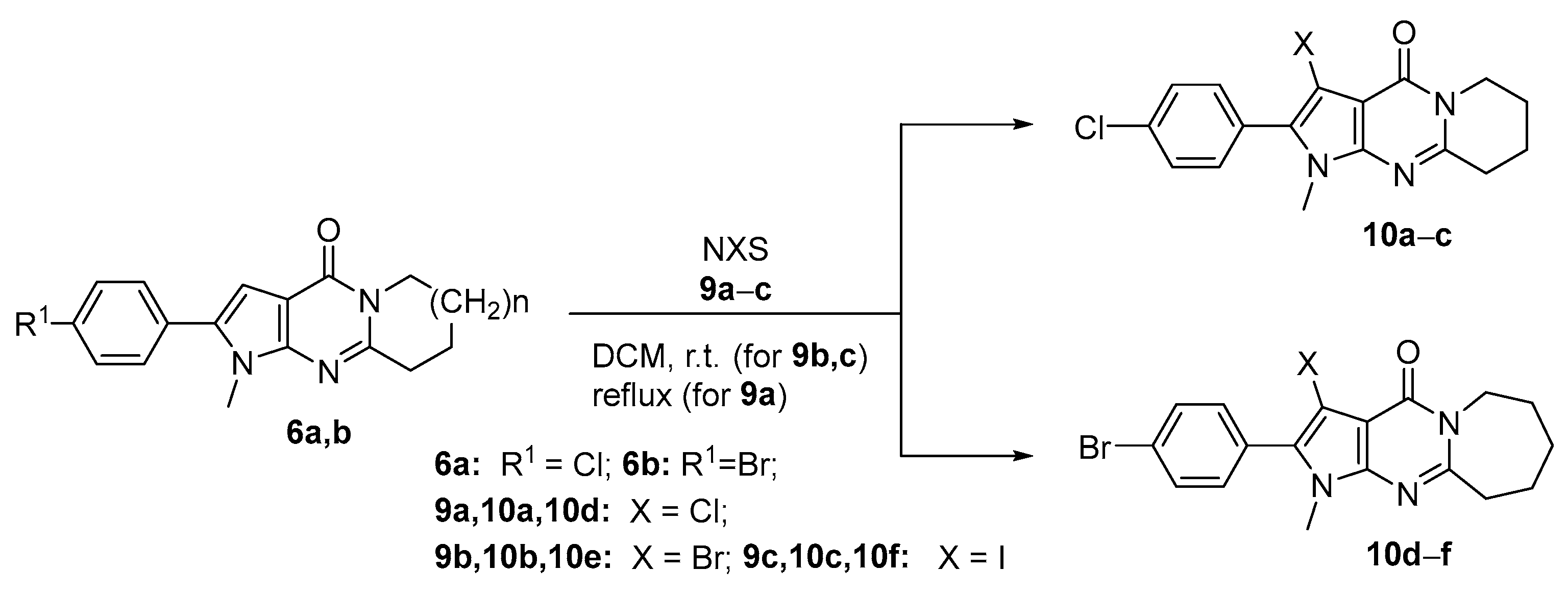
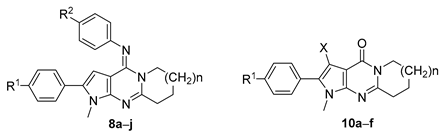
2.1. Synthesis
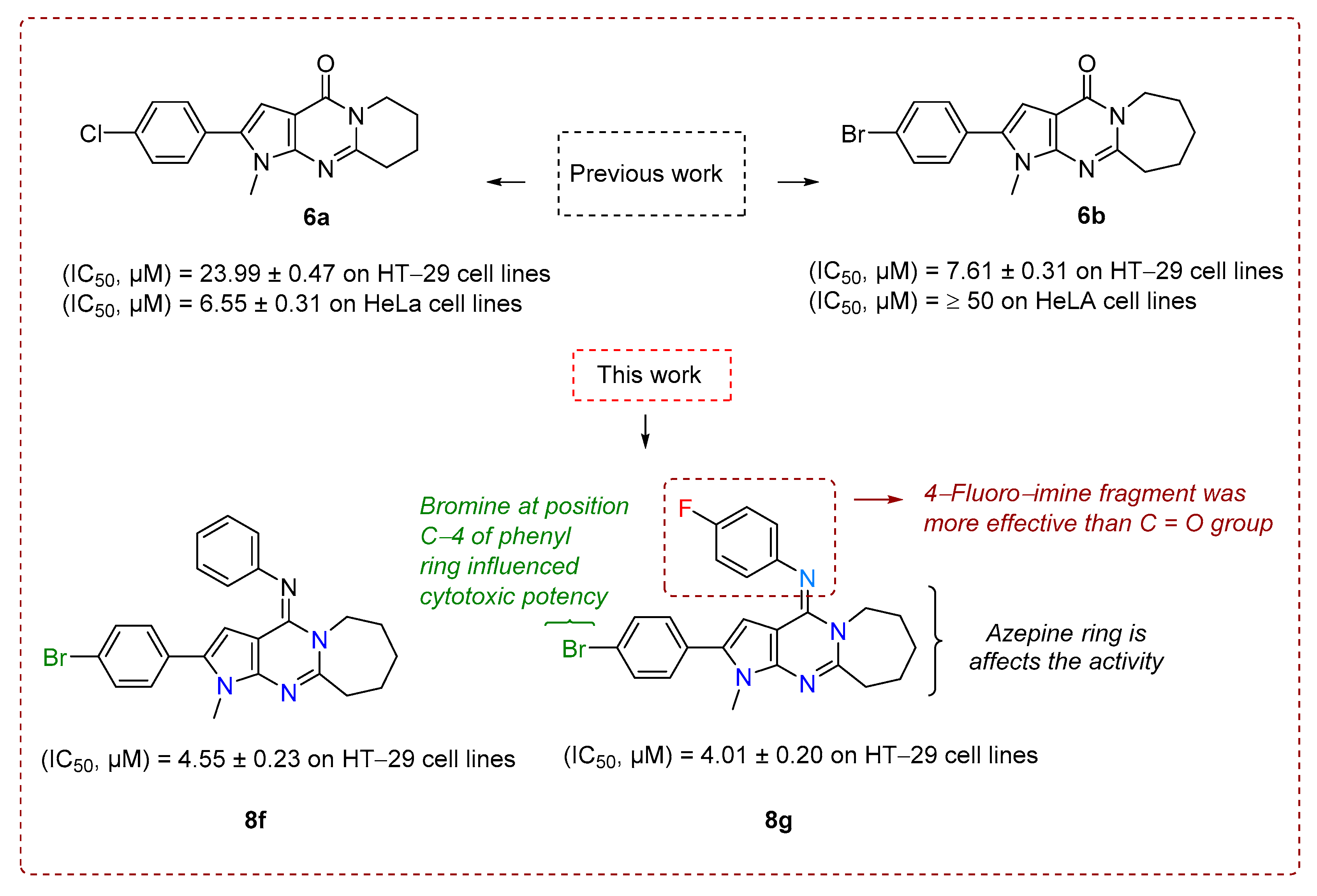
2.2. Antitumor Activity and Structure-Activity Relationship (SAR)
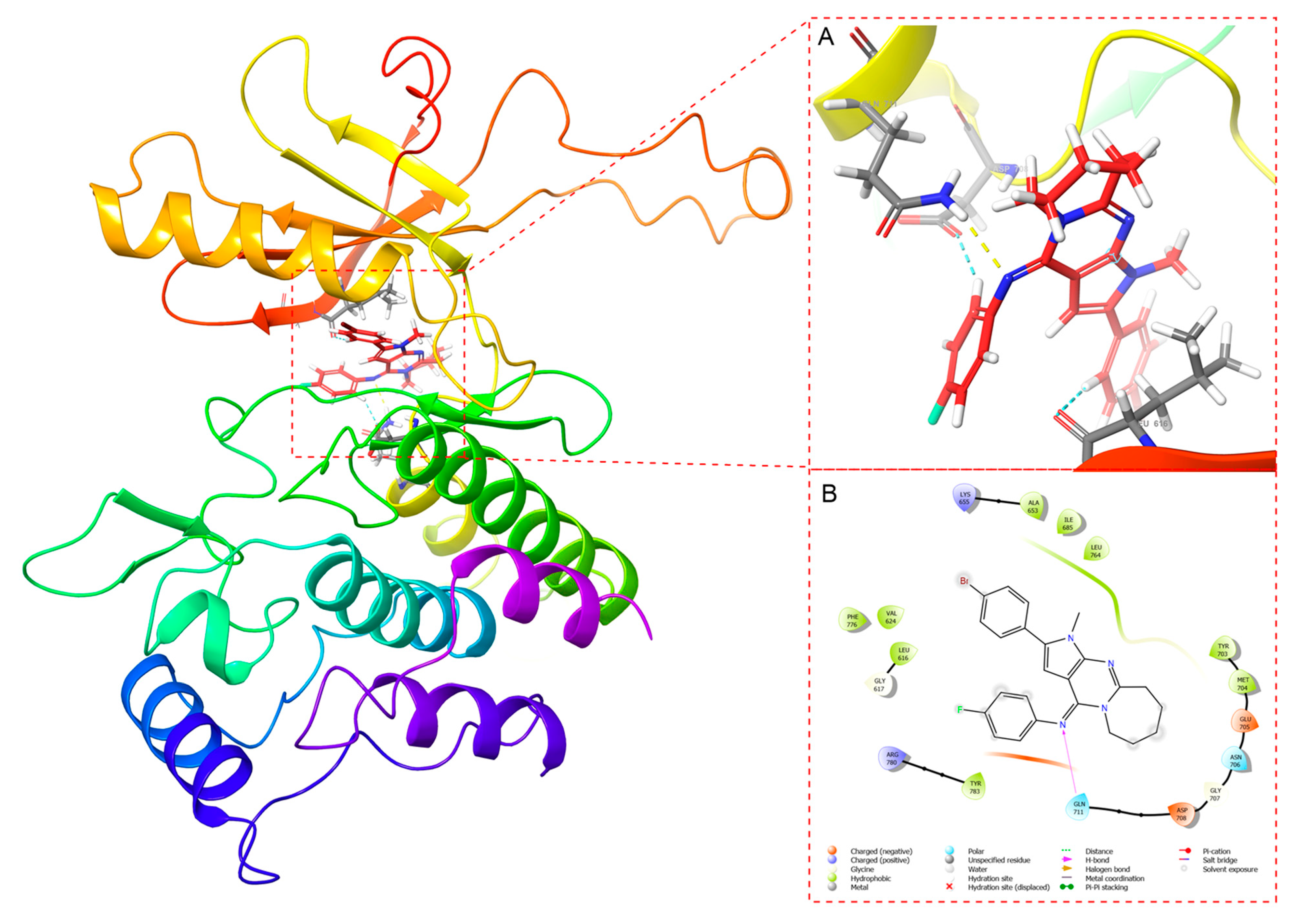
2.3. Docking Study
In Slico ADME Studies of Compounds 8f and 8g
3. Experimental Section
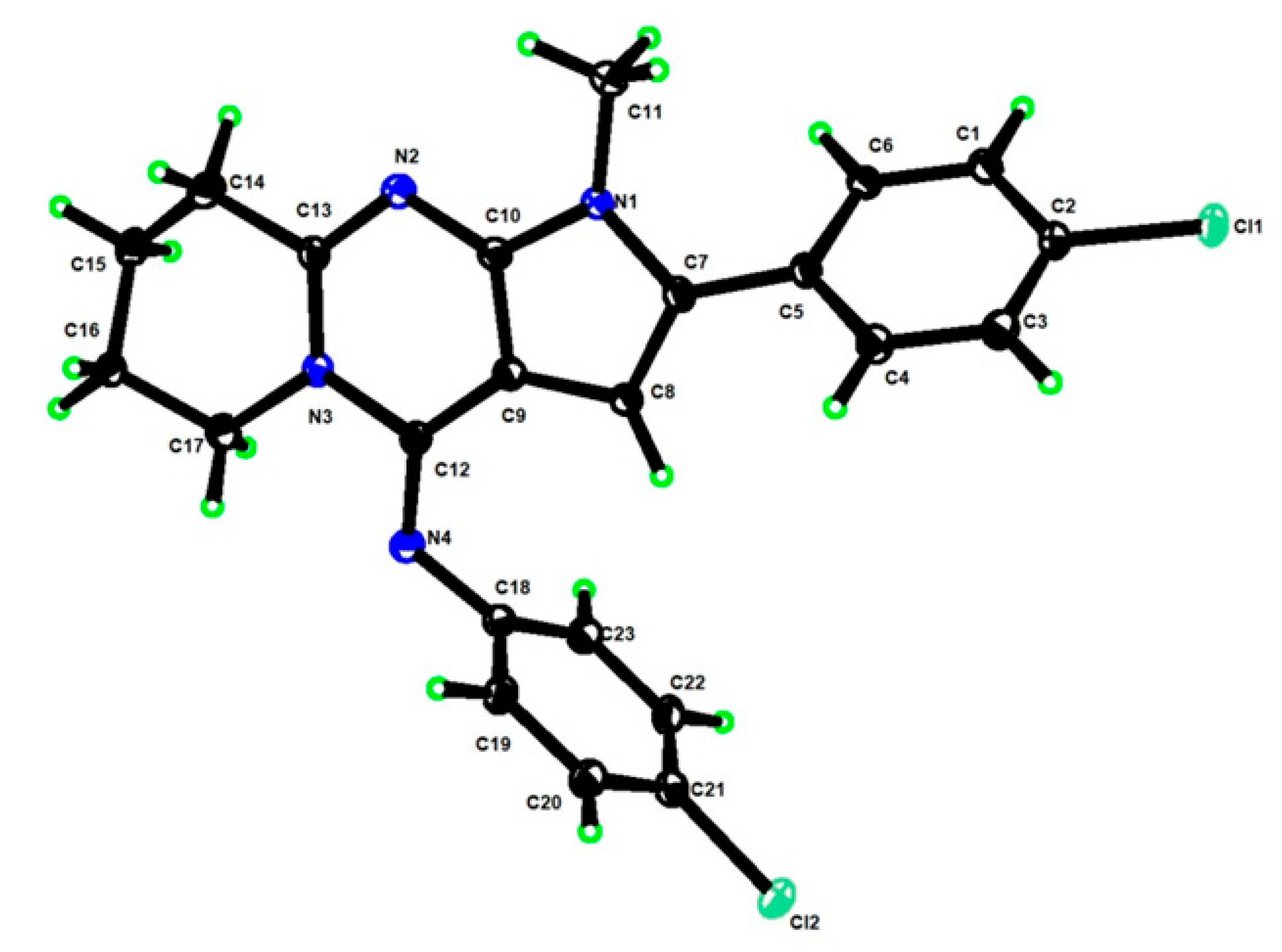
3.1. Chemistry
3.1.1. Materials and Methods
3.1.2. General Synthetic Procedures for 6a and 6b
3.1.3. Synthetic Procedure for the Pyrrolo[2,3-d]pyrimidines 8a–j
3.1.4. Synthetic Procedure for the Pyrrolo[2,3-d]pyrimidines 10a–f
3.2. Biology
Cytotoxic Activity Assay
3.3. Molecular Docking
ADME Pharmacokinetic Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ahmed, N.M.; Youns, M.M.; Soltan, M.K.; Said, A.M. Design, synthesis, molecular modeling and antitumor evaluation of novel indolyl-pyrimidine derivatives with EGFR inhibitory activity. Molecules 2021, 26, 1838. [Google Scholar] [CrossRef] [PubMed]
- Ruzi, Z.; Bozorov, K.; Nie, L.; Zhao, J.; Akber Aisa, H. Discovery of novel (E)-1-methyl-9-(3-methylbenzylidene)-6,7,8,9-tetrahydropyrazolo[3,4-d]pyrido[1,2-a]pyrimidin-4(1H)-one as DDR2 kinase inhibitor: Synthesis, molecular docking, and anticancer properties. Bioorg. Chem. 2023, 135, 106506. [Google Scholar] [CrossRef]
- Murtazaeva, Z.; Nasrullaev, A.; Buronov, A.; Gaybullaev, S.; Nie, L.; Numonov, S.; Khushnazarov, Z.; Turgunov, D.; Kuryazov, R.; Zhao, J.; et al. Imidazole Hybrids: A Privileged Class of Heterocycles in Medicinal Chemistry with New Insights into Anticancer Activity. Molecules 2025, 30, 2245. [Google Scholar] [CrossRef]
- Beloueche-Babari, M.; Casals Galobart, T.; Delgado-Goni, T.; Wantuch, S.; Parkes, H.G.; Tandy, D.; Harker, J.A.; Leach, M.O. Monocarboxylate transporter 1 blockade with AZD3965 inhibits lipid biosynthesis and increases tumour immune cell infiltration. Br. J. Cancer 2020, 122, 895–903. [Google Scholar] [CrossRef]
- Guan, X.; Rodriguez-Cruz, V.; Morris, M.E. Cellular Uptake of MCT1 Inhibitors AR-C155858 and AZD3965 and Their Effects on MCT-Mediated Transport of L-Lactate in Murine 4T1 Breast Tumor Cancer Cells. AAPS J. 2019, 21, 13. [Google Scholar] [CrossRef]
- Polanski, R.; Hodgkinson, C.L.; Fusi, A.; Nonaka, D.; Priest, L.; Kelly, P.; Trapani, F.; Bishop, P.W.; White, A.; Critchlow, S.E.; et al. Activity of the monocarboxylate transporter 1 inhibitor azd3965 in small cell lung cancer. Clin. Cancer Res. 2014, 20, 926–937. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.; Antunes, B.; Batista, A.; Pinto-Ribeiro, F.; Baltazar, F.; Afonso, J. In vivo anticancer activity of azd3965: A systematic review. Molecules 2022, 27, 181. [Google Scholar] [CrossRef]
- Sand, A.; Piacsek, M.; Donohoe, D.L.; Duffin, A.T.; Riddell, G.T.; Sun, C.; Tang, M.; Rovin, R.A.; Tjoe, J.A.; Yin, J. WEE1 inhibitor, AZD1775, overcomes trastuzumab resistance by targeting cancer stem-like properties in HER2-positive breast cancer. Cancer Lett. 2020, 472, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Wang, Y.; Hu, M.-Q.; Li, H.; Chen, X.; Qiang, G.; Sun, Y.; Zhu, Y.; Li, B. Discovery of pyrrolo[2,3-d]pyrimidine-based molecules as a Wee1 inhibitor template. Biorg. Med. Chem. Lett. 2022, 75, 128973. [Google Scholar] [CrossRef]
- Ozates, N.P.; Soğutlu, F.; Lermioglu Erciyas, F.; Demir, B.; Gunduz, C.; Shademan, B.; Avci, C.B. Effects of rapamycin and AZD3463 combination on apoptosis, autophagy, and cell cycle for resistance control in breast cancer. Life Sci. 2021, 264, 118643. [Google Scholar] [CrossRef]
- Goker Bagca, B.; Ozates, N.P.; Asik, A.; Caglar, H.O.; Gunduz, C.; Biray Avci, C. Temozolomide treatment combined with AZD3463 shows synergistic effect in glioblastoma cells. Biochem. Biophys. Res. Commun. 2020, 533, 1497–1504. [Google Scholar] [CrossRef] [PubMed]
- Kolinsky, M.P.; Rescigno, P.; Bianchini, D.; Zafeiriou, Z.; Mehra, N.; Mateo, J.; Michalarea, V.; Riisnaes, R.; Crespo, M.; Figueiredo, I.; et al. A phase I dose-escalation study of enzalutamide in combination with the AKT inhibitor AZD5363 (capivasertib) in patients with metastatic castration-resistant prostate cancer. Ann. Oncol. 2020, 31, 619–625. [Google Scholar] [CrossRef] [PubMed]
- Toren, P.; Kim, S.; Cordonnier, T.; Crafter, C.; Davies, B.R.; Fazli, L.; Gleave, M.E.; Zoubeidi, A. Combination AZD5363 with Enzalutamide Significantly Delays Enzalutamide-resistant Prostate Cancer in Preclinical Models. Eur. Urol. 2015, 67, 986–990. [Google Scholar] [CrossRef] [PubMed]
- Hong, C.R.; Buckley, C.D.; Wong, W.W.; Anekal, P.V.; Dickson, B.D.; Bogle, G.; Hicks, K.O.; Hay, M.P.; Wilson, W.R. Radiosensitisation of SCCVII tumours and normal tissues in mice by the DNA-dependent protein kinase inhibitor AZD7648. Radiother. Oncol. 2022, 166, 162–170. [Google Scholar] [CrossRef]
- Fok, J.H.L.; Ramos-Montoya, A.; Vazquez-Chantada, M.; Wijnhoven, P.W.G.; Follia, V.; James, N.; Farrington, P.M.; Karmokar, A.; Willis, S.E.; Cairns, J.; et al. AZD7648 is a potent and selective DNA-PK inhibitor that enhances radiation, chemotherapy and olaparib activity. Nat. Commun. 2019, 10, 5065. [Google Scholar] [CrossRef]
- Goldberg, F.W.; Finlay, M.R.V.; Ting, A.K.T.; Beattie, D.; Lamont, G.M.; Fallan, C.; Wrigley, G.L.; Schimpl, M.; Howard, M.R.; Williamson, B.; et al. The Discovery of 7-Methyl-2-[(7-methyl[1,2,4]triazolo[1,5- a]pyridin-6-yl)amino]-9-(tetrahydro-2 H-pyran-4-yl)-7,9-dihydro-8 H-purin-8-one (AZD7648), a Potent and Selective DNA-Dependent Protein Kinase (DNA-PK) Inhibitor. J. Med. Chem. 2020, 63, 3461–3471. [Google Scholar] [CrossRef]
- Wu, Q.; Hua, A.; Sun, Y.; Ma, C.; Tian, W.; Huang, C.; Yu, H.; Jiao, P.; Wang, S.; Tong, H.; et al. Determination and pharmacokinetic study of AZD-3759 in rat plasma by ultra performance liquid chromatography with triple quadrupole mass spectrometer. Thorac. Cancer 2018, 9, 1383–1389. [Google Scholar] [CrossRef]
- Li, W.; He, X.; Liu, H.; Zhu, J.; Zhang, H.M. Successful treatment after toxic epidermal necrolysis induced by AZD-9291 in a patient with non-small cell lung cancer: A case report. World J. Clin. Cases 2021, 9, 8846–8851. [Google Scholar] [CrossRef]
- Seela, F.; Budow, S.; Peng, X. 7-Deazapurine (Pyrrolo[2,3-d]pyrimidine) 2’-Deoxyribonucleosides: Syntheses and Transformations. Curr. Org. Chem. 2012, 16, 161–223. [Google Scholar] [CrossRef]
- Perlíková, P.; Hocek, M. Pyrrolo[2,3-d]pyrimidine (7-deazapurine) as a privileged scaffold in design of antitumor and antiviral nucleosides. Med. Res. Rev. 2017, 37, 1429–1460. [Google Scholar] [CrossRef]
- Jesumoroti, O.J.; Beteck, R.M.; Jordaan, A.; Warner, D.F.; Legoabe, L.J. Exploration of 4-aminopyrrolo[2,3-d]pyrimidine as antitubercular agents. Mol. Divers. 2023, 27, 753. [Google Scholar] [CrossRef] [PubMed]
- McCarty, R.M.; Bandarian, V. Biosynthesis of pyrrolopyrimidines. Bioorg. Chem. 2012, 43, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Ghorab, M.M.; Ragab, F.A.; Heiba, H.I.; Youssef, H.A.; El-Gazzar, M.G. Synthesis of novel pyrrole and pyrrolo[2,3-d]pyrimidine derivatives bearing sulfonamide moiety for evaluation as anticancer and radiosensitizing agents. Bioorg. Med. Chem. Lett. 2010, 20, 6316–6320. [Google Scholar] [CrossRef]
- Hassan Hilmy, K.M.; Khalifa, M.M.A.; Allah Hawata, M.A.; Aboalzeen Keshk, R.M.; El-Torgman, A.A. Synthesis of new pyrrolo[2,3-d]pyrimidine derivatives as antibacterial and antifungal agents. Eur. J. Med. Chem. 2010, 45, 5243–5250. [Google Scholar] [CrossRef]
- Mathison, C.J.N.; Yang, Y.; Nelson, J.; Huang, Z.; Jiang, J.; Chianelli, D.; Rucker, P.V.; Roland, J.; Xie, Y.F.; Epple, R.; et al. Antitarget Selectivity and Tolerability of Novel Pyrrolo[2,3- d]pyrimidine RET Inhibitors. ACS Med. Chem. Lett. 2021, 12, 1912–1919. [Google Scholar] [CrossRef]
- Xia, Z.; Huang, R.; Zhou, X.; Chai, Y.; Chen, H.; Ma, L.; Yu, Q.; Li, Y.; Li, W.; He, Y. The synthesis and bioactivity of pyrrolo[2,3-d]pyrimidine derivatives as tyrosine kinase inhibitors for NSCLC cells with EGFR mutations. Eur. J. Med. Chem. 2021, 224, 113711. [Google Scholar] [CrossRef]
- Gill, C.H.; Chate, A.V.; Shinde, G.Y.; Sarkate, A.P.; Tiwari, S.V. One-pot, four-component synthesis and SAR STUDIES of spiro[pyrimido[5,4-b]quinoline-10,5′-pyrrolo[2,3-d]pyrimidine] derivatives catalyzed by β-cyclodextrin in water as potential anticancer agents. Res. Chem. Intermed. 2018, 44, 4029–4043. [Google Scholar] [CrossRef]
- Al-Awar, R.; Isaak, M.; Chau, A.M.; Mamai, A.; Watson, I.; Poda, G.; Subramanian, P.; Wilson, B.; Uehling, D. Tricyclic inhibitors of the BCL6 BTB domain protein-protein interaction and uses thereof. WO2019119145, 27 June 2019. [Google Scholar]
- Song, B.; Nie, L.; Bozorov, K.; Kuryazov, R.; Zhao, J.; Aisa, H.A. Design, combinatorial synthesis and cytotoxic activity of 2-substituted furo[2,3-d]pyrimidinone and pyrrolo[2,3-d]pyrimidinone library. Mol. Divers. 2023, 27, 1767. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.; Nie, L.; Niu, C.; Mamatjan, A.; Bozorov, K.; Zhao, J.; Aisa, H.A. Synthesis and Biological Activities of Dihydrooxazolo[5,4-d]-pyrrolo[1,2-a]pyrimidinones. Chin. J. Org. Chem. 2022, 42, 543–556. [Google Scholar] [CrossRef]
- Liu, F.; Hou, X.; Nie, L.F.; Bozorov, K.; Decker, M.; Huang, G. A Convenient One-pot Synthesis of 2,3-Disubstituted Thieno[2,3-d]pyrimidin-4(3H)-ones from 2H-Thieno[2,3-d][1,3]oxazine-2,4(1H)-diones, Aromatic Aldehydes and Amines. SynOpen 2018, 2, 0207–0212. [Google Scholar] [CrossRef]
- Nasrullaev, A.; Bozorov, K.; Bobakulov, K.; Zhao, J.; Nie, L.F.; Turgunov, K.K.; Elmuradov, B.; Aisa, H.A. Synthesis, characterization, and antimicrobial activity of novel hydrazone-bearing tricyclic quinazolines. Res. Chem. Intermed. 2019, 45, 2287–2300. [Google Scholar] [CrossRef]
- Bozorov, K.; Zhao, J.Y.; Aisa, H.A. Recent advances in ipso-nitration reactions. ARKIVOC 2016, 2017, 41–66. [Google Scholar] [CrossRef]
- Elmuradov, B.Z.; Bozorov, K.A.; Shakhidoyatov, K.M. Thieno[2,3-d]pyrimidin-4-ones 1. Condensation of 2,3-dimethyl- and 2,3-tri-, 2,3-tetra-, and 2,3-pentamethylene-7,8-dihydro-pyrrolo[1,2-a]thieno[2,3-d]pyriminidin-4(6H)-ones with aromatic aldehydes and furfural. Chem. Heterocycl. Compd. 2011, 46, 1393–1399. [Google Scholar] [CrossRef]
- Song, B.; Nie, L.; Bozorov, K.; Kuryazov, R.; Aisa, H.A.; Zhao, J. Parallel synthesis of condensed pyrimidine-thiones and their antitumor activities. Res. Chem. Intermed. 2023, 49, 1327–1348. [Google Scholar] [CrossRef]
- Zeng, Y.; Nie, L.; Liu, L.; Bozorov, K.; Zhao, J. Design, synthesis, biological evaluation of a new tricyclicthiazolopy-rimidinone derivatives as acetylcholinesterase inhibitors. J. Heterocycl. Chem. 2024, 61, 1542–1553. [Google Scholar] [CrossRef]
- Lu, T.; Nie, L.; Tang, D.; Bozorov, K.; Zhao, J.; Aisa, H.A. Synthesis of tricyclic pyrazolopyrimidine arylidene ester derivatives and their cytotoxic and molecular docking evaluations. J. Heterocycl. Chem. 2024, 61, 651–668. [Google Scholar] [CrossRef]
- Kuryazov, R.S.; Mukhamedov, N.S.; Dushamov, D.A.; Okmanov, R.Y.; Shakhidoyatov, K.M.; Tashkhodzaev, B. Quinazolines. 3*. synthesis of 6-bromo-8-chloro- sulfonylquinazoline- 2,4(1H,3H)-dione and its interaction with nucleophilic reagents. Chem. Heterocycl. Compd. 2010, 46, 585–591. [Google Scholar] [CrossRef]
- Turgunov, D.; Nie, L.; Nasrullaev, A.; Murtazaeva, Z.; Wang, B.; Kholmurodova, D.; Kuryazov, R.; Zhao, J.; Bozorov, K.; Aisa, H.A. Synthesis of Novel 7-Phenyl-2,3-Dihydropyrrolo[2,1-b]Quinazolin-9(1H)-ones as Cholinesterase Inhibitors Targeting Alzheimer’s Disease Through Suzuki–Miyaura Cross-Coupling Reaction. Molecules 2025, 30, 2791. [Google Scholar] [CrossRef]
- Zhao, Y.; Rong, H.; Bozorov, K.; Zhang, X.; Song, B.; Liu, W. [4 + 2] Cycloaddition of α-bromotrifluoromethylhydrazone with alkenes: Synthesis of trifluoromethyltetrahydropyridazines. RSC Adv. 2025, 15, 19417–19420. [Google Scholar] [CrossRef]
- Zeng, Y.; Nie, L.; Liu, L.; Niu, C.; Li, Y.; Bozorov, K.; Zhao, J.; Shen, J.; Aisa, H.A. Design, synthesis, in vitro evaluation of a new pyrrolo[1,2-a]thiazolo[5,4-d]pyrimidinone derivatives as cholinesterase inhibitors against Alzheimer’s disease. J. Heterocycl. Chem. 2022, 59, 1086–1101. [Google Scholar] [CrossRef]
- Guo, H.; Nie, L.; Bozorov, K.; Aisa, H.A.; Zhao, J. Synthesis and Antitumor Activity of Novel Linear Tricyclic Compounds Derived from Purine. Heterocycles 2022, 104, 1085–1097. [Google Scholar]
- Zhong, H.-J.; Leung, K.-H.; Lin, S.; Chan, D.S.-H.; Han, Q.-B.; Chan, S.L.-F.; Ma, D.-L.; Leung, C.-H. Discovery of deoxyvasicinone derivatives as inhibitors of NEDD8-activating enzyme. Methods 2015, 71, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Jaouen, J.; Bailly, C. Alkaloids from Mackinlaya species and synthetic mackinazolinone derivatives: An overview. Biorg. Med. Chem. 2025, 117, 118018. [Google Scholar] [CrossRef]
- Adel, M.; Serya, R.A.T.; Lasheen, D.S.; Abouzid, K.A.M. Pyrrolopyrimidine, A Multifaceted Scaffold in Cancer Targeted Therapy. Drug Res (Stuttg) 2018, 68, 485–498. [Google Scholar] [CrossRef] [PubMed]
- Pathania, S.; Rawal, R.K. Pyrrolopyrimidines: An update on recent advancements in their medicinal attributes. Eur. J. Med. Chem. 2018, 157, 503–526. [Google Scholar] [CrossRef] [PubMed]
- Paez, J.G.; Jänne, P.A.; Lee, J.C.; Tracy, S.; Greulich, H.; Gabriel, S.; Herman, P.; Kaye, F.J.; Lindeman, N.; Boggon, T.J.; et al. EGFR Mutations in Lung Cancer: Correlation with Clinical Response to Gefitinib Therapy. Science 2004, 304, 1497–1500. [Google Scholar] [CrossRef]
- Buchdunger, E.; O.’Reilley, T.; Wood, J. Pharmacology of imatinib (STI571). Eur. J. Cancer 2002, 38, S28–S36. [Google Scholar] [CrossRef]
- Charette, A.B.; Grenon, M. Mild method for the synthesis of amidines by the electrophilic activation of amides. Tetrahedron Lett. 2000, 41, 1677–1680. [Google Scholar] [CrossRef]
- Ochiai, M.; Naito, M.; Miyamoto, K.; Hayashi, S.; Nakanishi, W. Imination of Sulfides and Sulfoxides with Sulfonylimino-λ3-Bromane under Mild, Metal-Free Conditions. Eur. J. 2010, 16, 8713–8718. [Google Scholar] [CrossRef]
- Vala, N.; Joshi, P.A.; Mishra, M. Catalytic activity of Mg–Al hydrotalcites and derived mixed oxides for imination reactions via an oxidative-dehydrogenation mechanism. New, J. Chem. 2020, 44, 8859–8868. [Google Scholar] [CrossRef]
- Devi Priya, D.; Lakshman, C.; Roopan, S.M. A review on various aspects of organic synthesis using Comins’ reagent. Mol. Divers. 2022, 26, 691–716. [Google Scholar] [CrossRef]
- Charette, A.B.; Mathieu, S.; Martel, J. Electrophilic Activation of Lactams with Tf2O and Pyridine: Expedient Synthesis of (±)-Tetraponerine T4. Org. Lett. 2005, 7, 5401–5404. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, D.; Bauer, A.; Lemmerer, M.; Maulide, N. Amide activation: An emerging tool for chemoselective synthesis. Chem. Soc. Rev. 2018, 47, 7899–7925. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Ash, J.; Kang, J.Y. Tf2O-Promoted Activating Strategy of Phosphate Analogues: Synthesis of Mixed Phosphates and Phosphinate. Org. Lett. 2018, 20, 4938–4941. [Google Scholar] [CrossRef] [PubMed]
- Lorpaiboon, W.; Bovonsombat, P. Halogen bond-induced electrophilic aromatic halogenations. Org. Biomol. Chem. 2021, 19, 7518–7534. [Google Scholar] [CrossRef]
- Coelho, N.M.; Wang, A.; McCulloch, C.A. Discoidin domain receptor 1 interactions with myosin motors contribute to collagen remodeling and tissue fibrosis. Biochim. Biophys. Acta 2019, 1866, 118510. [Google Scholar] [CrossRef]
- Mariadoss, A.V.A.; Wang, C.-Z. Exploring the Cellular and Molecular Mechanism of Discoidin Domain Receptors (DDR1 and DDR2) in Bone Formation, Regeneration, and Its Associated Disease Conditions. Int. J. Mol. Sci. 2023, 24, 14895. [Google Scholar] [CrossRef]
- Liu, L.; Hussain, M.; Luo, J.; Duan, A.; Chen, C.; Tu, Z.; Zhang, J. Synthesis and biological evaluation of novel dasatinib analogues as potent DDR1 and DDR2 kinase inhibitors. Chem. Biol. Drug Des. 2017, 89, 420–427. [Google Scholar] [CrossRef]
- Tan, X.; Li, C.; Yang, R.; Zhao, S.; Li, F.; Li, X.; Chen, L.; Wan, X.; Liu, X.; Yang, T.; et al. Discovery of Pyrazolo[3,4-d]pyridazinone Derivatives as Selective DDR1 Inhibitors via Deep Learning Based Design, Synthesis, and Biological Evaluation. J. Med. Chem. 2022, 65, 103–119. [Google Scholar] [CrossRef]
- Kuhn, B.; Ritter, M.; Benz, J.; Kocer, B.; Sarie, J.C.; Hochstrasser, R.; Rudolph, M.G.; Kadono, S.; Matsuura, T.; Murata, T.; et al. Novel potent and highly selective DDR1 inhibitors from integrated lead finding. Med. Chem. Res. 2023, 32, 1400–1425. [Google Scholar] [CrossRef]
- Sengupta, S.; Maji, L.; Das, P.K.; Teli, G.; Nag, M.; Khan, N.; Haque, M.; Matada, G.S.P. Explanatory review on DDR inhibitors: Their biological activity, synthetic route, and structure–activity relationship. Mol. Divers. 2025; Online ahead of print. [Google Scholar] [CrossRef]
- Ramachandran, B.; Kesavan, S.; Rajkumar, T. Molecular modeling and docking of small molecule inhibitors against NEK2. Bioinformation 2016, 12, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Panwar, U.; Singh, S.K. Atom-based 3D-QSAR, molecular docking, DFT, and simulation studies of acylhydrazone, hydrazine, and diazene derivatives as IN-LEDGF/p75 inhibitors. Struct. Chem. 2021, 32, 337–352. [Google Scholar] [CrossRef]
- Nada, H.; Lee, K.; Gotina, L.; Pae, A.N.; Elkamhawy, A. Identification of novel discoidin domain receptor 1 (DDR1) inhibitors using E-pharmacophore modeling, structure-based virtual screening, molecular dynamics simulation and MM-GBSA approaches. Comput. Biol. Med. 2022, 142, 105217. [Google Scholar] [CrossRef] [PubMed]
- Ginex, T.; Madruga, E.; Martinez, A.; Gil, C. MBC and ECBL libraries: Outstanding tools for drug discovery. Front. Pharmacol. 2023, 14, 1244317. [Google Scholar] [CrossRef]
- Pollastri, M.P. Overview on the Rule of Five. Curr. Protoc. Pharmacol. 2010, 49, 9.12.1–9.12.8. [Google Scholar] [CrossRef]
- Banks, W.A. The blood-brain barrier: Connecting the gut and the brain. Regul. Pept. 2008, 149, 11–14. [Google Scholar] [CrossRef] [PubMed]
- Yusuf, A.J.; Abdullahi, M.I.; Nasir, I.; Yunusa, A.; Musa, A.M.; Muhammad, A.A.; Adegboyega, A.E. Identification of possible antimalarial constituent(s) from the leaves of Ochna kibbiensis: A phytochemical, in vivo and in silico approaches. Phytomedicine Plus 2025, 5, 100764. [Google Scholar] [CrossRef]
- Almogaddam, M.A.; Shoaib, T.H.; Mohamed, S.G.A.; Mohamed, G.A.; Ibrahim, S.R.M.; Hussein, H.G.A.; Sindi, I.A.; Alzain, A.A. Computational screening identifies depsidones as promising Aurora A kinase inhibitors: Extra precision docking and molecular dynamics studies. Netw. Model. Anal. Health Inform. Bioinform. 2024, 13, 17. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||||
|---|---|---|---|---|---|---|---|
| Compd. | R1 | R2 | X | n a | Cell Lines (IC50, µM) | ||
| HeLa | MCF-7 | HT-29 | |||||
| 8a | Cl | H | − | 1 | ≥50 | ≥50 | 19.22 ± 0.91 |
| 8b | Cl | F | − | 1 | ≥50 | ≥50 | ≥50 |
| 8c | Cl | Cl | − | 1 | ≥50 | ≥50 | ≥50 |
| 8d | Cl | Br | − | 1 | ≥50 | ≥50 | ≥50 |
| 8e | Cl | CF3 | − | 1 | ≥50 | ≥50 | ≥50 |
| 8f | Br | H | − | 2 | ≥50 | ≥50 | 4.55 ± 0.23 |
| 8g | Br | F | − | 2 | ≥50 | ≥50 | 4.01 ± 0.20 |
| 8h | Br | Cl | − | 2 | ≥50 | ≥50 | ≥50 |
| 8i | Br | Br | − | 2 | ≥50 | ≥50 | ≥50 |
| 8j | Br | CF3 | − | 2 | ≥50 | ≥50 | ≥50 |
| 10a | Cl | − | Cl | 1 | 22.77 ± 0.98 | 27.52 ± 1.07 | 24.66 ± 1.13 |
| 10b | Cl | − | Br | 1 | ≥50 | 20.05 ± 0.93 | ≥50 |
| 10c | Cl | − | I | 1 | ≥50 | ≥50 | 34.16 ± 1.60 |
| 10d | Br | − | Cl | 2 | ≥50 | ≥50 | ≥50 |
| 10e | Br | − | Br | 2 | ≥50 | ≥50 | 15.97 ± 0.75 |
| 10f | Br | − | I | 2 | ≥50 | ≥50 | 19.04 ± 0.92 |
| DOX | − | − | − | − | 0.94 ± 0.016 | 0.18 ± 0.011 | 0.82 ± 0.029 |
| Compounds | IC50 (±SD, µM) | IC50 (HT-29), µM | SIHT-29 |
|---|---|---|---|
| 8f | 195 ± 0.07 | 4.55 ± 0.23 | 42.86 |
| 8g | 169 ± 0.10 | 4.01 ± 0.20 | 42.14 |
| Parameters | 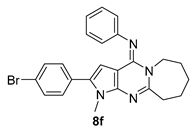 | 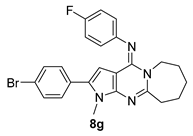 |
|---|---|---|
| Molecular weight a | 447.376 | 465.367 |
| Rule of five b | 1 | 1 |
| QPlogPo/w c | 6.947 | 7.186 |
| QPlogS d | −8.349 | −8.731 |
| QPlogHERG e | −6.218 | −6.094 |
| QPPCaco f | 7667.03 | 7667.03 |
| QPlogBB g | 0.513 | 0.627 |
| Human oral absorption h | 1 | 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, B.; Murtazaeva, Z.; Nie, L.; Kuryazov, R.; Gaybullaev, S.; Niu, C.; Bozorov, K.; Aisa, H.A.; Zhao, J. Pyrrolopyrimidines: Design, Synthesis and Antitumor Properties of Novel Tricyclic Pyrrolo [2,3-d]pyrimidine Derivatives. Molecules 2025, 30, 2917. https://doi.org/10.3390/molecules30142917
Song B, Murtazaeva Z, Nie L, Kuryazov R, Gaybullaev S, Niu C, Bozorov K, Aisa HA, Zhao J. Pyrrolopyrimidines: Design, Synthesis and Antitumor Properties of Novel Tricyclic Pyrrolo [2,3-d]pyrimidine Derivatives. Molecules. 2025; 30(14):2917. https://doi.org/10.3390/molecules30142917
Chicago/Turabian StyleSong, Buer, Zarifa Murtazaeva, Lifei Nie, Rustamkhon Kuryazov, Shukhrat Gaybullaev, Chao Niu, Khurshed Bozorov, Haji Akber Aisa, and Jiangyu Zhao. 2025. "Pyrrolopyrimidines: Design, Synthesis and Antitumor Properties of Novel Tricyclic Pyrrolo [2,3-d]pyrimidine Derivatives" Molecules 30, no. 14: 2917. https://doi.org/10.3390/molecules30142917
APA StyleSong, B., Murtazaeva, Z., Nie, L., Kuryazov, R., Gaybullaev, S., Niu, C., Bozorov, K., Aisa, H. A., & Zhao, J. (2025). Pyrrolopyrimidines: Design, Synthesis and Antitumor Properties of Novel Tricyclic Pyrrolo [2,3-d]pyrimidine Derivatives. Molecules, 30(14), 2917. https://doi.org/10.3390/molecules30142917