
Unveiling Vibrational Couplings in Model Peptides in Solution by a Theoretical Approach

 ,
,  , ,
, ,  ,
,  and
and 
Abstract
1. Introduction
2. Results and Discussion
2.1. Structural Characterization in Gas and Aqueous Solution from AIMD Simulations
2.2. Vibrational Analysis from AIMD Trajectories in Gas-Phase
2.3. Vibrational Analysis from AIMD Trajectories in Aqueous Solution
2.4. Hessian-Based vs. AIMD-Based Vibrational Analysis
3. Materials and Methods
3.1. Hessian-Based Vibrational Analysis
3.2. Generalized Normal Modes and Time-Resolved Vibrational Analysis
3.3. Computational Details
3.3.1. Hessian-Based Vibrational Analysis
3.3.2. Ab-Initio Molecular Dynamics Simulations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| A | Acceptor |
| AcAlaOMe | Methyl-2-Acetamidopropanoate |
| ADMP | Atom-centered density matrix propagation |
| C-PCM | Conductor-like polarizable continuum model |
| D | Donor |
| DFT | Density functional theory |
| FSRS | Femtosecond Stimulated Raman Spectroscopy |
| FT | Fourier transform |
| GNM | Generalized normal modes |
| IR | Infrared |
| MD | Molecular dynamics |
| MM | Molecular mechanics |
| NM | Normal modes |
| NPBC | Nonperiodic boundary conditions |
| PT2 | Perturbation theory to second order |
| QM | Quantum mechanics |
| RDF | Radial distribution function |
| tNMA | trans-N-Methyl-Acetamide |
| VPT2 | Second-order vibrational perturbation theory |
| WT | Wavelet transform |
References
- Meanwell, N.A. Improving Drug Design: An Update on Recent Applications of Efficiency Metrics, Strategies for Replacing Problematic Elements, and Compounds in Nontraditional Drug Space. Chem. Res. Toxicol. 2016, 29, 564–616. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Yang, C.; Chen, Y.; Ma, L.; Huang, K. Rational design of hybrid peptides: A novel drug design approach. Curr. Med. Sci. 2019, 39, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Huggins, D.J.; Sherman, W.; Tidor, B. Rational Approaches to Improving Selectivity in Drug Design. J. Med. Chem. 2012, 55, 1424–1444. [Google Scholar] [CrossRef]
- Chang, L.; Mondal, A.; Perez, A. Towards rational computational peptide design. Front. Bioinform. 2022, 2, 1046493. [Google Scholar] [CrossRef] [PubMed]
- Yaraghi, N.A.; Kisailus, D. Biomimetic Structural Materials: Inspiration from Design and Assembly. Annu. Rev. Phys. Chem. 2018, 69, 23–57. [Google Scholar] [CrossRef]
- Cao, Z.; Li, Y.; Wang, S.; Tang, B.; Guo, X.; Wang, L.; Zhao, W. Biomimetic synthesis of bis-α-substituent pyrrolidine alkaloids based on a proposed biosynthetic pathway. Tetrahedron Lett. 2016, 57, 2219–2221. [Google Scholar] [CrossRef]
- Donati, G.; De Nicola, A.; Munaò, G.; Byshkin, M.; Vertuccio, L.; Guadagno, L.; Le Goff, R.; Milano, G. Simulation of self-heating process on the nanoscale: A multiscale approach for molecular models of nanocomposite materials. Nanoscale Adv. 2020, 2, 3164–3180. [Google Scholar] [CrossRef]
- Li, T.; Li, Y.; Zhang, T. Materials, Structures, and Functions for Flexible and Stretchable Biomimetic Sensors. Accounts Chem. Res. 2019, 52, 288–296. [Google Scholar] [CrossRef]
- Huang, G.; Li, F.; Zhao, X.; Ma, Y.; Li, Y.; Lin, M.; Jin, G.; Lu, T.J.; Genin, G.M.; Xu, F. Functional and Biomimetic Materials for Engineering of the Three-Dimensional Cell Microenvironment. Chem. Rev. 2017, 117, 12764–12850. [Google Scholar] [CrossRef]
- Tuma, R. Raman spectroscopy of proteins: From peptides to large assemblies. J. Raman Spectrosc. 2005, 36, 307–319. [Google Scholar] [CrossRef]
- Benevides, J.M.; Overman, S.A.; Thomas, G.J., Jr. Raman, polarized Raman and ultraviolet resonance Raman spectroscopy of nucleic acids and their complexes. J. Raman Spectrosc. 2005, 36, 279–299. [Google Scholar] [CrossRef]
- Abramczyk, H.; Brozek-Pluska, B. Raman imaging in biochemical and biomedical applications. Diagnosis and treatment of breast cancer. Chem. Rev. 2013, 113, 5766–5781. [Google Scholar] [CrossRef] [PubMed]
- Andrushchenko, V.; Tsankov, D.; Wieser, H. Vibrational circular dichroism spectroscopy and the effects of metal ions on DNA structure. J. Mol. Struct. 2003, 661, 541–560. [Google Scholar] [CrossRef]
- Maharaj, V.; Rauk, A.; Van de Sande, J.; Wieser, H. Infrared absorption and vibrational circular dichroism spectra of selected deoxyoctanucleotides complexed with daunorubicin. J. Mol. Struct. 1997, 408, 315–318. [Google Scholar] [CrossRef]
- Gulotta, M.; Goss, D.J.; Diem, M. IR vibrational CD in model deoxyoligonucleotides: Observation of the B→Z phase transition and extended coupled oscillator intensity calcuations. Biopolym. Orig. Res. Biomol. 1989, 28, 2047–2058. [Google Scholar] [CrossRef]
- Werle, Y.; Kovermann, M. Fluorine labeling and 19F NMR spectroscopy to study biological molecules and molecular complexes. Chem. Eur. J. 2025, 31, e202402820. [Google Scholar] [CrossRef]
- Alderson, T.R.; Kay, L.E. NMR spectroscopy captures the essential role of dynamics in regulating biomolecular function. Cell 2021, 184, 577–595. [Google Scholar] [CrossRef]
- Guzmán-Embús, D.; Orrego Cardozo, M.; Vargas-Hernández, C. Genomic DNA characterization of pork spleen by Raman spectroscopy. J. Appl. Phys. 2013, 114. [Google Scholar] [CrossRef]
- Kukura, P.; McCamant, D.W.; Mathies, R.A. Femtosecond Stimulated Raman Spectroscopy. Annu. Rev. Phys. Chem. 2007, 58, 461–488. [Google Scholar] [CrossRef]
- Lynch, P.G.; Das, A.; Alam, S.; Rich, C.C.; Frontiera, R.R. Mastering femtosecond stimulated Raman spectroscopy: A practical guide. ACS Phys. Chem. Au 2023, 4, 1–18. [Google Scholar] [CrossRef]
- Frontiera, R.R.; Mathies, R.A. Femtosecond stimulated Raman spectroscopy. Laser Photonics Rev. 2011, 5, 102–113. [Google Scholar] [CrossRef]
- Frontiera, R.R.; Fang, C.; Dasgupta, J.; Mathies, R.A. Probing structural evolution along multidimensional reaction coordinates with femtosecond stimulated Raman spectroscopy. Phys. Chem. Chem. Phys. 2012, 14, 405–414. [Google Scholar] [CrossRef]
- Rubtsov, I.V.; Wang, J.; Hochstrasser, R.M. Vibrational Coupling between Amide-I and Amide-A Modes Revealed by Femtosecond Two Color Infrared Spectroscopy. J. Phys. Chem. A 2003, 107, 3384–3396. [Google Scholar] [CrossRef]
- Hayashi, T.; Mukamel, S. Two-dimensional vibrational lineshapes of amide III, II, I and A bands in a helical peptide. J. Mol. Liq. 2008, 141, 149–154. [Google Scholar] [CrossRef] [PubMed]
- Baiz, C.R.; Błasiak, B.; Bredenbeck, J.; Cho, M.; Choi, J.H.; Corcelli, S.A.; Dijkstra, A.G.; Feng, C.J.; Garrett-Roe, S.; Ge, N.H.; et al. Vibrational spectroscopic map, vibrational spectroscopy, and intermolecular interaction. Chem. Rev. 2020, 120, 7152–7218. [Google Scholar] [CrossRef]
- Ghosh, A.; Ostrander, J.S.; Zanni, M.T. Watching proteins wiggle: Mapping structures with two-dimensional infrared spectroscopy. Chem. Rev. 2017, 117, 10726–10759. [Google Scholar] [CrossRef] [PubMed]
- Saurabh, P.; Mukamel, S. Two-dimensional infrared spectroscopy of vibrational polaritons of molecules in an optical cavity. J. Chem. Phys. 2016, 144. [Google Scholar] [CrossRef]
- Woys, A.M.; Almeida, A.M.; Wang, L.; Chiu, C.C.; McGovern, M.; De Pablo, J.J.; Skinner, J.L.; Gellman, S.H.; Zanni, M.T. Parallel β-sheet vibrational couplings revealed by 2D IR spectroscopy of an isotopically labeled macrocycle: Quantitative benchmark for the interpretation of amyloid and protein infrared spectra. J. Am. Chem. Soc. 2012, 134, 19118–19128. [Google Scholar] [CrossRef]
- Maekawa, H.; Ge, N.H. Comparative study of electrostatic models for the amide-I and-II modes: Linear and two-dimensional infrared spectra. J. Phys. Chem. B 2010, 114, 1434–1446. [Google Scholar] [CrossRef]
- Woys, A.M.; Mukherjee, S.S.; Skoff, D.R.; Moran, S.D.; Zanni, M.T. A strongly absorbing class of non-natural labels for probing protein electrostatics and solvation with FTIR and 2D IR spectroscopies. J. Phys. Chem. B 2013, 117, 5009–5018. [Google Scholar] [CrossRef]
- Hunt, N.T. Using 2D-IR spectroscopy to measure the structure, dynamics, and intermolecular interactions of proteins in H2O. Accounts Chem. Res. 2024, 57, 685–692. [Google Scholar] [CrossRef] [PubMed]
- Nandi, N.; Roy, S.; Bagchi, B. Ultrafast solvation dynamics in water: Isotope effects and comparison with experimental results. J. Chem. Phys. 1995, 102, 1390–1397. [Google Scholar] [CrossRef]
- Nandi, N.; Bhattacharyya, K.; Bagchi, B. Dielectric relaxation and solvation dynamics of water in complex chemical and biological systems. Chem. Rev. 2000, 100, 2013–2046. [Google Scholar] [CrossRef]
- Bagchi, B. Molecular theory of nonpolar solvation dynamics. J. Chem. Phys. 1994, 100, 6658–6664. [Google Scholar] [CrossRef]
- Fleming, G.R.; Cho, M. Chromophore-solvent dynamics. Annu. Rev. Phys. Chem. 1996, 47, 109–134. [Google Scholar] [CrossRef]
- Bagchi, B. Water dynamics in the hydration layer around proteins and micelles. Chem. Rev. 2005, 105, 3197–3219. [Google Scholar] [CrossRef]
- Miyazawa, T.; Shimanouchi, T.; Mizushima, S.I. Normal vibrations of N-methylacetamide. J. Chem. Phys. 1958, 29, 611–616. [Google Scholar] [CrossRef]
- Tsuboi, M.; Onishi, T.; Nakagawa, I.; Shimanouchi, T.; Mizushima, S.I. Assignments of the vibrational frequencies of glycine. Spectrochim. Acta 1958, 12, 253–261. [Google Scholar] [CrossRef]
- Gómez, S.; Bottari, C.; Egidi, F.; Giovannini, T.; Rossi, B.; Cappelli, C. Amide Spectral Fingerprints are Hydrogen Bonding-Mediated. J. Phys. Chem. Lett. 2022, 13, 6200–6207. [Google Scholar] [CrossRef]
- Candelaresi, M.; Ragnoni, E.; Cappelli, C.; Corozzi, A.; Lima, M.; Monti, S.; Mennucci, B.; Nuti, F.; Papini, A.M.; Foggi, P. Conformational Analysis of Gly–Ala–NHMe in D2O and DMSO Solutions: A Two-Dimensional Infrared Spectroscopy Study. J. Phys. Chem. B 2013, 117, 14226–14237. [Google Scholar] [CrossRef]
- Ganim, Z.; Chung, H.S.; Smith, A.W.; DeFlores, L.P.; Jones, K.C.; Tokmakoff, A. Amide I two-dimensional infrared spectroscopy of proteins. Accounts Chem. Res. 2008, 41, 432–441. [Google Scholar] [CrossRef] [PubMed]
- DeFlores, L.P.; Ganim, Z.; Nicodemus, R.A.; Tokmakoff, A. Amide I’- II’ 2D IR spectroscopy provides enhanced protein secondary structural sensitivity. J. Am. Chem. Soc. 2009, 131, 3385–3391. [Google Scholar] [CrossRef]
- Watson, T.M.; Hirst, J.D. Density Functional Theory Vibrational Frequencies of Amides and Amide Dimers. J. Phys. Chem. A 2002, 106, 7858–7867. [Google Scholar] [CrossRef]
- Uversky, V.N.; Permiakov, E.A. Methods in Protein Structure and Stability Analysis: Vibrational Analysis; Nova Publishers: Hauppauge, NY, USA, 2007. [Google Scholar]
- Byler, D.M.; Susi, H. Examination of the secondary structure of proteins by deconvolved FTIR spectra. Biopolym. Orig. Res. Biomol. 1986, 25, 469–487. [Google Scholar] [CrossRef]
- Torii, H.; Tasumi, M.; Mantsch, H.; Chapman, D. Infrared Spectroscopy of Biomolecules; Mantsch, H.H., Chapman, D., Eds.; Wiley-Liss: New York, NY, USA, 1996; pp. 1–18. [Google Scholar]
- Torii, H.; Tasumi, M. Model calculations on the amide-I infrared bands of globular proteins. J. Chem. Phys. 1992, 96, 3379–3387. [Google Scholar] [CrossRef]
- Jackson, M.; Haris, P.I.; Chapman, D. Fourier transform infrared spectroscopic studies of lipids, polypeptides and proteins. J. Mol. Struct. 1989, 214, 329–355. [Google Scholar] [CrossRef]
- Mukamel, S. Multidimensional femtosecond correlation spectroscopies of electronic and vibrational excitations. Annu. Rev. Phys. Chem. 2000, 51, 691–729. [Google Scholar] [CrossRef] [PubMed]
- Palmer, M.H.; Biczysko, M.; Peterson, K.A.; Stapleton, C.S.; Wells, S.P. Structural and vibrational properties of iodopentafluorobenzene: A combined Raman and infrared spectral and theoretical study. J. Phys. Chem. A 2017, 121, 7917–7924. [Google Scholar] [CrossRef]
- Bloino, J.; Baiardi, A.; Biczysko, M. Aiming at an accurate prediction of vibrational and electronic spectra for medium-to-large molecules: An overview. Int. J. Quantum Chem. 2016, 116, 1543–1574. [Google Scholar] [CrossRef]
- Crisci, L.; Coppola, F.; Petrone, A.; Rega, N. Tuning ultrafast time-evolution of photo-induced charge-transfer states: A real-time electronic dynamics study in substituted indenotetracene derivatives. J. Comput. Chem. 2024, 45, 210–221. [Google Scholar] [CrossRef]
- Coppola, F.; Hussain, M.; Zhao, J.; El-Zohry, A.M.; Pastore, M. Key role of electronic and structural properties in regulating intersystem crossing: An in-depth investigation on naphthalene-diimide triads for thermally activated delayed fluorescence applications. J. Phys. Chem. C 2024, 128, 11998–12009. [Google Scholar] [CrossRef]
- Carfora, R.; Coppola, F.; Cimino, P.; Petrone, A.; Rega, N. A Cost-Effective Computational Strategy for the Electronic Layout Characterization of a Second Generation Light-Driven Molecular Rotary Motor in Solution. J. Comput. Chem. 2025, 46, e70023. [Google Scholar] [CrossRef] [PubMed]
- Hafner, J.; Wolverton, C.; Ceder, G. Toward computational materials design: The impact of density functional theory on materials research. MRS Bull. 2006, 31, 659–668. [Google Scholar] [CrossRef]
- Perrella, F.; Coppola, F.; Rega, N.; Petrone, A. An expedited route to optical and electronic properties at finite temperature via unsupervised learning. Molecules 2023, 28, 3411. [Google Scholar] [CrossRef]
- Alberto, M.E.; De Simone, B.C.; Marino, T.; Toscano, M.; Russo, N. Chalcogen effects in the photophysical properties of Dimethylamino-1, 8-naphthalimide dyes revealed by DFT investigation. J. Phys. Chem. A 2022, 126, 5167–5172. [Google Scholar] [CrossRef]
- Perrella, F.; Petrone, A.; Rega, N. Understanding Charge Dynamics in Dense Electronic Manifolds in Complex Environments. J. Chem. Theory Comput. 2023, 19, 626–639. [Google Scholar] [CrossRef]
- Wilson, E.B.; Decius, J.C.; Cross, P.C. Molecular Vibrations: The Theory of Infrared and Raman Vibrational Spectra; Courier Corporation: North Chelmsford, MA, USA, 2012. [Google Scholar]
- Barone, V. Characterization of the potential energy surface of the HO2 molecular system by a density functional approach. J. Chem. Phys. 1994, 101, 10666–10676. [Google Scholar] [CrossRef]
- Minichino, C.; Barone, V. From concepts to algorithms for the characterization of reaction mechanisms. H2CS as a case study. J. Chem. Phys. 1994, 100, 3717–3741. [Google Scholar] [CrossRef]
- Barone, V.; Minichino, C. From concepts to algorithms for the treatment of large amplitude internal motions and unimolecular reactions. J. Mol. Struct. Theochem 1995, 330, 365–376. [Google Scholar] [CrossRef]
- Rega, N. Vibrational analysis beyond the harmonic regime from ab-initio molecular dynamics. Theor. Chem. Accounts 2006, 116, 347–354. [Google Scholar] [CrossRef]
- Marx, D.; Hutter, J. Ab initio molecular dynamics: Theory and implementation. Mod. Methods Algorithms Quantum Chem. 2000, 1, 141. [Google Scholar]
- Schlegel, H.B.; Millam, J.M.; Iyengar, S.S.; Voth, G.A.; Daniels, A.D.; Scuseria, G.E.; Frisch, M.J. Ab Initio Molecular Dynamics: Propagating the Density Matrix with Gaussian Orbitals. J. Chem. Phys. 2001, 114, 9758–9763. [Google Scholar] [CrossRef]
- Iyengar, S.S.; Schlegel, H.B.; Millam, J.M.; Voth, G.A.; Scuseria, G.E.; Frisch, M.J. Propagating the Density Matrix with Gaussian Orbitals. II. Generalizations Based on Mass-Weighting, Idempotency, Energy Conservation and Choice of Initial Conditions. J. Chem. Phys. 2001, 115, 10291–10302. [Google Scholar] [CrossRef]
- Schlegel, H.B.; Iyengar, S.S.; Li, X.; Millam, J.M.; Voth, G.A.; Scuseria, G.E.; Frisch, M.J. Ab Initio Molecular Dynamics: Propagating the Density Matrix with Gaussian Orbitals. III. Comparison with Born-Oppenheimer Dynamics. J. Chem. Phys. 2002, 117, 8694–8704. [Google Scholar] [CrossRef]
- Strachan, A. Normal modes and frequencies from covariances in molecular dynamics or Monte Carlo simulations. J. Chem. Phys. 2004, 120, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Coppola, F.; Carfora, R.; Cimino, P.; Petrone, A.; Rega, N. Tetracyanoethylene Na+/K+ radical anion coordination sites unveiled via electronic and vibrational fingerprints. Theor. Chem. Accounts 2024, 143, 77. [Google Scholar] [CrossRef]
- Daubechies, I. Ten Lectures on Wavelets; Society for Industrial and Applied Mathematics: Philadelphia, PA, USA, 1992. [Google Scholar]
- Kaiser, G.; Hudgins, L.H. A Friendly Guide to Wavelets; Springer: Boston, MA, USA, 1994; Volume 300. [Google Scholar]
- Torrence, C.; Compo, C.P. A Practical Guide to Wavelet Analysis. Bull. Am. Meteorol. Soc. 1998, 79, 61. [Google Scholar] [CrossRef]
- Charbkaew, N.; Suwanasri, T.; Bunyagul, T.; Schnettler, A. Vibration signal analysis for condition monitoring of puffer-type high-voltage circuit breakers using wavelet transform. IEEJ Trans. Electr. Electron. Eng. 2012, 7, 13–22. [Google Scholar] [CrossRef]
- Saraydemir, Ş.; Taşpınar, N.; Eroğul, O.; Kayserili, H.; Dinçkan, N. Down syndrome diagnosis based on Gabor Wavelet Transform. J. Med. Syst. 2012, 36, 3205–3213. [Google Scholar] [CrossRef]
- Bello, J.P.; Daudet, L.; Abdallah, S.; Duxbury, C.; Davies, M.; Sandler, M.B. A tutorial on onset detection in music signals. IEEE Trans. Speech Audio Process. 2005, 13, 1035–1047. [Google Scholar] [CrossRef]
- Bharath, A.A.; Ng, J. A steerable complex wavelet construction and its application to image denoising. IEEE Trans. Image Process. 2005, 14, 948–959. [Google Scholar] [CrossRef] [PubMed]
- Svensson, M.; Krüger, N.A. Mortality and economic fluctuations. J. Popul. Econ. 2012, 25, 1215–1235. [Google Scholar] [CrossRef]
- Donati, G.; Petrone, A.; Caruso, P.; Rega, N. The mechanism of a green fluorescent protein proton shuttle unveiled in the time-resolved frequency domain by excited state ab initio dynamics. Chem. Sci. 2018, 9, 1126–1135. [Google Scholar] [CrossRef]
- Chiariello, M.G.; Donati, G.; Raucci, U.; Perrella, F.; Rega, N. Structural origin and vibrational fingerprints of the ultrafast excited state proton transfer of the pyranine-acetate complex in aqueous solution. J. Phys. Chem. B 2021, 125, 10273–10281. [Google Scholar] [CrossRef]
- Coppola, F.; Cimino, P.; Perrella, F.; Crisci, L.; Petrone, A.; Rega, N. Electronic and vibrational manifold of tetracyanoethylene–chloronaphthalene charge transfer complex in solution: Insights from TD-DFT and ab initio molecular dynamics. J. Phys. Chem. A 2022, 126, 7179–7192. [Google Scholar] [CrossRef]
- Coppola, F.; Cimino, P.; Petrone, A.; Rega, N. Evidence of excited-state vibrational mode governing the photorelaxation of a charge-transfer complex. J. Phys. Chem. A 2024, 128, 1620–1633. [Google Scholar] [CrossRef] [PubMed]
- Coppola, F.; Cimino, P.; Raucci, U.; Chiariello, M.G.; Petrone, A.; Rega, N. Exploring the Franck–Condon region of a photoexcited charge transfer complex in solution to interpret femtosecond stimulated Raman spectroscopy: Excited state electronic structure methods to unveil non-radiative pathways. Chem. Sci. 2021, 12, 8058–8072. [Google Scholar] [CrossRef]
- Donati, G.; Petrone, A.; Rega, N. Multiresolution continuous wavelet transform for studying coupled solute–solvent vibrations via ab initio molecular dynamics. Phys. Chem. Chem. Phys. 2020, 22, 22645–22661. [Google Scholar] [CrossRef] [PubMed]
- Chiariello, M.G.; Raucci, U.; Coppola, F.; Rega, N. Unveiling anharmonic coupling by means of excited state ab initio dynamics: Application to diarylethene photoreactivity. Phys. Chem. Chem. Phys. 2019, 21, 3606–3614. [Google Scholar] [CrossRef]
- Papamokos, G.V.; Demetropoulos, I.N. Vibrational Frequencies of Amides and Amide Dimers: The Assessment of PW91XC Functional. J. Phys. Chem. A 2004, 108, 7291–7300. [Google Scholar] [CrossRef]
- Wang, J.; Hochstrasser, R.M. Anharmonicity of Amide Modes. J. Phys. Chem. B 2006, 110, 3798–3807. [Google Scholar] [CrossRef] [PubMed]
- Gaigeot, M.P.; Martinez, M.; Vuilleumier, R. Infrared spectroscopy in the gas and liquid phase from first principle molecular dynamics simulations: Application to small peptides. Mol. Phys. 2007, 105, 2857–2878. [Google Scholar] [CrossRef]
- Ji, Y.; Yang, X.; Ji, Z.; Zhu, L.; Ma, N.; Chen, D.; Jia, X.; Tang, J.; Cao, Y. DFT-calculated IR spectrum amide I, II, and III band contributions of N-methylacetamide fine components. ACS Omega 2020, 5, 8572–8578. [Google Scholar] [CrossRef]
- Warshel, A.; Levitt, M. Theoretical studies of enzymic reactions: Dielectric, electrostatic and steric stabilization of the carbonium ion in the reaction of lysozyme. J. Mol. Biol. 1976, 103, 227–249. [Google Scholar] [CrossRef]
- Field, M.J.; Bash, P.A.; Karplus, M. A combined quantum mechanical and molecular mechanical potential for molecular dynamics simulations. J. Comput. Chem. 1990, 11, 700–733. [Google Scholar] [CrossRef]
- Petrone, A.; Perrella, F.; Coppola, F.; Crisci, L.; Donati, G.; Cimino, P.; Rega, N. Ultrafast photo-induced processes in complex environments: The role of accuracy in excited-state energy potentials and initial conditions. Chem. Phys. Rev. 2022, 3. [Google Scholar] [CrossRef]
- Lessing, J.; Roy, S.; Reppert, M.; Baer, M.; Marx, D.; Jansen, T.L.C.; Knoester, J.; Tokmakoff, A. Identifying residual structure in intrinsically disordered systems: A 2D IR spectroscopic study of the GVGXPGVG peptide. J. Am. Chem. Soc. 2012, 134, 5032–5035. [Google Scholar] [CrossRef]
- Schweitzer-Stenner, R. Different degrees of disorder in long disordered peptides can be discriminated by vibrational spectroscopy. J. Phys. Chem. B 2013, 117, 6927–6936. [Google Scholar] [CrossRef]
- Schweitzer-Stenner, R. The combined use of amide I bands in polarized Raman, IR, and vibrational dichroism spectra for the structure analysis of peptide fibrils and disordered peptides and proteins. J. Raman Spectrosc. 2021, 52, 2479–2499. [Google Scholar] [CrossRef]
- Zheng, X.; Ni, Z.; Pei, Q.; Wang, M.; Tan, J.; Bai, S.; Shi, F.; Ye, S. Probing the Molecular Structure and Dynamics of Membrane-Bound Proteins during Misfolding Processes by Sum-Frequency Generation Vibrational Spectroscopy. ChemPlusChem 2024, 89, e202300684. [Google Scholar] [CrossRef]
- Tan, J.; Zhang, B.; Luo, Y.; Ye, S. Ultrafast Vibrational Dynamics of Membrane-Bound Peptides at the Lipid Bilayer/Water Interface. Angew. Chem. 2017, 129, 13157–13161. [Google Scholar] [CrossRef]
- Pavela, O.; Juhász, T.; Tóth, L.; Czajlik, A.; Batta, G.; Galgóczy, L.; Beke-Somfai, T. Mapping of the lipid-binding regions of the antifungal protein NFAP2 by exploiting model membranes. J. Chem. Inf. Model. 2024, 64, 6557–6569. [Google Scholar] [CrossRef]
- Myshakina, N.S.; Ahmed, Z.; Asher, S.A. Dependence of amide vibrations on hydrogen bonding. J. Phys. Chem. B 2008, 112, 11873–11877. [Google Scholar] [CrossRef] [PubMed]
- Rubtsov, I.; Wang, J.; Hochstrasser, R. Dual frequency 2D-IR of peptide amide-A and amide-I modes. J. Chem. Phys. 2003, 118, 7733–7736. [Google Scholar] [CrossRef]
- Kuznetsova, L.; Furer, V.; Maklakov, L. Infrared intensities of N-methylacetamide associates. J. Mol. Struct. 1996, 380, 23–29. [Google Scholar] [CrossRef]
- De Marco, L.; Thämer, M.; Reppert, M.; Tokmakoff, A. Direct observation of intermolecular interactions mediated by hydrogen bonding. J. Chem. Phys. 2014, 141, 034502. [Google Scholar] [CrossRef]
- Lee, S.H.; Mirkin, N.G.; Krimm, S. A quantitative anharmonic analysis of the amide A band in α-helical poly(L-alanine). Biopolymers 1999, 49, 195–207. [Google Scholar] [CrossRef]
- Mirkin, N.G.; Krimm, S. Ab Initio Vibrational Analysis of Hydrogen-Bonded trans- and cis-N-Methylacetamide. J. Am. Chem. Soc. 1991, 113, 9742–9747. [Google Scholar] [CrossRef]
- Candelaresi, M.; Pagliai, M.; Lima, M.; Righini, R. Chemical Equilibrium Probed by Two-Dimensional IR Spectroscopy: Hydrogen Bond Dynamics of Methyl Acetate in Water. J. Phys. Chem. A 2009, 113, 12783–12790. [Google Scholar] [CrossRef]
- Cai, S.; Singh, B.R. A distinct utility of the amide III infrared band for secondary structure estimation of aqueous protein solutions using partial least squares methods. Biochemistry 2004, 43, 2541–2549. [Google Scholar] [CrossRef]
- Barth, A.; Zscherp, C. What vibrations tell about proteins. Q. Rev. Biophys. 2002, 35, 369–430. [Google Scholar] [CrossRef]
- Krimm, S.; Bandekar, J. Vibrational spectroscopy and conformation of peptides, polypeptides, and proteins. Adv. Protein Chem. 1986, 38, 181–364. [Google Scholar]
- Kubelka, J.; Keiderling, T.A. Ab Initio Calculation of Amide Carbonyl Stretch Vibrational Frequencies in Solution with Modified Basis Sets. 1. N-Methyl Acetamide. J. Phys. Chem. A 2001, 105, 10922–10928. [Google Scholar] [CrossRef]
- Andrushchenko, V.; Anderson, D.T.; Kaminský, J.; Horniíček, J.; Paulson, L.O.; Bouř, P. Solvent dependence of the N-methylacetamide structure and force field. J. Phys. Chem. A 2009, 113, 9727–9736. [Google Scholar] [CrossRef] [PubMed]
- Raucci, U.; Perrella, F.; Donati, G.; Zoppi, M.; Petrone, A.; Rega, N. Ab-initio molecular dynamics and hybrid explicit-implicit solvation model for aqueous and nonaqueous solvents: GFP chromophore in water and methanol solution as case study. J. Comput. Chem. 2020, 41, 2228–2239. [Google Scholar] [CrossRef] [PubMed]
- Cossi, M.; Rega, N.; Scalmani, G.; Barone, V. Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model. J. Comput. Chem. 2003, 24, 669–681. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Ditchfield, R.; Hehre, W.J.; Pople, J.A. Self-Consistent Molecular Orbital Methods. 9. Extended Gaussian-type basis for molecular-orbital studies of organic molecules. J. Chem. Phys. 1971, 54, 724–728. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision A.02; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Barrientos-Salcedo, C.; Lara-Rodríguez, M.; Campos-Fernández, L.; Legorreta-Herrera, M.; Soto-Cruz, I.; Soriano-Correa, C. DFT calculations of electronic structure evaluation and intermolecular interactions of p53-derived peptides with cytotoxic effect on breast cancer. Theor. Chem. Accounts 2021, 140, 121. [Google Scholar] [CrossRef]
- Jono, R.; Watanabe, Y.; Shimizu, K.; Terada, T. Multicanonical ab inito QM/MM molecular dynamics simulation of a peptide in an aqueous environment. J. Comput. Chem. 2010, 31, 1168–1175. [Google Scholar] [CrossRef]
- Berhanu, W.M.; Mikhailov, I.A.; Masunov, A.E. Are density functional theory predictions of the Raman spectra accurate enough to distinguish conformational transitions during amyloid formation? J. Mol. Model. 2010, 16, 1093–1101. [Google Scholar] [CrossRef] [PubMed]
- Gaigeot, M.P.; Besley, N.A.; Hirst, J.D. Modeling the infrared and circular dichroism spectroscopy of a bridged cyclic diamide. J. Phys. Chem. B 2011, 115, 5526–5535. [Google Scholar] [CrossRef] [PubMed]
- De La Rosa, A.F.; Hernández-Meléndez, O.; Saniger-Blesa, J.M.; Nicolás-Vázquez, M.I.; Bárzana, E. Theoretical–experimental prediction of the selectivity between polyamidoamine dendrimers and bioactive peptides derived from amaranth seeds. New J. Chem. 2023, 47, 12170–12181. [Google Scholar] [CrossRef]
- Perrella, F.; Petrone, A.; Rega, N. Second-Order Mass-Weighting Scheme for Atom-Centered Density Matrix Propagation Molecular Dynamics. J. Chem. Theory Comput. 2024, 20, 8820–8832. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Development Version H.37; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Yao, L.; Vogeli, B.; Ying, J.; Bax, A. NMR determination of amide N-H equilibrium bond length from concerted dipolar coupling measurements. J. Am. Chem. 2008, 130, 16518–16520. [Google Scholar] [CrossRef]
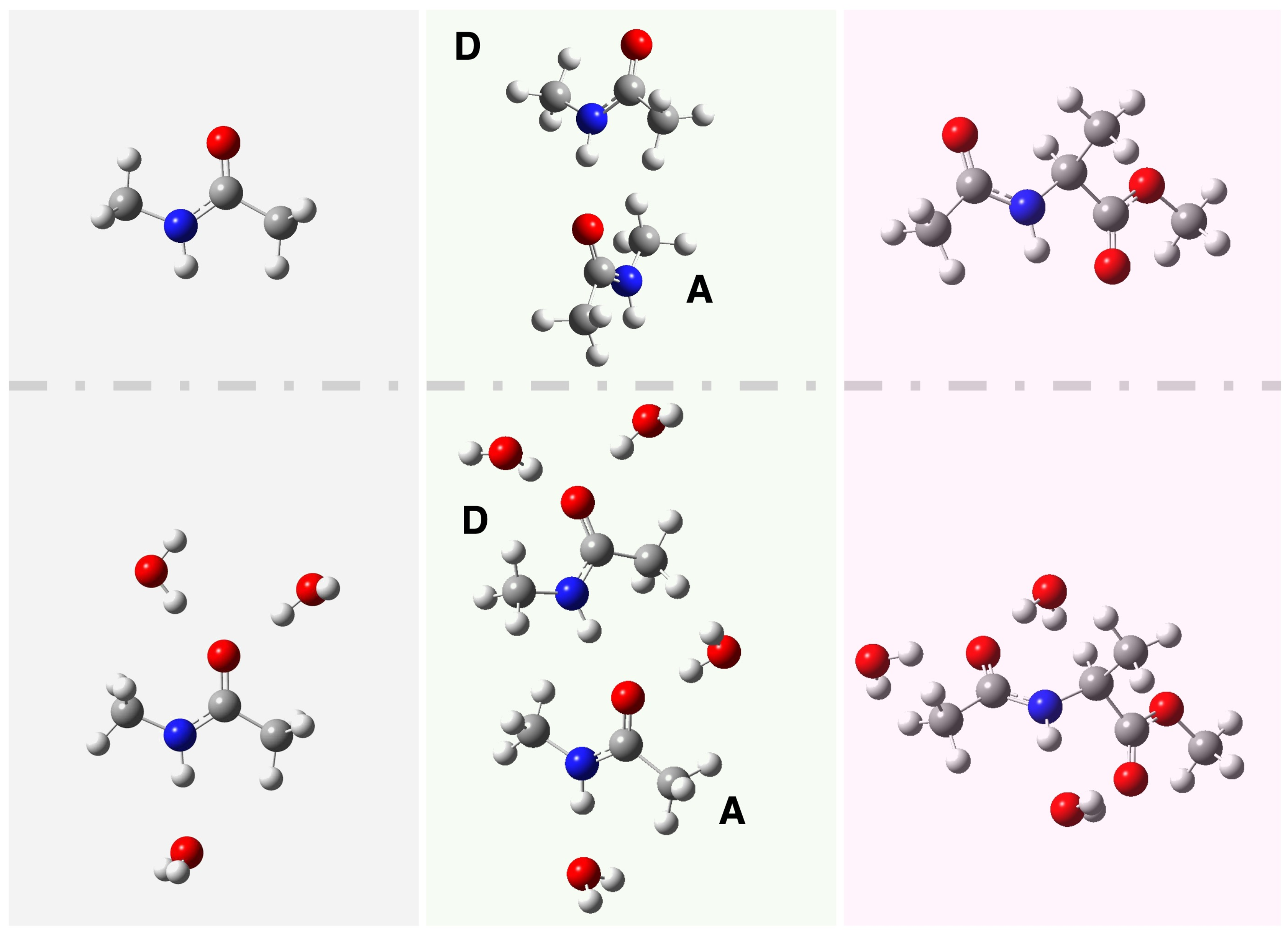

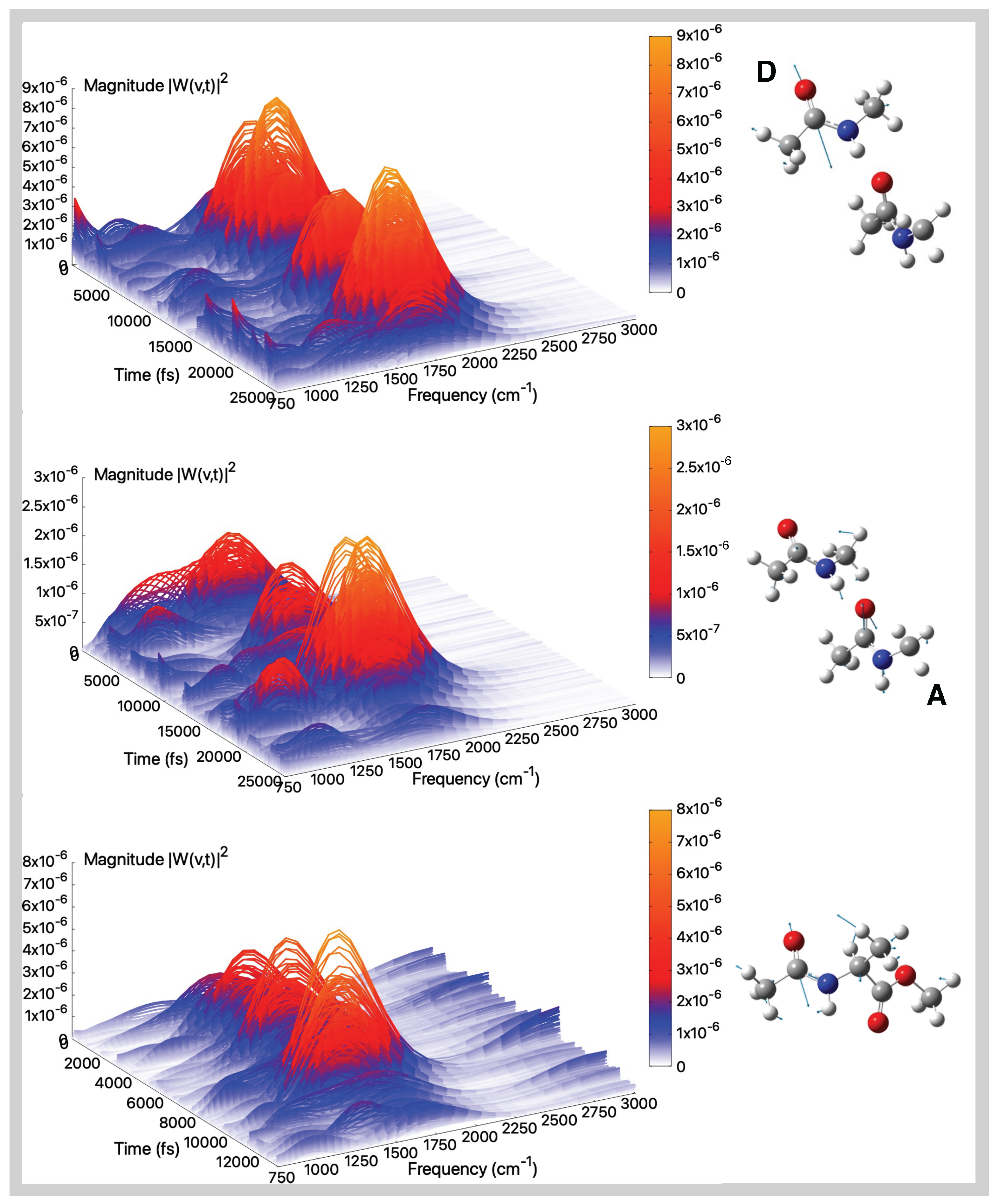


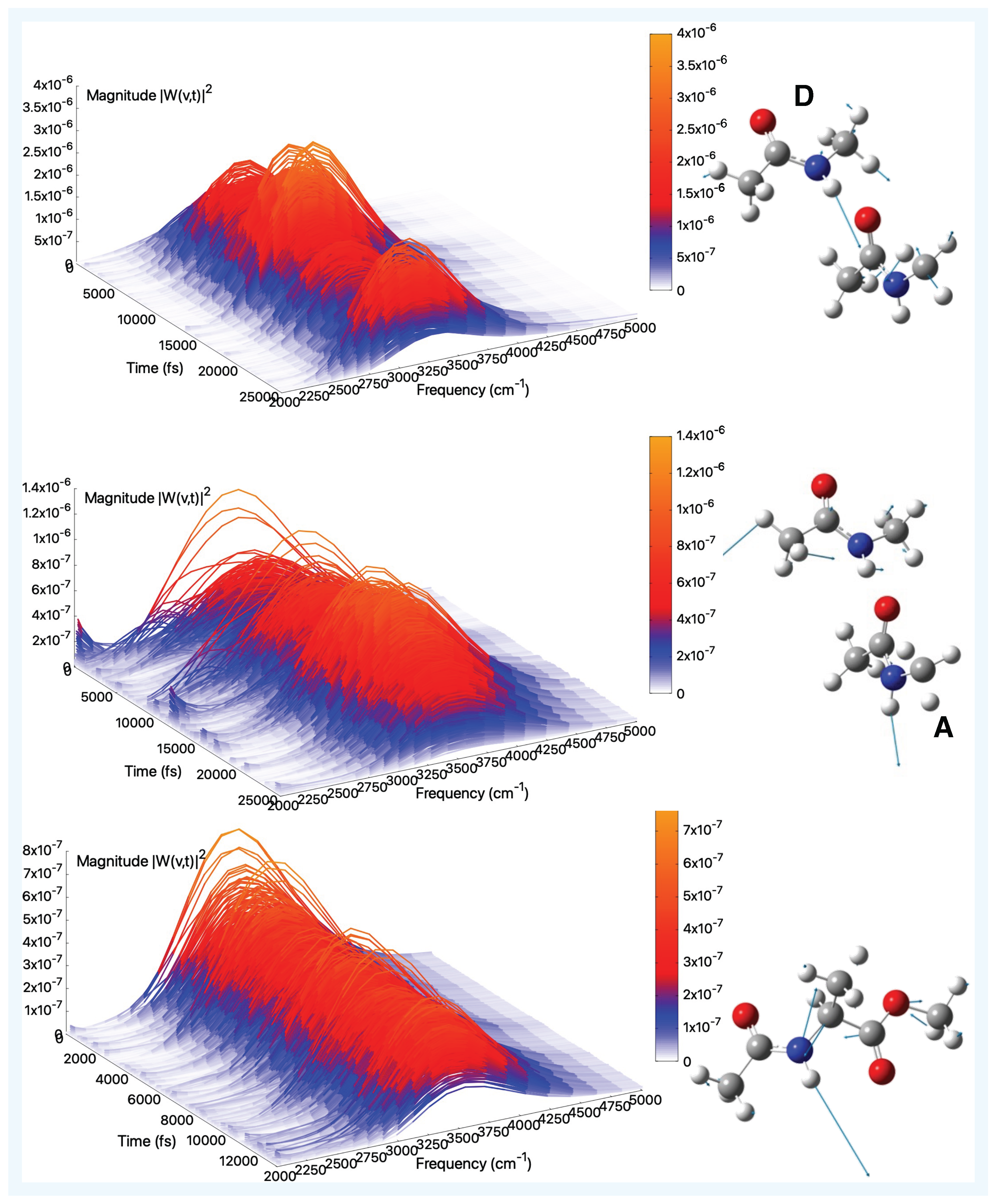

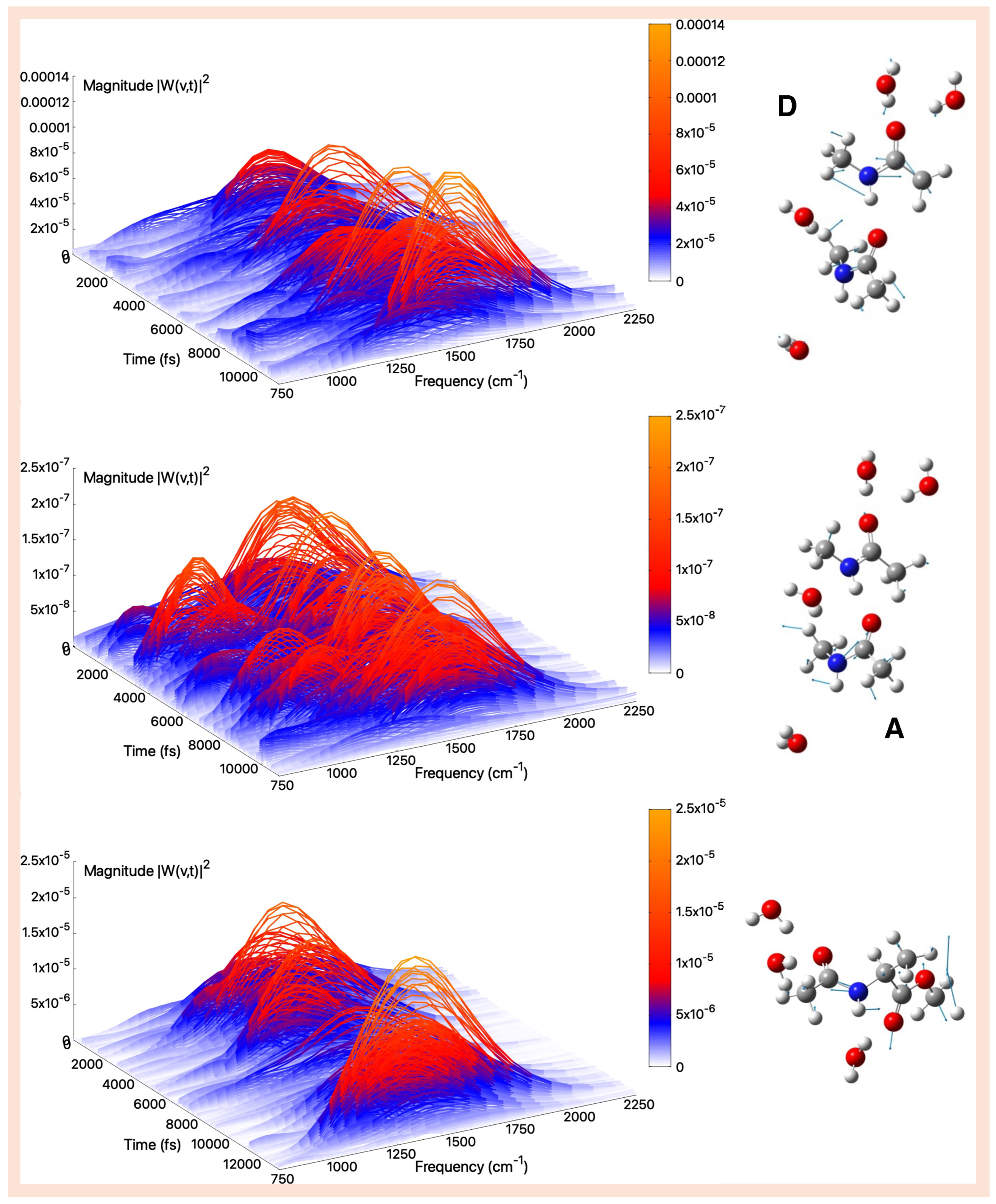



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gas Phase (AIMD) | Aqueous Solution (AIMD) | |||||
|---|---|---|---|---|---|---|
| C=O | C–N | N–H | C=O | C–N | N–H | |
| tNMA | 1.224 | 1.379 | 1.013 | 1.245 | 1.348 | 1.016 |
| (0.011) | (0.029) | (0.025) | (0.019) | (0.024) | (0.007) | |
| tNMA dimer | (A) 1.231 | (A) 1.368 | (A) 1.011 | (A) 1.244 | (A) 1.351 | (A) 1.017 |
| (0.018) | (0.027) | (0.016) | (0.015) | (0.019) | (0.028) | |
| (D) 1.228 | (D) 1.369 | (D) 1.017 | (D) 1.243 | (D) 1.353 | (D) 1.020 | |
| (0.013) | (0.021) | (0.029) | (0.022) | (0.022) | (0.020) | |
| AcAlaOMe | 1.227 | 1.374 | 1.014 | 1.239 | 1.361 | 1.018 |
| (0.018) | (0.025) | (0.017) | (0.022) | (0.028) | [0.010) | |
| Gas Phase | |||
|---|---|---|---|
| tNMA | tNMA Dimer | AcAlaOMe | |
| Amide I | |||
| Static | 1757 | 1743 (A); 1744 (D) | 1737 |
| Dynamics | 1723 | 1698 (A); 1715 (D) | 1708 |
| Refs | 1735 a (calc), 1731 b (exp) | 1700 c (exp) | 1678, 1665 d (exp) |
| Amide II | |||
| Static | 1517 | 1534 (A); 1537 (D) | 1496 |
| Dynamics | 1497 | 1515 (A); 1505 (D) | 1470 |
| Refs | 1522 a (calc), 1499 b (exp) | 1530 c (exp), 1555 e (exp) | / |
| Amide III | |||
| Static | 1254 | 1262 (A); 1259 (D) | 1232 |
| Dynamics | 1244 | 1247 (A); 1243 (D) | 1215 |
| Refs | 1231 a (calc), 1265 b (exp) | 1300 c (exp), 1275 c (exp) | / |
| Amide A | |||
| Static | 3492 | 3484 (A); 3383 (D) | 3451 |
| Dynamics | 3511 | 3533(A); 3167 (D) | 3508 |
| Refs | / | 3374 c (exp), 3318 c (exp), 3498 f (exp) | 3452 d (exp) |
| Aqueous Solution | |||
|---|---|---|---|
| tNMA | tNMA Dimer | AcAlaOMe | |
| Amide I | |||
| Static (C-PCM) | 1728 | 1683 (A); 1702 (D) | 1701 |
| Static (Cluster) | 1588 | 1651 (A); 1617 (D) | 1658 |
| Dynamics | 1620 e | 1655 (A); 1686 (D) | 1680-1723 |
| Refs | 1620 c (exp), 1690 a (calc) | 1655 d (exp) | 1672 b (calc) |
| Amide II | |||
| Static (C-PCM) | 1548 | 1529 (A); 1556 (D) | 1493 |
| Static (Cluster) | 1568 | 1596 (A); 1601 (D) | 1629 |
| Dynamics | 1510 e | 1551 (A); 1549 (D) | 1510 |
| Refs | 1560 c (exp), 1523 a (calc) | 1555 d (exp) | 1514 b (calc) |
| Amide III | |||
| Static (C-PCM) | 1284 | 1259 (A); 1285 (D) | 1231 |
| Static (Cluster) | 1306 | 1312 (A); 1289 (D) | 1337 |
| Dynamics | 1267 e | 1294 (A); 1281 (D) | 1290 |
| Refs | 1300 c (exp), 1272 a (calc) | / | / |
| Amide A | |||
| Static (C-PCM) | 3491 | 3480 (A); 3299 (D) | 3460 |
| Static (Cluster) | 3340 | 3309 (A); 3342 (D) | 3143 |
| Dynamics | 3455 | 3490 (A); 3423, 3441 (D) | 3447 |
| Refs | 3300 c (exp) | 3340 d (exp) | 3464 b (calc) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coppola, F.; Perrella, F.; Petrone, A.; Donati, G.; Marinelli, L.; Rega, N. Unveiling Vibrational Couplings in Model Peptides in Solution by a Theoretical Approach. Molecules 2025, 30, 2854. https://doi.org/10.3390/molecules30132854
Coppola F, Perrella F, Petrone A, Donati G, Marinelli L, Rega N. Unveiling Vibrational Couplings in Model Peptides in Solution by a Theoretical Approach. Molecules. 2025; 30(13):2854. https://doi.org/10.3390/molecules30132854
Chicago/Turabian StyleCoppola, Federico, Fulvio Perrella, Alessio Petrone, Greta Donati, Luciana Marinelli, and Nadia Rega. 2025. "Unveiling Vibrational Couplings in Model Peptides in Solution by a Theoretical Approach" Molecules 30, no. 13: 2854. https://doi.org/10.3390/molecules30132854
APA StyleCoppola, F., Perrella, F., Petrone, A., Donati, G., Marinelli, L., & Rega, N. (2025). Unveiling Vibrational Couplings in Model Peptides in Solution by a Theoretical Approach. Molecules, 30(13), 2854. https://doi.org/10.3390/molecules30132854